Abstract
According to the cancer stem cell (CSC) theory, therapies that do not target the CSC compartment have limited, if any, chances to eradicate established tumors. While cytotoxic T lymphocytes (CTLs) have the potential to recognize and kill single neoplastic cells within a tissue, whether CSCs can be targeted by the immune system during spontaneous or vaccination-elicited responses is poorly defined. Here, we provide experimental evidence showing that CSC lines established from the prostate of transgenic adenocarcinoma of the mouse prostate (TRAMP) mice expressed prostate cancer-associated antigens, MHC Class I and II molecules as well as ligands for natural killer (NK) cell receptors. Indeed, CSC were targets for both NK cell- and CTL-mediated cytotoxicity, both in vitro and in vivo. The administration of dendritic cells pulsed with irradiated CSCs induced a tumor-specific immune response that was more robust than that induced by dendritic cells pulsed with differentiated tumor cells, delayed tumor growth in mice challenged with prostate CSCs and caused tumor regression in TRAMP mice. Thus, CSC are targeted by both innate and adaptive immune responses and might be exploited for the design of novel immunotherapeutic approaches against cancer.
Introduction
Prostate cancer has a dramatic social impact, being the most frequently diagnosed cancer and the second cause of cancer-related mortality among men in economically more developed countries.Citation1 Despite significant progress in the fields of surgery, radiotherapy and chemotherapy, therapeutic options for locally advanced or metastatic prostate cancer remain limited. Androgen deprivation-based therapies currently represent the most effective alternative against metastatic disease but often lead to androgen-independent tumor progression and therapeutic failure (corresponding to the death of patients) within a few years.Citation2 The primary option for castration-resistant disease is chemotherapy, which however has a limited impact on overall survival and is associated with relevant morbidity.Citation3,Citation4 Several promising molecules have recently been approved by the Food and Drug Administration to treat castration-resistant prostate cancer.Citation5
It has been proposed that chemotherapy is not able to eradicate tumors because it is poorly effective against cancer stem cells (CSCs), which may be quiescent and/or develop drug resistance.Citation6 An alternative approach for the treatment of advanced prostate cancer is antigen-specific immunotherapy,Citation7 aimed at eliminating even single cancer cells by inducing a systemic immune response against tumor-associated antigens (TAAs). A relevant advantage of antitumor vaccination with respect to chemotherapy is the lack of relevant toxic side effects.
Several reports have shown that CSCs can be recognized and killed by cytotoxic T lymphocytes (CTLs),Citation8,Citation9 NK cellsCitation10 and γδ T cells in vitro.Citation11 Furthermore, CSCs can be used as antigen sources to elicit dendritic cells (DC)-mediated tumor specific immune responses.Citation12-Citation14 However, little is known about the immunogenicity of CSCs in vivo.
Although prostate CSCs have been isolated from human specimens,Citation15-Citation17 their paucity and indolent growthCitation18 make their immunological characterization particularly difficult, raising the need for appropriate animal models. By applying the NeuroSphere Assay (NSA)Citation19,Citation20 to neoplastic prostate tissues from transgenic adenocarcinoma of the mouse prostate (TRAMP) mice,Citation21 which spontaneously develop a prostatic malignancy that closely resemble the human pathology,Citation22 we generated several cell lines that manifest features generally associated with CSCs, including an endless self-renewal capacity, multi-lineage differentiation and an elevated tumorigenic potential.Citation23 Gene expression analyses showed a remarkable similarity among CSC replicates originating from the neoplastic epithelium (mPIN and adenocarcinoma; n = 6, hereafter named PAC-SCs) and a substantial difference with TRAMP-C1 cells,Citation24 a serum-dependent fully differentiated prostate adenocarcinoma cell line derived from TRAMP mice.Citation23 A remarkable difference in gene expression profiles was also observed between PAC-SCs and CSC lines obtained from prostatic neuroendocrine (NE) tumors (n = 3; hereafter named PNE-SCs) that occasionally develop in TRAMP mice,Citation22 presumably owing to the different nature of epithelial and NE tumors. Indeed, the origin of NE tumors in TRAMP mice is still debated, although the former are thought to arise independently from adenocarcinomas.Citation25,Citation26 In the present study, we analyzed the antigenic and immunogenic properties of these prostatic adenocarcinoma- and prostatic neuroendocrine tumor-derived CSCs in vitro and in vivo.
Results
Phenotypic characterization of CSCs obtained from TRAMP mice
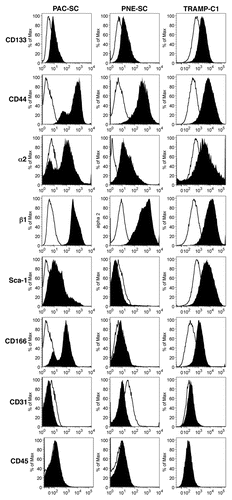
A major issue in CSC biology is whether specific markers exist that allow their unambiguous identification. Thus, PAC- and PNE-SC lines were investigated for the cell-surface expression of several CSC-associated markers, including CD44, CD133, CD166, Sca-1 as well as integrins α2 and β1.Citation15,Citation16,Citation27 Both PAC-SCs and PNE-SCs revealed low but consistent levels of CD133 and expressed to high levels CD44 as well as integrins α2 and β1 (). Only PAC-SCs stained positive for Sca-1 and CD166 expression (). TRAMP-C1 cells expressed all the markers tested above, although some of them were expressed with a different intensity. In particular, the expression of CD44, CD166 and intergrin β1 (to a lesser extent) was higher in PAC-SC than in TRAMP-C1 cells (). All together, these data underline the lack of markers that unequivocally identify prostate CSCs obtained from TRAMP mice. What differentiates prostate CSCs from TRAMP-C1 cells is that the former are not serum-dependent and are not terminally differentiated, but they self-renew and are able to give rise to heterogeneous cancer cell lineages.Citation23,Citation28 As expected, PAC-SCs, PNE-SCs and TRAMP-C1 cells all failed to express lineage markers such as CD31 and CD45 ().
Figure 1. Phenotypic characterization of cancer stem cells. Prostate adenocarcinoma-derived stem cells (PAC-SCs, left column), prostatic neuroendocrine tumor-derived stem cells (PNE-SCs, middle column) and TRAMP-C1 cells (right column) were harvested, dissociated to single-cell suspension, stained with FITC-, PE- or APC-conjugated antibodies specific for the indicated markers and analyzed by cytofluorimetry. Histograms illustrate the expression of specific markers (black profiles). White profiles represent isotype controls. Each panel is representative of three independent experiments.
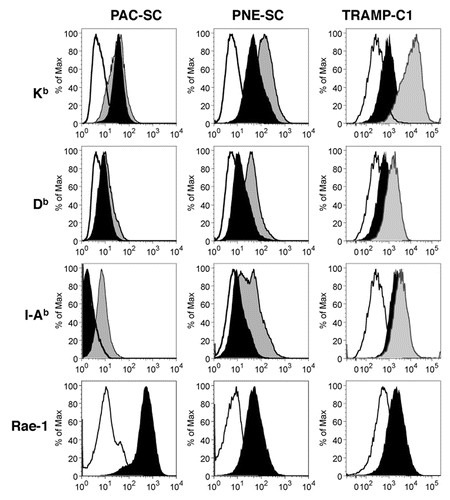
To determine whether cellular effectors of the innate and adaptive immune responses could potentially recognize CSCs, we investigated the expression of MHC molecules and NK-cell receptor ligands on the CSC surface. Both PAC- and PNE-SCs expressed MHC Class I (Kb and Db) molecules comparably to TRAMP-C1 cells, while only PNE-SCs and TRAMP-C1 cells also expressed MHC Class II (I-Ab) molecules (). While the administration of interferon γ (IFNγ) to PAC-SCs only caused the upregulation of I-Ab molecules, the levels of all MHC molecules were increased on the surface of PNE-SCs and TRAMP-C1 cells upon IFNγ stimulation (). All CSC lines also expressed the NK cell-receptor ligand Rae-1Citation29 (), suggesting that—at least under specific conditions—these cells can be recognized by the innate immune system.
Figure 2. Prostate adenocarcinoma-derived and prostatic neuroendocrine tumor-derived stem cell express MHC molecules and other relevant immunologic markers. Prostate adenocarcinoma-derived stem cells (PAC-SCs, left column), prostatic neuroendocrine tumor-derived stem cells (PNE-SCs, middle column) and TRAMP-C1 cells (right column) were cultured in standard medium (black profiles) or in the presence of interferon γ (IFNγ, gray profiles) for 48 h, then harvested and analyzed as described in the legend to . Histograms illustrate the expression of specific markers (black or gray profiles). White profiles represent isotype controls. Each panel is representative of three independent experiments.
CSCs express prostate cancer-associated antigens and can be targeted by antigen-specific CTLs and NK cells
Besides MHC Class I and II molecules, CSCs must express TAAs for being recognizable by tumor-specific T cells. Thus, we next evaluated the expression of prostate cancer-specific antigens on our CSC lines. By RT-PCR, we detected the expression of six transmembrane epithelial antigen of the prostate 1 (STEAP), prostate stem cell antigen (PSCA) and prostatic acid phosphatase (PAP) on both PAC- and PNE-SCs. In this setting, we either used TRAMP-C1 cells or TRAMP prostate cells from as positive controls (Fig. S1A). SV40gp6 large T antigen (Tag) was expressed by PNE-SCs as well as by prostate CSCs obtained from the seminal vesicles of tumor-bearing TRAMP mice, but not by PAC-SCs (Fig. S1A). PAC- and PNE-SCs also clearly expressed the breast cancer resistance protein (BCRP) (Fig. S1A), a marker of pluripotent hematopoietic, muscle, neural stem cells and prostate CSCs.Citation30 The expression of STEAP, PSCA and Tag was confirmed by quantitative RT-PCR () and by immunofluorescence microscpopy (Fig. S1B). Because the origin of tumors originating in the seminal vesicles of TRAMP mice has not yet been clarified, and some authors consider them as independent carcinosarcomatous tumors,Citation31 the corresponding CSCs were abandoned and experiments were conducted only with PAC- and PNE-SCs.
Figure 3. Prostate adenocarcinoma-derived and prostatic neuroendocrine tumor-derived stem cell express prostate-specific antigens and can be targeted in vitro by antigen-specific cytotoxic T lymphocytes and natural killer cells. (A) Expression of STEAP, PSCA or Tag was measured in prostate adenocarcinoma-derived stem cells (PAC-SCs), prostatic neuroendocrine tumor-derived stem cells (PNE-SCs) or TRAMP-C1 cells by real-time PCR and the ΔΔCT method. Expression data are normalized to TRAMP-C1 (for STEAP and PSCA) or B6/K0 (for Tag) cells. (B) Upper left panel. Tag-specific CD8+ T-cell blasts were tested for their cytotoxic activity against PAC-SCs, PNE-SCs, unpulsed (−) or Tag-IV404–411-pulsed RMA cells (+) in a standard 51Cr release assay (effector to target ratio of 50:1). Upper right panel, PSCA-specific CD8+ T-cell blasts were challenged with PAC-SCs, PNE-SCs, unpulsed (−) or PSCA83–91-pulsed RMA cells (+) and analyzed for intracellular interferon γ (IFNγ) production by cytofluorimetry. Data are reported as the percentage of CD44+IFNγ+ cells among CD8+ T cells in each experimental condition tested. Values reported in each column are subtracted of background noise (i.e., IFNγ production against the irrelevant target RMA). Lower panels, representative dot plots. Dot plots are gated on CD8+ T cells. The percentage of CD44+IFNγ+ cells is indicated in each plot. (B) Natural killer (NK) (left panel) and lymphokine-activated killer (LAK) (right panel) cells were tested for their cytotoxic activity against PAC-SCs, PNE-SCs, RMA-S (+) or RMA cells (−) in a standard 51Cr release assay (effector to target ratios of 40:1 or 50:1, in the left and right panel, respectively). (D) 2 × 106 PAC-SCs (left panel) or PNE-SCs (right panel) were injected in 200 μL DMEM into NOD-SCID, nude, wild type (WT) or TRAMP mice (at least 10 mice/group). Mice were monitored for tumor formation and survival, and sacrificed when lesions reached 100 mm2 of area or after 6 mo of observation. Data were compared with ANOVA followed by Tukey’s tests: *p < 0.05; **p < 0.01; ***p < 0.001. Each panel is representative of at least two independent experiments.
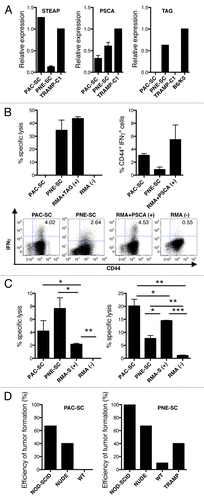
The expression of Tag by PNE-SCs rendered them recognizable by Tag-IV404–411-specific CTLs,Citation32 as shown in standard 51Cr release assays (, upper left panel). To address whether CSCs can be targeted by CTLs specific for TAAs that are commonly found in human prostate cancer, T-cell blasts from mice vaccinated with DCs pulsed with the immunodominant CTL epitope PSCA83–91Citation33 were challenged with CSCs or control tumor cells in a short-term (4 h) IFNγ production assay.Citation34 As shown in (upper right and lower panels), both PAC- and PNE-SCs elicited specific IFNγ production by CD8+ T-cell blasts.
Because both PAC- and PNE-SCs expressed the NK cell- receptor ligand Rae-1 (), we investigated if CSCs would constitute targets for NK-cell cytotoxicity. In an ex vivo assay, we found a modest but consistent lysis of both PAC- and PNE-SCs by purified NK cells (, left panel). In addition, lymphokine-activated killer (LAK) cells generated upon interleukin-2 (IL-2) stimulation in vitro,Citation35 exhibited a very robust cytolytic activity against CSCs (, right panel), confirming that CSCs can be targeted in vitro by effectors of both adaptive and innate immune responses.
Finally, we investigated if CSCs would be targeted by immune effectors also in vivo, by injecting them subcutaneously into mice bearing selected immunodeficiencies. When injected with Matrigel™ plugs, which confer growth support and may prevent immune recognition, all prostate CSCs tested generated tumors in 100% of wild type (WT) mice ( and data not shown). However, Matrigel™ may also contain undefined factors that potentially modulate immunosurveillance. Thus, CSCs were injected subcutaneously in the absence of Matrigel™. In spite of an immunoediting processCitation36 that might have selected for less immunogenic CSCs already during the spontaneous formation of tumors in TRAMP mice from which the CSCs were originally obtained, PAC-SC-induced tumors occurred more frequently in NOD-SCID mice (67%), lacking both B and T cells and exhibiting impaired NK-cell functions, than in nude mice (40%), bearing functional NK cells (, left panel). Being the lack of functional NK cells the substantial immunological difference between NOD-SCID and nude mice, these data indicate that CSCs are susceptible to NK-mediated immunosurveillance in vivo. No tumors developed when PAC-SCs were injected in fully immunocompetent mice (, left panel), pointing to a role for the adaptive immune system in anticancer immunosurveillance in this model.
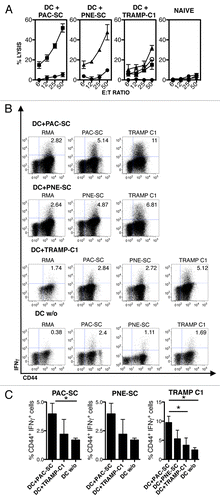
Figure 4. Vaccination with dendritic cells pulsed with prostate adenocarcinoma-derived or prostatic neuroendocrine tumor-derived stem cells elicit an antigen-specific cytotoxic T-lymphocyte response. (A–C) C57BL/6 mice were injected intradermally with 5 × 105 dendritic cells (DCs) pulsed with prostate adenocarcinoma-derived stem cells (DC+PAC-SC), prostatic neuroendocrine tumor-derived stem cells (DC+PNE-SC), TRAMP-C1 cells (DC+TRAMP-C1), unpulsed DCs (DC w/o) or 200 μL of PBS (naive) and killed one week later. (A) Splenocytes were stimulated in vitro and 5-d-old blasts were assessed for cytotoxic activity against PAC-SCs (black squares), PNE-SCs (black triangles), TRAMP-C1 cells (white circles) or RMA cells (black circles) in standard 51Cr release assays. (B) Alternatively, blasts were challenged as indicated and analyzed for intracellular interferon γ (IFNγ) production by cytofluorometry. Dot plots are gated on CD8+ T cells. The percentage of CD44+IFNγ+ cells is indicated in each plot. (C) Histograms depict the quantification of IFNγ production by CD8+ T cells against PAC-SCs (left panel) PNE-SCs (middle panel) or TRAMP-C1 cells (right panel). Values are subtracted of background noise (i.e., IFNγ production against the irrelevant target RMA). Each panel is representative of at least three independent experiments. Data were compared with Student's t tests: *p < 0.05.
Similarly, tumor incidence in mice challenged with PNE-SCs inversely correlated with immune competence (i.e., 100% in NOD-SCID mice, 67% in nude mice, and 10% in WT mice; , right panel). As tumor development and progression in TRAMP mice is associated with the induction of a profound state of selective tolerance against Tag, and tumor-bearing TRAMP mice no longer mount a Tag-specific CTL response upon DC-Tag vaccination,Citation32 we also asked whether tumor-specific tolerance impacted on the susceptibility of TRAMP mice to develop tumors induced by Tag-expressing PNE-SCs (). Indeed, up to 40% of the non-previously immunized 16 week-old TRAMP mice challenged with PNE-SCs developed a tumor (, right panel). Collectively these results suggest that prostate CSC are targeted both by CTLs and NK cells in vivo.
CSCs are sources of antigens for the induction of a tumor-specific immune response
To investigate if CSCs are an adequate source of antigens for the induction of tumor-specific immune responses, DCs were cultured overnight together with PAC-SCs, PNE-SCs or TRAMP-C1 cells undergoing cell death upon γ-irradiation (Fig. S2; DC+PAC-SCs, DC+PNE-SCs or DC+TRAMP-C1, respectively), matured with lipopolysaccharide (LPS) and injected in WT mice, as a model of optimal immunization.Citation37 One week later, splenocytes from immunized mice were restimulated in vitro and tested for the cytotoxic activity and IFNγ production. Blasts from DC+PAC-SC-immunized mice specifically killed PAC-SCs (lytic units: 64.5 ± 30.5 × 106; ), but not syngeneic RMA thymoma cells (). Lysis did not rely on priming in vitro, because blasts from naïve mice failed to exert cytotoxic effects in this system (). In addition, blasts from mice vaccinated with DC+PAC-SCs specifically produced IFNγ upon challenge with PAC-SCs (, upper panels and , left panel). The same blasts produced IFNγ upon challenge with TRAMP-C1 cells (, upper panels and , left panel), suggesting that PAC-SCs and TRAMP-C1 cells share (at least some) TAAs. Conversely, blasts obtained from mice immunized with unpulsed DCs exhibited negligible IFNγ production (, lower panels and ).
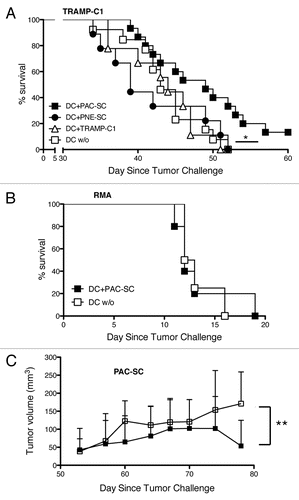
Figure 5. Dendritic cells pulsed with prostate adenocarcinoma-derived stem cells delay the growth of transplantable prostate tumors. (A) C57BL/6 male mice were immunized with 5 × 105 dendritic cells (DCs) pulsed with either prostate adenocarcinoma-derived stem cells (DC+PAC-SC; n = 16, black squares), either prostatic neuroendocrine tumor-derived stem cells (DC+PNE-SC; n = 10, black circles), either TRAMP-C1 cells (DC+TRAMP-C1; n = 10 white triangles) or nothing (DC w/o; n = 14, white squares) and challenged one week later with 2.5 × 106 TRAMP-C1 cells s.c.. Mice were monitored twice a week and sacrificed when the tumor size reached a surface area ≥ 100 mm2. Data are reported in a Kaplan–Maier plot. Statistical comparisons were performed by means of the logrank tests: DC+PAC-SC vs DC+TRAMP-C1, p = 0.044 (*); DC+PAC-SC vs DC w/o, p = 0.029 (*); DC+PAC-SC vs DC+PNE-SC, p = 0.024 (*) All the other comparisons were not statistically significant. (B) C57BL/6 mice were immunized either with DC+PAC-SC (n = 5; black squares) or DC w/o (n = 5; white squares), challenged one week later with 7 × 104 RMA cells and monitored as described in (A). (C) C57BL/6 males were challenged with 2 × 106 PAC-SCs admixed with Matrigel™ s.c. and immunized two weeks later with DC+PAC-SC (n = 10, black squares) or DC w/o (n = 10, white squares). Mice were monitored three times a week for tumor formation and progression, and were killed 78 d after tumor challenge. Data are reported as average ± SD of tumor volume (mm3). Statistical comparisons were performed by means of Student's t-tests: **p < 0.01.
Similarly, but to a lower extent, blasts from mice immunized with DC+PNE-SCs selectively killed PNE-SCs (lytic units: 23.9 ± 19.3 × 106; ) and produced IFNγ when stimulated with PNE-SCs or TRAMP-C1 cells (, middle panel). Interestingly, blasts recovered from mice immunized with DC+TRAMP-C1 were less efficient in killing PAC-SCs, PNE-SCs or TRAMP-C1 cells () and released significantly lower amounts of IFNγ upon specific stimulation with PAC-SCs, PNE-SCs or TRAMP-C1 cells (, right panel) when compared with DC+PAC-SC-elicited blasts, suggesting that PAC-SCs are a better source of TAAs than TRAMP-C1 cells.
Immunization with DCs exposed to dying prostatic adenocarcinoma-derived CSCs elicits a tumor-specific immune response and delays the growth of transplantable prostate tumors
To investigate whether the immune response induced by DCs pulsed with CSCs is effective in vivo, WT mice were immunized with DC+PAC-SCs, DC+PNE-SCs, DC+TRAMP-C1 or unpulsed DCs and challenged one week later with TRAMP-C1 cells. Mice immunized with DC+PAC-SCs exhibited a significant delay in tumor growth and a considerable improvement in survival when compared with mice vaccinated with DC+PNE-SCs, DC+TRAMP-C1 cells or unpulsed DCs (), supporting the hypothesis that CSCs elicit more effective tumor-specific immune responses than differentiated tumor cells. Mice immunized with DC+PAC-SCs were not protected against the growth of RMA thymoma cells (), proving the specificity of vaccination-elicited immune responses. Finally, the infusion of DC+PAC-SCs was assessed as therapeutic vaccination strategy in C56BL/6 mice bearing subcutaneous PAC-SC-derived tumors. To this aim, WT mice were challenged subcutaneously with PAC-SCs admixed with Matrigel™ and immunized two weeks later with DC+PAC-SCs or with unpulsed DCs, as a negative control. When mice were killed (day 78), PAC-SC-elicited tumors were significantly smaller in mice vaccinated with DC+PAC-SCs than in mice unpulsed DCs ().
Taken together, these results suggest that PAC-SCs express antigens shared by prostate CSCs and terminally differentiated tumor cells, and hence can be used for both prophylactic and therapeutic vaccinations. In addition, PAC-SCs are likely to express antigens that are not expressed by TRAMP-C1 cells, and may therefore elicit immune responses that more efficiently target prostate CSCs than those triggered by differentiated tumor cells.
Immunization with DCs pulsed with dying prostatic adenocarcinoma-derived CSCs promotes tumor-specific immune responses and tumor regression in TRAMP mice
The results obtained with DC+PAC-SC vaccination in mice bearing TRAMP-C1- or PAC-SC-derived tumors prompted us to test this approach also in TRAMP mice. Six weeks-old TRAMP mice were immunized once with DC+PAC-SCs or unpulsed DCs and sacrificed at week 16. Mice vaccinated with DC+PAC-SCs exhibited an immune response specific for PAC-SCs and TRAMP-C1 cells that was not detectable in mice immunized with unpulsed DCs (Fig. S3). Moreover, when TRAMP mice of 16 weeks, an age at which they manifest developed prostate cancers and are fully tolerant to Tag,Citation32,Citation38 were immunized with DC+PAC-SCs and sacrificed one week later, a specific immune response against PAC-SCs and TRAMP-C1 cells was still detectable (Fig. S3). Thus, TRAMP mice do not become entirely tolerant to at least some of the antigens expressed by PAC-SCs.
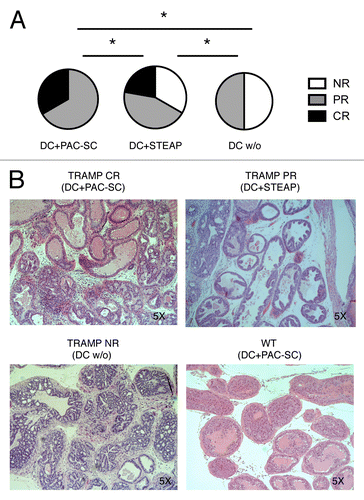
We have previously reported that concomitant tumor- and minor histocompatibility antigen-specific T-cell responses in the context of non-myeloablative allotransplantation from MHC-compatible donors cooperate in rejecting advanced autochthonous tumors in TRAMP mice, provided that transplanted mice are vaccinated with Tag-pulsed DCs immediately upon donor lymphocyte infusion (DLI).Citation34 Thus, we selected this setting to evaluate the potential antitumor efficacy of DC+PAC-SC vaccination in TRAMP mice. To this aim, 16 week-old male TRAMP mice received non-myeloablative (600 Rad) total body irradiation (TBI), and a day later they were transplanted with 1 × 107 bone marrow cells from naïve C57BL/6 female donors. Two weeks later, mice received 6 × 107 splenocytes from naive C57BL/6 female donors (DLI). A day after DLI, mice were immunized with donor-derived DC+PAC-SCs, unpulsed DCs or DC pulsed with the immunodominant CTL epitope of STEAP (DC+STEAP)Citation39 and boosted after additional 3 weeks. We compared DC+PAC-SCs to DC+STEAP and not DC+TRAMP-C1 because the administration of DC+TRAMP-C1 was less effective than that of DC+PAC-SCs in WT mice (; ). Instead, DC-STEAP induced a consistent antigen-specific immune response (Fig. S4). Mice were sacrificed one week after boosting for disease scoring on their urogenital apparata (UGA). As shown in , all transplanted TRAMP mice that received DC+PAC-SCs showed a measurable response. In particular, 33% of these animals showed complete regression (CR) (), as indicated by the presence of well-lined normal epithelial cells and expanded thick-walled tubules with hyalinosis of the fibromuscular wall (which is suggestive of former disease) (, upper left panel). The remaining TRAMP mice receiving DC+PAC-SCs showed signs of partial regression (PR) (), which we previously defined as areas of CR scattered among acini affected by adenocarcinoma.Citation34 Interestingly, only 22% of mice treated with DC+STEAP showed CR, 44% of them underwent PR (, upper right panel) and 33% of these animals had no evidence of tumor regression (NR) (). Half (50%) of the mice treated with unpulsed DCs manifested a PR, presumably due to the DLI, while the remaining half of these animals had no tumor regression (). Indeed, the prostate tubules of TRAMP mice vaccinated with unpulsed DCs appeared enlarged by the presence of well-differentiated adenocarcinomaa with an average disease score of 4+ (, lower left panel), a situation that was comparable with that of non-vaccinated transplanted TRAMP mice.Citation34 Thus, DC+PAC-SC vaccination drives the eradication of spontaneous prostate cancers in TRAMP mice and is more efficient that the administration of STEAP-pulsed DCs.
Figure 6. Dendritic cells pulsed with prostate adenocarcinoma-derived stem cells cooperate with hematopoietic stem cell transplantation and donor lymphocyte infusion in inducing tumor regression in TRAMP mice. (A and B) Sixteen week-old TRAMP mice received total body irradiation (TBI), 1 × 107 bone marrow cells from C57BL/6 female donors (hematopoietic stem cell transplantation, HSCT), 6 × 107 splenocytes from non-presensitized C57BL/6 female donors (donor lymphocyte infusion, DLI), two vaccinations with dendritic cells (DCs) pulsed with either prostate adenocarcinoma-derived stem cells (DC+PAC-SC; n = 6), either STEAP (CD+STEAPn = 9) or nothing (DC w/o; n = 6), and were killed one week after the last vaccination. The urogenital apparata (UGA) of euthanized mice were collected and embedded in paraffin. Slides were stained with hematoxylin and eosin (H&E) (B) and scored by a trained pathologist in a blind fashion (A). Statistical comparisons were performed by means of χ2 tests: *p < 0.05
Discussion
Here, we demonstrate that CSCs isolated from TRAMP mice are antigenic and can be recognized and killed in vitro by CTLs, NK and LAK cells. We also investigated if CSC are targets of immunosurveillance in vivo by challenging immunocompetent and selectively immunodeficent mice with prostate CSCs. It is rather difficult to generate tumors by injecting prostate CSCs, presumably owing to the site of injection, the presence of an existing supporting stroma, the rate of CSC proliferation and the ability of CSCs to favor the generation of an appropriate niche. Thus, a few authors have succeeded in this sense. Abou-Kheir and colleagues injected prostate CSCs orthotopically in nude mice and subcutaneously in NOD-SCID mice to generate tumors.Citation40 However, most frequently prostate CSCs have been combined with urogenital sinus mesenchyme (UGSM) cells, admixed with Matrigel™ or collagen and injected subcutaneously or under the renal capsule to SCID or nude mice.Citation27,Citation41,Citation42 We also obtained evidence that Matrigel™ supports the subcutaneous growth of tumors originating from prostate CSCs, and this is to our knowledge the first report showing the growth of prostate CSC-induced tumors in immunocompetent mice. However, UGSM cells and Matrigel™ may interfere with the immune system by releasing immunosuppressive factors or by impeding the access of immune effectors to malignant cells. To circumvent this issue, CSCs were injected subcutaneously in the absence of UGSM or Matrigel™ support. Consistent with our in vitro results, experiments in immunocompetent and selectively immunodeficient mice clearly showed that the tumorigenicity of CSCs inversely correlate with the presence of effectors of the innate and adaptive immune systems. Indeed, none and only 10% of fully immunocompetent C57BL/6 mice developed tumors when challenged with PAC-SCs and PNE-SCs, respectively. These results may appear surprising in view of the fact that we obtained prostate CSCs from immunocompetent mice, in which immunosurveillance should have already selected for less immunogenic cell variants. Perhaps, in their natural niche CSCs are partially protected by the attacks of the immune system, and undergo limited, if any, immunoediting. Thus, when injected subcutaneously without a stromal support, CSCs are more susceptible to immunosurveillance. We are investigating the possibility that PNE-SCs generate a favorable niche more rapidly than PAC-SCs, which however appears to protect them from the immune system only to limited extents. Indeed, the frequency of PNE-SC-generated tumors increased to 40% in Tag-tolerant TRAMP mice. Finally, the frequency of both PAC- and PNE-SC-generated tumors progressively increased in nude mice and NOD/SCID mice. Taken together and within the limitations of heterotopic transplantable models, these results suggest not only that tumor-specific CTLs are involved in prostate cancer immunoediting in TRAMP mice already at the level of CSCs, but also that an NK cell-mediated immunosurveillance against CSC might be active in this model. Thus, strategies aimed at increasing the antitumor functions of innate immune effects, such as administration of specific cytokines,Citation43 should promote the eradication of prostate tumors.
Prostate CSCs expressed prostate cancer-associated antigens such as PSCA and STEAP, which are selectively upregulated in human prostate cancersCitation44,Citation45 as well as in prostate tumors developing in TRAMP mice.Citation24,Citation46 Hence, CTLs specific for these antigens, if not tolerized by growing cancers, should target both CSCs and tumor cells in vivo. Indeed, both PSCA and STEAP have been successfully exploited in vaccination strategies that have been shown to significantly increase the survival of TRAMP mice.Citation33,Citation39,Citation47 Because CSCs can be targeted by NK as well as T cells and express TAAs that are already exploited in the clinics, one may argue that traditional anticancer vaccines should target both CSCs and more differentiated tumor cells. So why have anticancer vaccines a limited efficacy in patients?Citation43 At least in part, this might depend on the poor general status of patients that have been enrolled so far in clinical trials, and/or on previous therapies that may have irreversibly undermined the patient’s immune system. The tumor itself may also establish a robust state of local and systemic immunosuppression.Citation43 Finally, CSCs, like mesenchimal stem cells,Citation48 might be protected by an immunosuppressive niche and/or be immunosuppressive by themselves.Citation8,Citation49 Thus, CSC-targeting vaccines should be combined with strategies aimed at neutralizing the immunosuppressive microenvironment of the tumor and the CSC niche in particular.
When we used prostate CSCs as a source of antigens for DC-based vaccination, we obtained CSC- and tumor-specific immune responses, although PAC-SC-pulsed DCs turned out to be more immunogenic than their PNE-SC-pulsed counterparts in vivo. This correlated with the ability of CSC-pulsed DCs to delay the growth of TRAMP-C1 tumors. Indeed, only mice vaccinated with PAC-SC-pulsed DCs exhibited a significantly increase in overall survival. This may be explained at least in part by the different nature of epithelial and NE tumors from which PAC-SCs and PNE-SCs were obtained, respectively, suggesting that adenocarcinoma and prostatic NE tumors share only a few TAAs. As a corollary of this hypothesis, PNE-SC-pulsed DCs should be more effective than their PAC-SC-pulsed counterparts against NE tumors.
Our data also support the idea that CSCs express antigens shared with differentiated tumor cells (e.g., TRAMP-C1 cells) and unique TAAs. Indeed, the administration of PAC-SC-pulsed DCs evoked recall responses to TRAMP-C1 tumor cells and vice versa, suggesting that PAC-SCs and TRAMP-C1 cells do share some TAAs recognized by CTLs. However, blasts from mice vaccinated with PAC-SC-pulsed DCs produced significantly higher amounts of IFNγ upon stimulation with either PAC-SCs or TRAMP-C1 cells and exhibited higher cytotoxicity against PAC-SCs than blasts obtained from mice immunized with TRAMP-C1-pulsed DCs. This suggests the existence of unique TAAs expressed by CSCs. Furthermore, PAC-SC-pulsed but not TRAMP-C1-pulsed DCs delayed the growth of TRAMP-C1 cells in vivo. Taken together, our data support the notion that DCs pulsed with CSCs elicit an antitumor immune responses that is both qualitatively and quantitatively superior to that induced by DCs pulsed with differentiated tumor cells.
The administration of PAC-SC-pulsed DCs elicited a measurable immune response also in tumor-bearing TRAMP mice, underscoring the potential of this strategy and indicating that the tolerance against (at least some of) the antigens expressed by PAC-SCs is not as profound as that reported for Tag.Citation32 This is in agreement with previously reported clinical findings.Citation50,Citation51 DC+PAC-SC immunization, when associated to hematopoietic stem cell transplantation and DLI, was also effective in TRAMP mice affected by well-established autochthonous tumors. This protocol promoted a clinical response in all treated mice. The relevant contribution of PAC-SC-pulsed DCs to the success of the treatment is underlined by the fact that only 50% of transplanted mice receiving unpulsed DCs experienced a PR, most likely as a consequence of the DLI, and none of such animals exhibited signs of CR. In a similar setting, DCs pulsed with the immunodominant CTL epitope STEAP186–193 promoted a clinical response that was inferior to that elicited by PAC-SC-pulsed DCs, suggesting that the latter strategy induces an immune response specific for multiple epitopes. Active or adoptive immunotherapeutic strategies aimed at targeting antigens shared by CSCs and differentiated tumor cells should be particularly effective.
While effective, our vaccination strategy did not cure mice challenged with CSCs or differentiated tumor cells, and tumor eradication was not observed in all treated TRAMP mice. Strategies aimed at neutralizing tumor-induced immunosuppressionCitation43 and at favoring the persistence of endogenous or adoptively-transferred tumor-specific T cellsCitation52 are expected to increase the therapeutic efficacy of PAC-SC-pulsed DCs. Further studies are warranted to specifically address this issue.
In conclusion, we demonstrated that CSCs can be targeted by the immune system in vitro and in vivo, and that they are a relevant source of antigens to elicit antitumor immune responses. These data might be useful to design more effective immunotherapeutic strategies against cancer.
Materials and Methods
Mice, cell lines and reagents
Heterozygous TRAMP miceCitation21 were generated by breeding wild-type C57BL/6 male mice and heterozygous female TRAMP mice and were typed for Tag expression by PCR-based screening assay, as previously described.Citation32 C57BL/6, NOD-SCID and nude mice were purchased from Charles River Italy. Animals were housed in a pathogen-free animal facility and treated in accordance with the European Community guidelines and with the approval of the Institutional Ethical Committee.
TRAMP-C1 cells derived from a TRAMP tumor,Citation24 and B6/K-0 embrionic kidney cells expressing TagCitation53 were cultured in DMEM (Lonza) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen), 2 mM L-glutamine, 150 U/mL streptomycin and 200 U/mL penicillin (Cambrex). For tumor challenge, 2.5 × 106 TRAMP-C1 cells were injected in the right flank of male WT mice. RMA, a Rauscher virus-induced thymoma,Citation54 and RMA-S cells, a subclone of RMA that is defective in antigen presentation,Citation55 were cultured in RPMI-1640 (Lonza) supplemented with 10% heat-inactivated FBS (Invitrogen), 2 mM L-glutamine, 150 U/mL streptomycin and 200 U/mL penicillin (Cambrex). The T-cell medium (TCM) was composed by RPMI supplemented with 8% FBS (Invitrogen), 2 mM L-glutamine, 150 U/mL streptomycin, 200 U/mL penicillin (Cambrex), 10 mM HEPES buffer, 10 mM sodium pyruvate and 5 μM β-mercaptoetanol (Gibco-Invitrogen). Prostate CSCs were isolated and cultured in a serum-free medium containing epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF2) as described in Mazzoleni et al.Citation23 Briefly, tissue samples collected from the prostate of TRAMP mice affected by high grade prostatic intraepithelial neoplasia or adenocarcinoma were microscopically dissected and enzimatically digested with collagenase IV (Whortington; 1600 units/mL) for 1 h at 37 °C. Following the removal of small undigested tissue fragments and differential centrifugation, single-cell suspension was seeded in a serum-free medium containing EGF and FGF2.Citation20 Cultures were passaged every 2–10 d, according to the stage of origin. Long-term self-renewal analysis was performed as described in ref. Citation20. For in vivo tumor formation experiments, 2 × 106 PAC- or PNE-SCs were injected in the right flank of male NOD-SCID, nude, C57BL/6 or 16 week-old TRAMP mice. Unless specified, all reagents were from Sigma-Aldrich. Peptides were kindly provided by Renato Longhi (CNR).
Flow cytometry
Single-cell suspensions were incubated with the FcR-blocking reagent (BD Biosciences), labeled with fluorochrome-conjugated monoclonal antibodies or isotype controls (all from BD-Biosciences or BioLegend) and acquired on a BD FACSCanto® cytofluorimeter. Data were analyzed using the FlowJo software. For the intracellular detection of IFNγ, blasts were incubated for 4 h with target cells, in the presence of brefeldin A (Sigma) for the last 3 h, as previously described.Citation32 Cells were then stained for surface markers, fixed with 2% paraformaldehyde (Sigma), and permeabilized with 0.5% saponin (Sigma), before incubation with an anti-IFNγ antibody. To favor blast recognition, TRAMP-C1 cells were cultured in the presence of 40 IU/mL IFNγ for 48 h.
Semi-quantitative RT-PCR and real-time PCR
Total RNA from prostate CSC lines was extracted using the RNeasy Micro and Mini kit (Qiagen). cDNA was obtained by using the Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (Promega). The abundance of β actin-coding cDNAs was used to normalize PCR data. The conditions for the PCR were as follows: 94 °C for 30 sec, annealing temperature (optimized for each primer set) for 1 min, 72 °C for 2 min, 40 cycles. The following primers (Primm) were used: Tag (FWD: CTTGTCAGTGAGGTCCAGATACCTACAG, REV: AGGCATTCCACCACTGCTCCCATTCATC, annealing temperature 58 °C); PSCA (FWD: TTCTCCTGCTGGCCACCTAC, REV: GCAGCTCATCCCTTCACAAT, annealing temperature 58 °C), STEAP (FWD: GGTGGCTGAAGCCGTACTAT, REV: GGATGATATGATGGCAGCGAC, annealing temperature 58 °C), BCRP (FWD: AAATGGAGCACCTCAACCTG, REV: CCCATCACAACGTCATCTTG, annealing temperature 58 °C), PAP (FWD: TCTGGAGAAGTTTGCGGACGTACTGGA, REV: TCAGTTCTGCTACCCAGCGCGTTCTAAC, annealing temperature 54 °C). PCR products were visualized upon separation on a 1.5% or 2.5% agarose gel stained with Sybr-safe (Invitrogen). Real-Time PCR was performed in a total volume of 20 μL using the SYBR Green PCR Master Mix (Applied Biosystems) and 3 μL of cDNA. Specific primers for Tag (FWD: ATGGAAGACTCAGGGCATGAA, REV: TCTACAAATGTGGTATGGCTGATTATG), PSCA (FWD: TCATCTGTGCTGTGCATGAAT, REV: GCTCACTGCAACCATGAAGA) and STEAP (FWD: GTCACTGATCTCCATGACTGCT, REV: GTGGGACTGGGAGTCCGT) were obtained from Primm. The cycler was set as follow: 10 min at 95 °C, followed by 40 cycles at 95 °C for 15 sec and 60 °C for 1 min. To normalize mRNA expression data, L-19 was amplified as a house-keeping gene (FWD: CTGAAGGTCAAAGGGAATGTG, REV: GGACAGAGTCTTGTGATCTC).
Immunocytochemistry (ICC)
Prostate CSC lines were seeded for 48 h at a density of 25.000 cells/cm2 onto Matrigel™-coated (BD-Biosciences) glass coverslips (Ø 12 mm). ICC was performed as previously described.Citation20 Anti-mouse STEAP and PSCA antibodies were from Santa Cruz Biotech, anti-Tag antibodies were from BD-Biosciences. Sections were examined under an Axioscope 40FL microscope (Carl Zeiss).
Immunization protocols and in vivo experiments
DCs were prepared as described elsewhereCitation56 culturing bone marrow precursors for 7 d with 5 ng/mL interleukin-4 (IL-4) and 25 ng/mL granulocyte macrophage colony-stimulating factor (GM-CSF) (R&D Systems). At day 6, DCs were pulsed overnight at a 1:3 ratio with irradiated PAC- or PNE-SCs (50 Gy) or TRAMP-C1 (100 Gy), or left unpulsed, matured in the last 8 h with 1 μg/mL LPS (Sigma), washed and suspended at 2.5 × 106/mL in PBS. Five × 105 DCs were injected i.d. into WT or TRAMP mice. Alternatively, on day 7 of culture, LPS-matured DCs were pulsed for 1 h with 2 μg/mL Tag-IV404–411,Citation53 PSCA83–91Citation33 or STEAP186–193Citation39 peptides and injected i.d. into mice. Mice were sacrificed one week later, and their splenocytes were re-stimulated in vitro for 5 d in the presence of irradiated PAC- or PNE-SCs (10:1 ratio), or of Tag-IV404–411 (1 μg/mL), PSCA83–91 (4 μg/mL) or STEAP186–193 peptides (4 μg/mL), and then tested for IFNγ production and cytotoxic activity as previously described.Citation32,Citation34 For preventive vaccination experiments, mice were challenged with 2.5 × 106 TRAMP-C1 cells s.c. one week after immunization with DC+PAC-SCs, DC+PNE-SCs, DC+TRAMP-C1 or unpulsed DCs. Mice were monitored twice a week and tumor size was measured by two perpendicular diameters and major thickness with a caliper. Animals were killed when the tumor reached a volume ≥ 550 mm3. In the therapeutic vaccination setting, DC+PAC-SCs or unpulsed DCs were injected in C57BL/6 mice that had been challenged with 2 × 106 PAC-SCs diluted 1:1 in Matrigel™ High Concentration (BD-Biosciences; 354248) s.c. two weeks before. Mice were killed 80 d later, and their tumors were measured as described above.
In vitro cytotoxicity assay
Five days upon in vitro restimulation, splenocytes were tested for their cytolytic activity in a standard 4 h 51Cr release assay.Citation32 51Cr release of target cells alone was always < 25% of maximal 51Cr release (target cells in 0.25 M SDS). Lytic units (LUs) were determined as the number of effector cells capable to kill 30% of target cells, and were expressed as 106. NK cells were isolated from the spleen of WT or Rag1−/− mice with anti-DX5 magnetic beadsCitation57 (Miltenyi Biotec). LAK cells were induced by culturing WT splenocytes with 1600 IU/mL IL-2 (R&D Systems) for 7 d.Citation35 Both cell types were used as effector cells for in vitro standard 4 h 51Cr release assay, as described for T-cell blasts.
Hematopoietic stem cell transplantation and tumor specific vaccination
Sixteen week-old TRAMP mice were sub-lethally irradiated (600 rad) and, the day after, they received 1 × 107 viable bone marrow cells i.v. A DLI consisting of 6 × 107 splenocytes was provided 2 weeks later. The following day, mice were immunized with DC+PAC-SCs, unpulsed DCs or DCs pulsed with the STEAP186–193 peptide as described above. Mice received a boost 3 weeks later and were sacrificed after one additional week. Their UGA were embedded in paraffin, processed for immunohistochemistry and scored on coded samples in a blind manner by a pathologist, as previously described.Citation32,Citation34 Briefly, a score of 0 was given to prostates showing CR. A score of 4, corresponding to non-responding tumors, was attributed to lesions characterized by (1) acinar enlargement due to the proliferation of neoplastic cells exhibiting increased nuclear to cytoplasm ratio, (2) nuclear hyperchromasia, (3) cribriform structures invading the lumen and (4) marked proliferation of smooth muscle stromal cells with penetration of malignant Tag+ cells through the basement membrane of the glands into the surrounding stroma. Prostates with areas of CR scattered among acini affected by adenocarcinoma were considered as partially responding.
Statistical analyses
Statistical analyses were performed using the Log-rank, Student’s t, χ2, ANOVA and Tukey’s tests. Statistical significance was defined as: *p < 0.05, **p < 0.01, ***p < 0.001.
| Abbreviations: | ||
| CSC | = | cancer stem cells |
| CTL | = | cytotoxic T lymphocyte |
| DC | = | dendritic cells |
| DLI | = | donor lymphocyte infusion |
| HSCT | = | hematopoietic stem cell transplantation |
| NE | = | neuroendocrine |
| NK | = | natural killer |
| PAC-SC | = | prostatic adenocarcinoma-derived stem cell |
| PNE-SC | = | prostatic NE tumor-derived stem cell |
| TAA | = | tumor-associated antigen |
| TBI | = | total body irradiation |
| TRAMP | = | transgenic adenocarcinoma of the mouse prostate |
| WT | = | wild type |
Additional material
Download Zip (1.2 MB)Acknowledgments
Grant sponsor: Associazione Italiana per la Ricerca sul Cancro (AIRC); Ministero della salute. Elena Jachetti has been awarded a fellowship from AIRC/FIRC. We thank Paolo Dellabona and Maria Pia Protti (San Raffaele Scientific Institute, Milan, Italy) for critical revision of the manuscript. We are indebted with Renato Longhi for peptide synthesis (CNR, Milan, Italy).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary Material
Supplementary materials may be found here:
http://www.landesbioscience.com/journals/oncoimmunology/article/24520
Notes
† These authors contributed equally to this work.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10 - 29; http://dx.doi.org/10.3322/caac.20138; PMID: 22237781
- Smith MR, Kaufman D, George D, Oh WK, Kazanis M, Manola J, et al. Selective aromatase inhibition for patients with androgen-independent prostate carcinoma. Cancer 2002; 95:1864 - 8; http://dx.doi.org/10.1002/cncr.10844; PMID: 12404279
- Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al, TAX 327 Investigators. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004; 351:1502 - 12; http://dx.doi.org/10.1056/NEJMoa040720; PMID: 15470213
- Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr., Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004; 351:1513 - 20; http://dx.doi.org/10.1056/NEJMoa041318; PMID: 15470214
- Antonarakis ES, Eisenberger MA. Expanding treatment options for metastatic prostate cancer. N Engl J Med 2011; 364:2055 - 8; http://dx.doi.org/10.1056/NEJMe1102758; PMID: 21612475
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8:755 - 68; http://dx.doi.org/10.1038/nrc2499; PMID: 18784658
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al, IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010; 363:411 - 22; http://dx.doi.org/10.1056/NEJMoa1001294; PMID: 20818862
- Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res 2010; 16:800 - 13; http://dx.doi.org/10.1158/1078-0432.CCR-09-2730; PMID: 20103663
- Brown CE, Starr R, Martinez C, Aguilar B, D’Apuzzo M, Todorov I, et al. Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res 2009; 69:8886 - 93; http://dx.doi.org/10.1158/0008-5472.CAN-09-2687; PMID: 19903840
- Pietra G, Manzini C, Vitale M, Balsamo M, Ognio E, Boitano M, et al. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int Immunol 2009; 21:793 - 801; http://dx.doi.org/10.1093/intimm/dxp047; PMID: 19491215
- Todaro M, D’Asaro M, Caccamo N, Iovino F, Francipane MG, Meraviglia S, et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J Immunol 2009; 182:7287 - 96; http://dx.doi.org/10.4049/jimmunol.0804288; PMID: 19454726
- Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, Suarez-Merino B, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res 2006; 66:10247 - 52; http://dx.doi.org/10.1158/0008-5472.CAN-06-2048; PMID: 17079441
- Xu Q, Liu G, Yuan X, Xu M, Wang H, Ji J, et al. Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells 2009; 27:1734 - 40; http://dx.doi.org/10.1002/stem.102; PMID: 19536809
- Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res 2012; 72:1853 - 64; http://dx.doi.org/10.1158/0008-5472.CAN-11-1400; PMID: 22473314
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005; 65:10946 - 51; http://dx.doi.org/10.1158/0008-5472.CAN-05-2018; PMID: 16322242
- Guzmán-Ramírez N, Völler M, Wetterwald A, Germann M, Cross NA, Rentsch CA, et al. In vitro propagation and characterization of neoplastic stem/progenitor-like cells from human prostate cancer tissue. Prostate 2009; 69:1683 - 93; http://dx.doi.org/10.1002/pros.21018; PMID: 19644960
- Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-κB signalling. Nat Commun 2011; 2:162; http://dx.doi.org/10.1038/ncomms1159; PMID: 21245843
- Wang ZA, Shen MM. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene 2011; 30:1261 - 71; http://dx.doi.org/10.1038/onc.2010.530; PMID: 21119602
- Rietze RL, Reynolds BA. Neural stem cell isolation and characterization. Methods Enzymol 2006; 419:3 - 23; http://dx.doi.org/10.1016/S0076-6879(06)19001-1; PMID: 17141049
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 2004; 64:7011 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-04-1364; PMID: 15466194
- Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, et al. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A 1995; 92:3439 - 43; http://dx.doi.org/10.1073/pnas.92.8.3439; PMID: 7724580
- Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, Rubin MA, et al. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res 2004; 64:2270 - 305; http://dx.doi.org/10.1158/0008-5472.CAN-03-0946; PMID: 15026373
- Mazzoleni S, Jachetti E, Morosini S, Grioni M, Piras I, Pala M, et al. Gene signatures distinguish stage-specific prostate cancer stem cells from transgenic adenocarcinoma of the mouse prostate (TRAMP) lesions and predict the malignancy of human tumors.. Stem Cells Trans Med 2013;
- Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res 1997; 57:3325 - 30; PMID: 9269988
- Chiaverotti T, Couto SS, Donjacour A, Mao JH, Nagase H, Cardiff RD, et al. Dissociation of epithelial and neuroendocrine carcinoma lineages in the transgenic adenocarcinoma of mouse prostate model of prostate cancer. Am J Pathol 2008; 172:236 - 46; http://dx.doi.org/10.2353/ajpath.2008.070602; PMID: 18156212
- Huss WJ, Gray DR, Tavakoli K, Marmillion ME, Durham LE, Johnson MA, et al. Origin of androgen-insensitive poorly differentiated tumors in the transgenic adenocarcinoma of mouse prostate model. Neoplasia 2007; 9:938 - 50; http://dx.doi.org/10.1593/neo.07562; PMID: 18030362
- Mulholland DJ, Xin L, Morim A, Lawson D, Witte O, Wu H. Lin-Sca-1+CD49fhigh stem/progenitors are tumor-initiating cells in the Pten-null prostate cancer model. Cancer Res 2009; 69:8555 - 62; http://dx.doi.org/10.1158/0008-5472.CAN-08-4673; PMID: 19887604
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006; 66:9339 - 44; http://dx.doi.org/10.1158/0008-5472.CAN-06-3126; PMID: 16990346
- Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol 2000; 1:119 - 26; http://dx.doi.org/10.1038/77793; PMID: 11248803
- Huss WJ, Gray DR, Greenberg NM, Mohler JL, Smith GJ. Breast cancer resistance protein-mediated efflux of androgen in putative benign and malignant prostate stem cells. Cancer Res 2005; 65:6640 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-04-2548; PMID: 16061644
- Yeh IT, Reddick RL, Kumar AP. Malignancy arising in seminal vesicles in the transgenic adenocarcinoma of mouse prostate (TRAMP) model. Prostate 2009; 69:755 - 60; http://dx.doi.org/10.1002/pros.20924; PMID: 19170049
- Degl’Innocenti E, Grioni M, Boni A, Camporeale A, Bertilaccio MT, Freschi M, et al. Peripheral T cell tolerance occurs early during spontaneous prostate cancer development and can be rescued by dendritic cell immunization. Eur J Immunol 2005; 35:66 - 75; http://dx.doi.org/10.1002/eji.200425531; PMID: 15597325
- Garcia-Hernandez MdeL, Gray A, Hubby B, Klinger OJ, Kast WM. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res 2008; 68:861 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-07-0445; PMID: 18245488
- Hess Michelini R, Freschi M, Manzo T, Jachetti E, Degl’Innocenti E, Grioni M, et al. Concomitant tumor and minor histocompatibility antigen-specific immunity initiate rejection and maintain remission from established spontaneous solid tumors. Cancer Res 2010; 70:3505 - 14; http://dx.doi.org/10.1158/0008-5472.CAN-09-4253; PMID: 20388780
- Capobianco A, Manfredi AA, Monno A, Rovere-Querini P, Rugarli C. Melanoma and lymphoma rejection associated with eosinophil infiltration upon intratumoral injection of dendritic and NK/LAK cells. J Immunother 2008; 31:458 - 65; http://dx.doi.org/10.1097/CJI.0b013e318174a512; PMID: 18463539
- Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011; 29:235 - 71; http://dx.doi.org/10.1146/annurev-immunol-031210-101324; PMID: 21219185
- Ronchetti A, Rovere P, Iezzi G, Galati G, Heltai S, Protti MP, et al. Immunogenicity of apoptotic cells in vivo: role of antigen load, antigen-presenting cells, and cytokines. J Immunol 1999; 163:130 - 6; PMID: 10384108
- Degl’Innocenti E, Grioni M, Capuano G, Jachetti E, Freschi M, Bertilaccio MT, et al. Peripheral T-cell tolerance associated with prostate cancer is independent from CD4+CD25+ regulatory T cells. Cancer Res 2008; 68:292 - 300; http://dx.doi.org/10.1158/0008-5472.CAN-07-2429; PMID: 18172322
- Garcia-Hernandez MdeL, Gray A, Hubby B, Kast WM. In vivo effects of vaccination with six-transmembrane epithelial antigen of the prostate: a candidate antigen for treating prostate cancer. Cancer Res 2007; 67:1344 - 51; http://dx.doi.org/10.1158/0008-5472.CAN-06-2996; PMID: 17283172
- Abou-Kheir WG, Hynes PG, Martin PL, Pierce R, Kelly K. Characterizing the contribution of stem/progenitor cells to tumorigenesis in the Pten-/-TP53-/- prostate cancer model. Stem Cells 2010; 28:2129 - 40; http://dx.doi.org/10.1002/stem.538; PMID: 20936707
- Zhou Z, Flesken-Nikitin A, Nikitin AY. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res 2007; 67:5683 - 90; http://dx.doi.org/10.1158/0008-5472.CAN-07-0768; PMID: 17553900
- Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009; 461:495 - 500; http://dx.doi.org/10.1038/nature08361; PMID: 19741607
- Rigamonti N, Bellone M. Prostate cancer, tumor immunity and a renewed sense of optimism in immunotherapy. Cancer Immunol Immunother 2012; 61:453 - 68; http://dx.doi.org/10.1007/s00262-012-1216-6; PMID: 22331081
- Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci U S A 1998; 95:1735 - 40; http://dx.doi.org/10.1073/pnas.95.4.1735; PMID: 9465086
- Hubert RS, Vivanco I, Chen E, Rastegar S, Leong K, Mitchell SC, et al. STEAP: a prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc Natl Acad Sci U S A 1999; 96:14523 - 8; http://dx.doi.org/10.1073/pnas.96.25.14523; PMID: 10588738
- Yang D, Holt GE, Velders MP, Kwon ED, Kast WM. Murine six-transmembrane epithelial antigen of the prostate, prostate stem cell antigen, and prostate-specific membrane antigen: prostate-specific cell-surface antigens highly expressed in prostate cancer of transgenic adenocarcinoma mouse prostate mice. Cancer Res 2001; 61:5857 - 60; PMID: 11479226
- Gray A, de la Luz Garcia-Hernandez M, van West M, Kanodia S, Hubby B, Kast WM. Prostate cancer immunotherapy yields superior long-term survival in TRAMP mice when administered at an early stage of carcinogenesis prior to the establishment of tumor-associated immunosuppression at later stages. Vaccine 2009; 27:Suppl 6 G52 - 9; http://dx.doi.org/10.1016/j.vaccine.2009.09.106; PMID: 20006141
- Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol 2008; 8:726 - 36; http://dx.doi.org/10.1038/nri2395; PMID: 19172693
- Schatton T, Schütte U, Frank NY, Zhan Q, Hoerning A, Robles SC, et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res 2010; 70:697 - 708; http://dx.doi.org/10.1158/0008-5472.CAN-09-1592; PMID: 20068175
- Alves PM, Faure O, Graff-Dubois S, Cornet S, Bolonakis I, Gross DA, et al. STEAP, a prostate tumor antigen, is a target of human CD8+ T cells. Cancer Immunol Immunother 2006; 55:1515 - 23; http://dx.doi.org/10.1007/s00262-006-0165-3; PMID: 16622681
- Thomas-Kaskel AK, Zeiser R, Jochim R, Robbel C, Schultze-Seemann W, Waller CF, et al. Vaccination of advanced prostate cancer patients with PSCA and PSA peptide-loaded dendritic cells induces DTH responses that correlate with superior overall survival. Int J Cancer 2006; 119:2428 - 34; http://dx.doi.org/10.1002/ijc.22097; PMID: 16977630
- Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 2012; 12:269 - 81; http://dx.doi.org/10.1038/nri3191; PMID: 22437939
- Mylin LM, Bonneau RH, Lippolis JD, Tevethia SS. Hierarchy among multiple H-2b-restricted cytotoxic T-lymphocyte epitopes within simian virus 40 T antigen. J Virol 1995; 69:6665 - 77; PMID: 7474076
- Ljunggren HG, Kärre K. Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J Exp Med 1985; 162:1745 - 59; http://dx.doi.org/10.1084/jem.162.6.1745; PMID: 3877776
- Townsend A, Ohlén C, Bastin J, Ljunggren HG, Foster L, Kärre K. Association of class I major histocompatibility heavy and light chains induced by viral peptides. Nature 1989; 340:443 - 8; http://dx.doi.org/10.1038/340443a0; PMID: 2666863
- Camporeale A, Boni A, Iezzi G, Degl’Innocenti E, Grioni M, Mondino A, et al. Critical impact of the kinetics of dendritic cells activation on the in vivo induction of tumor-specific T lymphocytes. Cancer Res 2003; 63:3688 - 94; PMID: 12839960
- Granucci F, Zanoni I, Pavelka N, Van Dommelen SL, Andoniou CE, Belardelli F, et al. A contribution of mouse dendritic cell-derived IL-2 for NK cell activation. J Exp Med 2004; 200:287 - 95; http://dx.doi.org/10.1084/jem.20040370; PMID: 15289500