Abstract
We evaluated a neutralizing anti-TGFβ antibody (GC1008) in cancer patients with malignant pleura mesothelioma (MPM). The goal of this study was to assess immunoregulatory effects in relation to clinical safety and clinical response. Patients with progressive MPM and 1–2 prior systemic therapies received GC1008 at 3mg/kg IV over 90 min every 21 d as part of an open-label, two-center Phase II trial. Following TGFβ blockade therapy, clinical safety and patient survival were monitored along with the effects of anti-TGFβ antibodies on serum biomarkers and peripheral blood mononuclear cells (PBMC). Although designed as a larger trial, only 13 patients were enrolled when the manufacturer discontinued further development of the antibody for oncology indications. All participants tolerated therapy. Although partial or complete radiographic responses were not observed, three patients showed stable disease at 3 mo. GC1008 had no effect in the expression of NK, CD4+, or CD8+ T cell activating and inhibitory markers, other than a decrease in the expression of 2B4 and DNAM-1 on NK cells. However, serum from 5 patients showed new or enhanced levels of antibodies against MPM tumor lysates as measured by immunoblotting. Patients who produced anti-tumor antibodies had increased median overall survival (OS) (15 vs 7.5 mo, p < 0.03) compared with those who did not. To our knowledge, these data represent the first immune analysis of TGFβ- blockade in human cancer patients.
Introduction
Transforming growth factor β (TGFβ) is a 25,000 Da homodimeric protein synthesized and secreted by various normal cells, including macrophages, neutrophils, platelets, subsets of activated lymphocytes, and most transformed cells.Citation1–Citation3 In normal epithelial cells, TGFβ is a potent growth inhibitor and promoter of cellular differentiation through a variety of complex signaling pathways.Citation3,Citation4 Conversely, as tumors develop and progress, they frequently lose their negative growth response to TGFβ, and often produce large amounts of this cytokine. In this setting, TGFβ becomes a potential powerful tumor promoter due to its abilities to stimulate angiogenesis, alter the stromal environment, and importantly, cause local and systemic immunosuppression.Citation1-Citation7
Although the effect of TGFβ blockade has been extensively studied in murine models and in in vitro studies with human cells, there is virtually nothing known about how TGFβ inhibition would affect the human immune system in cancer patients. Based on murine and human in vitro data, it is thought that the overall effect of TGFβ on immune responses is a composite effect on several cellular functions such as T cell proliferation, apoptosis, antigen presentation and differentiation.Citation5-Citation7 Although TGFβ promotes T cells differentiation into T regulatory cells (Tregs),Citation8 its effect on immunoregulatory molecules such as programmed cell death 1 (PDCD1, best known as PD-1) is not known. TGFβ has also been reported to exert a suppressive effect on cells of the innate compartmentCitation5,Citation6 by repressing NK cell proliferation and cytotoxic functionCitation9,Citation10 through inhibition of activating receptors such as natural cytotoxicity triggering receptor 3 (NCR3, also known as NKp30) and killer cell lectin-like receptor subfamily K, member 1 (KLRK1, best known as NKG2D) and of components of the cytotoxic apparatus (i.e., perforin, granzymes and cytotoxins).Citation11-Citation14
Despite two decades of study in preclinical models and in in vitro systems that have identified TGFβ as a promising potential anti-cancer target,Citation3,Citation15,Citation16 few human trials targeting TGFβ for cancer have been conducted and reported (reviewed in Refs. Citation3 and Citation16). One reason for this may be that TGFβ has very complex and context-dependent actions, and thus its inhibition may not only lead to the proposed tumor suppression, but could also affect wound healing, epithelial homeostasis, and inflammation, or could even lead to tumor promotion.Citation1-Citation3
Over the past 10 to 15 y, a large number of biotech and pharmaceutical companies developed potential systemic anti-TGFβ blocking agents (antibodies, soluble receptors, and ALK inhibitors), however, most of these programs have been abandoned, presumably because of the potential difficulties with side effects and the complex regulatory pathway that would be needed for approval.Citation3,Citation16 To our knowledge, the only manuscript published to date, in non-abstract form, describing systemic TGFβ blockade focuses on the use of an antibody, GC1008, in patients with focal segmental glomerulosclerosis,Citation17 however no immunologic data were presented. GC1008 (fresolimumab) is a human IgG4 kappa monoclonal antibody capable of neutralizing all mammalian isoforms of TGFβ (i.e., β1, β2, and β3) with high-affinity.Citation18
We and others have preclinical dataCitation19-Citation22 to support the use of TGFβ blockade in animal models of malignant pleural mesothelioma (MPM), a highly lethal cancer with few effective therapies.Citation23 There is considerable evidence to suggest that MPM may be amenable to immunotherapies.Citation19 Significant levels of TGFβ are produced by murine and rat MPM cell linesCitation24,Citation25 and human MPM cell lines,Citation24,Citation26,Citation27 while high levels of TGFβ have also been documented in tumors of patients with MPMCitation28,Citation29 and in pleural effusions in MPM patients.Citation30
In light of these observations, we designed a Phase II trial of GC1008 in patients with previously treated progressive MPM. Unfortunately, the trial was terminated after only 13 patients were enrolled when the manufacturer discontinued further development of the antibody for oncology indications. Although this small number of patients precluded any definitive conclusions about the clinical efficacy and safety of this approach, we were able to obtain serum and peripheral blood mononuclear cells (PBMC) from the majority of these patients before and after receiving the anti-TGFβ antibody. We thus report, to the best of our knowledge, the first data regarding the immunologic effects of TGFβ blockade in human cancer patients. In particular, we assessed the production of tumor-targeting antibodies, the phenotype of circulating T cells, as well as a series of potential serum biomarkers of GC1008 efficacy.
Results
Patient demographics
Baseline patient characteristics are summarized in and shown for individual patients in Table S1. The study comprised a total of 13 patients (11 males and 2 females), of which 7 were treated at the University of Pennsylvania and 6 at the University of Chicago. All were evaluable for response and toxicity. The median age was 69 y with a range from 46 to 81 y. Eleven patients had epithelial histology, one had biphasic, and one had sarcomatoid histology. The median level of serum mesothelin reactive protein (SMRP) at entry was 2.9 nM with a range from 0.9 to 30.6 nM. Eastern Cooperative Oncology Group (ECOG) performance status was 0 in 10/13 patients and 1 in 3/13.
Table 1. Patient demographics (n = 13)
Treatment delivered
Thirty-six cycles of GC1008 were delivered to the 13 patients. Patients received a median of 2 cycles with a range of 2 to 6 (Table S1). Although the modified Gehan Stage 1 stopping criterion of 1/13 patients with 3 mo progression-free survival (PFS) was exceeded (see below), the trial was ended after 13 patients when the manufacturer discontinued further development of the antibody for oncology indications.
Efficacy
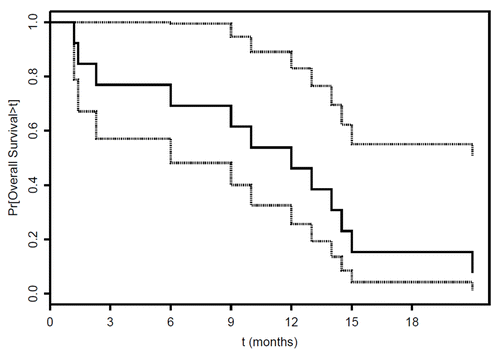
Three out of the 13 subjects treated with GC1008 (23%) met the primary objective of the study (PFS 3 mo), with times to progression of 4.1, 4.2 and 9 mo. Stable disease (SD) was seen in all 3 of these subjects. The median time to progression was 1.4 mo (95% CI 1.2-∞). The median overall survival was 12 mo (). Eight patients received post-study chemotherapy, one patient received radiotherapy, and 4 patients received no further therapy (). At the time of submission of the manuscript, one patient was still alive over 25 mo after his last cycle of GC1008 (Table S1).
Figure 1. Overall survival of GC1008-treated malignant pleural mesothelioma patients. Kaplan-Meier plot of overall survival for 13 patients with malignant pleural mesothelioma (with 95% confidence limits, dotted lines). Median overall survival was 12 mo.
Safety and toxicity profile
Treatment-related toxicities were generally mild (Table S2). Grade 3/4 toxicities included nausea (1 patient), constipation (1 patient), and ileus (1 patient). Two medical events of interest were noted. The risk for each of such events was documented in the informed consent. The first event involved a 69 y old male who received 2 doses of GC1008 and was found to have a dramatic increase in tumor burden on a 6 week staging CT scan (Fig. S1) that exceeded the pace of disease progression prior to enrollment. The second event involved a 77 y old male who received 5 doses of GC1008 and developed two erythematous, verrucous papules in the right shaft and dorsum of the penis. The lesions were biopsied and pathology was reported as “atypical keratinocytic proliferation” (Fig. S2). The skin lesions were classified as a Grade 2 toxicity. Final diagnosis of the pathology by the site pathologists was squamous cell carcinoma. The central pathologist of the trial made a final diagnosis of “atypical keratoacanthoma.”
Pharmacokinetic evaluations
Measurements of GC1008 antibody levels were performed in five patients to confirm previous pharmacokinetic experience.Citation17 Antibody levels were undetectable before therapy. Serum levels on Day 8 averaged 31.1 µg/ml (range 28–34 µg/ml). Levels on Day 21, immediately before the next dose, remained high with an average of 16.2 µg/ml (range 13–19.7 ug/ml). Antibodies to GC1008 were not detected in any patient.
Biochemical evaluations
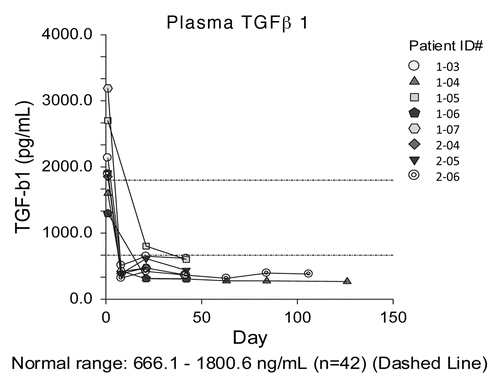
The mean baseline plasma level of TGFβ was 2447 pg/ml (range 1298–6038; individual values are reported in Table S1), which is elevated compared with the average level in normal subjects (1051 pg/ml; n = 42, Genzyme internal validation tests). Baseline levels of TGFβ did not correlate with time-to-progression or overall survival. After treatment with GC1008, TGFβ levels detected by ELISA markedly decreased (), consistent with the specificity of GC1008.
Figure 2. Plasma levels of TGFβ. Plasma transforming growth factor β (TGFβ) levels as detected by ELISA are plotted vs. time after the initiation of GC1008-based immunotherapy for 8 malignant pleural mesothelioma patients.
ELISA assays were performed on sera or plasma from each patient over time including: serum osteopontin, serum hyaluronan, serum MMP-7, serum IL-6, plasma CCL18, plasma VEGF, and plasma PAI-1. There was no consistent change in these markers after treatment with GC1008.
GC1008-elicited anti-tumor immune responses
To assess the ability of GC1008 to induce anti-tumor immune responses, pre-treatment serum and serum obtained 30–60 d after antibody infusion was reacted with purified proteins or protein lysates from human mesothelioma cell lines on nylon membranes (immunoblotting). This time point was chosen since samples were available for all patients and our previous work has shown that peak anti-tumor antibody response to Ad.INF occurred at 1–2 mo after instillation (see methods). Anti-tumor immunoreactivty was assessed by two blinded observers who noted the presence of clear increases in intensity of preexisting bands or new bands as exemplified in . Immunoblots for all 13 of the patients are shown in Figure S3.
Figure 3. Antitumor humoral immune responses. Extracts from four different mesothelioma cell lines were separated by PAGE, transferred to nitrocellulose, and immunoblotted with diluted (1:1500) sera obtained from two malignant pleural mesothelioma patients (1–03 and 1–04) before and 6-weeks after the initiation of GC1008-based immunotherapy. Black boxes highlight increase reactivity as observed in samples obtained in the course of GC1008-based immunotherapy.
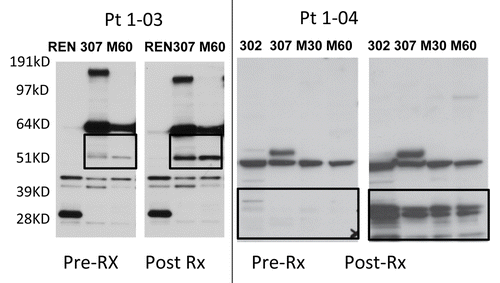
No subjects developed antibodies against the known mesothelioma tumor antigens mesothelin, SV40 T-antigen, or osteopontin. However, serum from five of the 13 subjects (Patients 1–03,1–04,1–05, 1–07 and 2–06) showed new or enhanced antibodies against MPM tumor lysates (Table S1; Figure S3). We repeated the immunoblots in patients 103 and 104 where we had samples from later time points. In Pt 1–03 (who received 2 doses of antibody), the new bands identified at ~51kD () were present at 7 weeks post-therapy, appeared to peak in intensity 19 weeks after therapy, and then diminished at the 31 week time point (Fig. S4A). In Pt 1–04 (who received 6 doses of antibody), new bands were identified at ~28 kD, peaked at 4 weeks after the start of therapy and were largely gone by 11 weeks (Fig. S4B). In the patients exhibiting anti-MPM humoral responses, we also tested pre- and post-treatment serum specimens for their ability to bind to live MPM cell lines by flow cytometry, but detected no binding.
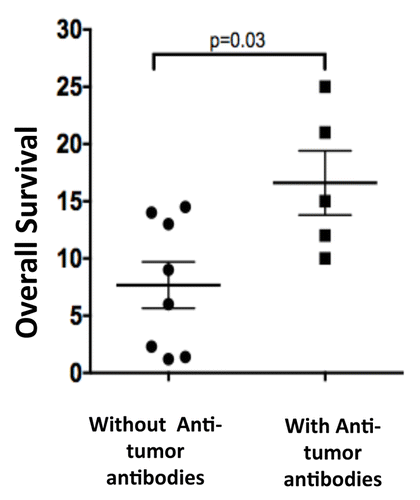
Although there appeared to be no correlation between humoral responses against MPM cell lysates and baseline TGFβ levels, a correlation between clinical responses and the presence of antitumor antibodies was noted. Two of 3 patients exhibiting stable disease developed anti-tumor antibody responses (Table S1). The five subjects who produced anti-tumor antibodies had a statistically significant (p = 0.03) increase in survival compared with the eight patients who did not produce anti-tumor antibodies (). The median OS in the 5 patients who produced anti-tumor antibodies was 15 mo (average OS 16.6 mo), while the median OS in the 8 patients who did not produce anti-tumor antibodies was only 7.5 mo (average OS 7.6 mo).
Figure 4. Relationship between overall survival and the generation of tumor-targeting antibodies. The overall survival (in months) of 5 malignant pleural mesothelioma (MPM) patients who formed antitumor antibodies in response to GC1008-based is compared with that of 8 MPM patients who failed to do so. Data were compared with a Log-rank test, p value is indicated.
Immunological evaluations of PBMC
Given the literature from murine models and in vitro human studies suggesting that TGFβ might inhibit the activity of NK cells and CD8+ T cells, while stimulating the production of CD4+ T-regulatory cells,Citation11-Citation14,Citation32 we measured a panel of NK cell and T cell surface activation and inhibitory markers in PBMC. Of 11 patients with available pre- and post-treatment PBMC samples that were available for flow cytometry, 10 had a sufficient number of lymphocytes for immunological evaluations (patient 2–02 did not have sufficient lymphocytes and hence was excluded from the analysis) (Fig. S5).
To evaluate the immunological profile of NK and T cells, we assayed the expression levels of activation and inhibition markers in 6 healthy individuals (matched with MPM patients in terms of age, gender, and smoking habits) as well as in MPM patients before and after GC1008-based therapy (; Table S3). NK-cell activation markers included NKG2D, NKp30, CD244 (also known as 2B4) and CD266 (best known as DNAM1), while inhibition markers included killer cell lectin-like receptor subfamily D, member 1 (KLRD1, also known as CD94) and killer cell lectin-like receptor subfamily C (KLRC1, better known as NKG2A)Citation31 We observed no significant differences between the healthy subjects and MPM patients in terms of NKG2D, DNAM1, CD94 and NKG2A expression on the surface of NK cells. Conversely, the expression levels of 2B4 and NKp30 on NK cells was reduced by approximately 50% in MPM patients as compared with control individuals. The administration of GC1008 did not alter any of these markers, with the exception of a significant decrease in the levels of 2B4 and DNAM1 (; Table S3).
Table 2. Phenotypic characterization of immune cell subsets by cytofluorometric analysis.
On T cells, we examined expression of the negative immunoregulatory molecule PD-1 and the activation receptors CD38 and NKG2D. PD-1 and NKG2D were lower and CD38 was higher in CD8+ T cells from patients before treatment when compared with controls. However, none of these markers were significantly changed following treatment with GC1008 (; Table S3). Conversely, both CD38 and NKG2D were higher in CD4+ T cells in patients before treatment when compared with controls, but again, treatment with GC1008 had no effect (; Table S3). Finally, the percentage of Tregs (defined as CD4+/FoxP3+ % of CD4+ T cells; see ref. Citation33) was 8.7% in controls and 4.8% in mesothelioma patients (p < 0.001), while no significant change was observed in patients after GC1008 treatment (; Table S3; Figure S6).
Discussion
The primary objectives of this Phase II clinical trial were to examine the safety and the clinical efficacy of GC1008. Unfortunately, the small number of patients enrolled precludes any definitive conclusions for these endpoints. However, with these limitations in mind, there are a number of “first in human” observations that should be of value to the field and in the design of future trials.
On the basis of previous animal studies and two Phase 1 trials,Citation21-Citation23 we used a moderately high dose (3 mg/kg) of anti-TGFβ antibody in our trial. This dose resulted in very high levels of detectable GC1008 in the serum of treated subjects immediately after injection that persisted up to 3 weeks after injection and resulted in a marked fall of TGFβ levels in the blood (), presumably due to antibody binding and clearance. The antibody thus performed well in its ability to reduce serum TGFβ levels.
We found GC1008 to be generally well tolerated by MPM patients with mild fatigue, nausea, and anorexia being the most common toxicities. There were two events of medical interest. One patient developed keratoacanthoma-type skin lesions, an event consistent with prior non-human primate studiesCitation18 and the experience with GC1008 in melanoma.Citation34 TGFβ has been linked to the development of skin carcinomas in animal models of chemical-induced skin carcinogenesis.Citation35 We also observed one patient who exhibited rapid tumor growth after being given GC1008. Although rapid growth of mesotheliomasCitation36 and other malignancies is not clinically unusual, the marked tumor growth in this patient raises the question that GC1008-induced TGFβ blockade may have accelerated tumor progression in this case. It is conceivable- that in a small minority of tumors- TGFβ inhibitory pathways are still intact and that removal of these signals by the antibody results in “release of inhibition” with subsequent rapid tumor growth. Experience with additional patients given long-term TGFβ blockade will be needed to determine how frequently these tumor-accelerating events might occur and to what extent this will limit use of TGFβ blockade (especially for non-malignant conditions).
With regard to clinical efficacy, and again acknowledging the caveats mentioned above, we observed no partial or complete radiographic responses, and the median time to progression was short (1.4 mo), with 10 patients progressing after only two cycles of antibody therapy. Stable disease was noted after 4 cycles in 3 of the 13 patients (23%). Nevertheless, despite the lack of radiographic response and short PFS time, the 12 mo median overall survival was notable, with one patient still alive more than 25 mo after treatment. These clinical responses to GC1008 compare favorably to other Phase II trials in previously-treated advanced MPM,Citation37-Citation41 suggesting that further study of TGFβ inhibition in MPM may be worthwhile.
Although the small number of patients studied limited clinical conclusions, the pre-treatment and post-treatment samples did provide an opportunity to evaluate some of the systemic effects of TGFβ blockade in human cancer patients for the first time. One objective was to assess a number of potential blood biomarkers of TGFβ blocking effect including serum osteopontin, serum hyaluronan, serum MMP-1, serum MMP-7, serum IL-6, as well as plasma CCL18, plasma VEGF, and plasma PAI-1. We were not able to detect any changes after therapy which was unexpected as sera levels of TGFβ did decrease. This suggests that acute changes in TGFβ levels in the blood of cancer patients are not associated with the set of biomarker changes that we predicted might be observed based on animal models.
We were also interested in studying potential immunologic effects of TGFβ blockade. One hypothesis that might explain potential antineoplastic activity of GC1008 is that blockade of TGFβ would release tumor-induced immunosuppression and induce of anti-tumor immune responses. Of note, NK cell function is attenuated in cancer patientsCitation32 and that, at least in vitro, TGFβ downregulates key activating NK cell receptors such as NKp30 and NKG2D, and 2B4.Citation11,Citation32 Furthermore, at least in vitro, anti-TGFβ antibodies can upregulate these receptors from patients with chronic viral infection.Citation42 In our study, MPM patients showed reduced levels of the NK activation markers 2B4 and NKp30 as compared with controls, whereas no difference was observed for the activating receptors NKG2D, and DNAM-1, nor in the inhibitory receptor CD94/NKG2A. However, when assessed 3 weeks after injection of GC1008, upregulation of these receptors was not observed. Overall, these findings confirm an inhibition of NK activation in untreated MPM patients when compared with controls, but we were unable to show any upregulation of these receptors following treatment with anti-TGFβ antibody.
To study non-specific T cell activation, we assessed the levels of PD-1, NKG2D and CD38 on CD4+ and CD8+ T cells. At baseline, we noted significantly lower levels of PD-1 and NKG2D in MPM vs. control patients on CD8+ T cells and significantly higher levels of NKG2D on CD4+ T cells. Furthermore, CD38 on CD4+ and CD8+ T cells was higher in patients when compared with controls. However, there were no significant changes in any of these T cell activation markers were elicited after GC1008 therapy.
Finally, as TGFβ has been implicated in the formation of Tregs and blockade of TGFβ in animal models inhibited their formation,Citation5,Citation43,Citation44 we measured the frequency of circulating CD4+ Tregs (marked by expression of FOXP3Citation33) in the blood of healthy volunteers and MPM patients before and after treatment with GC1008. In contrast to a previous study reporting a similar proportion of CD4+/CD25+ Tregs in peripheral blood of mesothelioma patients and normal healthy controls,Citation45 we found that the levels of circulating Tregs in controls were higher than that seen in MPM patients. A follow-up study in a large patient cohort is required to validate this observation. Nonetheless, of greater relevance to our study, we observed no significant change in the frequency of circulating Tregs upon the administration of GC1008.
Despite our inability to detect changes in PBMC markers expected on the basis of immunoregulatory pathways described in preclinical models, we present three pieces of data to support the hypothesis that TGFβ blockade induced anti-tumor immune response in some of our patients. First, the interesting discordance between short PFS times with increased overall survival times seen in our study is consistent with a growing understanding of immune-based responses, as this pattern has been recently described as characteristic of that seen in vaccines or immune checkpoint blockers.Citation46,Citation47 Second, we were able to detect direct evidence of humoral anti-tumor immune responses. Without a clearly identified tumor antigen in a vaccine setting, it is very difficult to detect specific anti-tumor lymphocyte responses.Citation48 Using a polyclonal immunogene therapy activator (namely, the intratumoral injection of an adenovirus expressing Type I interferon), we have previously quantified the induction of humoral responses against known mesothelioma-associated antigens (i.e., mesothelin, SV40T and WT1) or mesothelioma cell lysates, invariably observing the induction of novel or pre-existing anti-mesothelioma antibodies after immunotherapy.Citation49 A number of other investigators have also used humoral responses to identify anti-tumor responses.Citation50-Citation52 Although the therapeutic importance of these antibodies has not been conclusively established, their appearance indicates that, at least, the humoral activity of CD4 and B cells has been stimulated. Utilizing this approach in our current study, we compared the presence and intensity of immunoreactive bands obtained by exposing MPM cell lysates to pre- vs. post-treatment patient sera, revealing the induction of anti-mesothelioma antibodies in 5 out of 13 patients (; Table S1; Figure S3). Third, we found that the five subjects who did produce anti-tumor antibodies had a statistically significant increase in survival compared with the eight patients who did not produce anti-tumor antibodies (). Taken together, our data are consistent with the hypothesis that blockade of TGFβ can lead to local anti-tumor immune responses, in at least a subpopulation of subjects, and that induction of anti-tumor immunity may be associated with increased survival.
Although the clinical responses of GC1008 used as a single agent were not impressive, we speculate that clinical efficacy might be augmented by using TGFβ blockade in combination with other therapies. There is evidence to indicate that combination of TGFβ blockade with chemotherapy could be synergisticCitation53,Citation54; of note, a combination trial in brain cancer using an ALK5 inhibitor plus or minus lomustine is underway (ClinicalTrials.gov NCT01582269). However, our own preclinical studies,Citation55,Citation56 those of others,Citation57-Citation59 along with the data from this trial suggesting that induction of an anti-tumor immune response may be associated with longer survival, raises the possibility that addition of a second form of immunotherapy (e.g., an anti-tumor vaccine or adoptive T cell transfer) in combination with TGFβ blockade might result in synergy.
In summary, despite the limitations of our trial (including the small sample size and lack of post-treatment tumor samples), we were able to preliminarily address a number of questions regarding the clinical and immunological effects of TGFβ blockade in cancer patients. GC1008 was generally well tolerated in pretreated MPM patients; however, there was a suggestion that blockade of TGFβ might support the growth of susceptible premalignant or malignant cells in a minority of subjects. We also observed stable disease in 3 subjects, all with prior disease progression, and the median overall survival compared favorably to prior single-agent studies in pretreated MPM. Few of the expected changes in the phenotype of circulating T cells were seen, however evidence for humoral anti-tumor immunity was seen in about 40% of the subjects and this response correlated with OS. The heterogeneous clinical responses to anti-TGFβ therapy, either delaying or accelerating disease progression, may indicate a multifactorial role for TGFβ in this context, implying that the clinical outcome of TGFβ-blocking agents may be determined several factors, including the presence of TGFβ-independent immunosuppressive mechanisms. Importantly, our data do not exclude the possibility that anti-TGFβ antibodies may be effectively combined with immunotherapeutic regimens that activate cellular immunity (i.e., anticancer vaccines, antibodies specific for CTLA4 or PD-1), resulting in the elicitation of potent antitumor immune responses.
Materials and Methods
Trial design
This trial was designed as a 40 patient two-center (University of Pennsylvania and University of Chicago), open-label, Phase II trial for patients with relapsed MPM. The ClinicalTrials.gov registration number is NCT01112293. The original objectives were the determination of progression-free survival (PFS) at 3 mo, as well as toxicity and safety of GC1008 in pretreated MPM patients. Secondary endpoints included radiographic response assessment and overall survival (OS). Laboratory objectives included identification of biomarkers of effects of TGFβ blockade and assessment of immune responses against the tumor. The protocol was approved by the University of Pennsylvania Medical Center and University of Chicago Institutional Review Boards and the Food and Drug Administration. Written informed consent, in accordance with the Declaration of Helsinki protocols, was obtained from each patient at the time of screening. An independent safety monitor reviewed safety data after the first three subjects were enrolled. The study was sponsored by the University of Pennsylvania and was funded primarily by the Richard Schulze Family Foundation. The study drug, GC1008, was provided by Genzyme (now Sanofi Oncology).
Eligibility
Eligibility required: 1) pathologically documented MPM, 2) at least one, but no more than two prior systemic therapies, of which at least one contained pemetrexed, 3) documented progressive disease evaluated by Modified RECIST criteria, 4) ECOG Performance status of 0 or 1, 5) serum albumin ≥ 2.5 mg/dL, 6) normal hematologic, hepatic, renal and coagulation parameters, and 7) ability to understand and give informed consent.
Treatment protocol
Based upon previous pharmacokinetic studies,Citation17 GC1008 was administered at a dose of 3 mg/kg every three weeks as a one-hour IV infusion. This dose was chosen based on non-human primate studiesCitation18 and the data from the previous Phase I trial in cancer,Citation34 where maximally tolerated dose of up to 15 mg/kg had been established, but clinical responses were observed in patients at doses of 1 mg/kg or less. In the absence of disease progression or unacceptable toxicity, subsequent courses were repeated every 21 d. Tumor assessments by CT scanning took place every two cycles (every 6 weeks) using Modified RECIST criteriaCitation60 by an experienced thoracic oncologist at each visit.
Patients with confirmed stable disease or responses were continued on treatment every three weeks until either progressive disease (PD) or unacceptable toxicity emerged. Subjects who were stable or responding, but elected to halt therapy, were assessed every three mo thereafter until documented disease progression.
Biochemical measurements
For correlative studies, blood was obtained at baseline and immediately before GC1008 infusions. We obtained plasma samples for detection of TGFβ, platelet factor-4 (PF4), vascular endothelial growth factor (VEGF), CCL-18, and plasminogen activator-1 (PAI-1) levels in CTAD tubes (Becton-Dickinson, Franklin Lakes, NJ) to prevent platelet activation. PF4 is released in significant amounts from platelets as a result of activation and was therefore used as a surrogate marker to assess the quality of samples in which the presence of TGFβ may be due to platelet degranulation related to sample processing.Citation61 Serum markers were collected in SST tubes (Becton-Dickinson) to assess anti-tumor antibody responses (see below), serum mesothelin-related protein (SMRP) levels, osteopontin, hyaluronic acid, matrix metalloprotease-1 (MMP-1), MMP-7, PAI-1, and IL-6. SMRP was measured using an ELISA kit provided by Fujirebio, Inc. (Malvern, PA) All other markers were measured at Genzyme using commercially available ELISA kits. GC1008 concentrations were determined using a validated solid-phase enzyme-linked immunosorbent assay with colorimetric detection.Citation17 Samples were obtained to measure circulating levels of antibodies against GC1008 at baseline and 2 mo after therapy and were measured at Genzyme.Citation17
Immunoblots
To detect anti-TGFβ-induced humoral responses against tumor antigens, we performed immunoblotting against purified proteins and extracts from allogeneic mesothelioma cell lines. Purified SV40 large T antigen protein was purchased from Chimerx. Purified mesothelin was purchased from Raybiotech. Purified recombinant human osteopontin was purchased from Peprotech. Cell lines were derived from patient pleural fluid samples from previous clinical trials and were grown in culture as previously described.Citation49 Extracts from cells or purified proteins were prepared and immunoblotted with patient serum (diluted at 1:1500) from time points before treatment, and 6 weeks to 6 mo after treatment as previously described.Citation49 Multiple exposures were obtained and comparisons were made on the exposures in which the major bands detected on pre-treatment blots were of equal intensity in post-treatment blots. Two independent blinded observers visually scanned each blot to detect new bands or bands that appeared markedly increased in the post-treatment serum. There was complete consensus between reviewers. The blots used for comparisons are shown in Figure S3.
Phenotypic characterization of immune cell subsets by flow cytometry
Cryopreserved peripheral blood mononuclear cells (PBMC) collected prior to treatment and immediately prior to the second injection of GC1008 (21 d after the first dose) available for 11/13 patients were thawed, and natural killer cell (NK) and T cell subsets, as well as activating and inhibitory receptors on these cells, were assessed using mAbs against CD3, CD4, CD8, CD14, CD19, CD20, CD56, CD16, NKG2D, NKG2A, CD94, 2B4, DNAM-1, NKp30, PD1, CD38, CD25 and FOXP3. All mAbs were from BD Biosciences and R&D Systems. PBMCs from a set of six control subjects (without cancer) who were matched for age, gender, and smoking. were also studied. Briefly, PBMC samples were thawed, adjusted to 3 × 106 cells/ml in culture media and 100 μl (3 × 105 cells) were placed into sterile FACS tubes (one for each stain/condition). Subsequently, cells were incubated for 10 min at RT with 10% human serum and 10% serum corresponding to each of the Abs used and then stained with cell surface mAb or corresponding surface isotype mAb for 30 min on ice (see Table S4). Cells for staining with FoxP3 were permeabilized first. Cells were then washed with FACS washing buffer (1 × PBS supplemented with 0.1% BSA and 0.02% NaN3, supplemented with 10% human serum and 10% serum corresponding to each of the Abs used), incubated with 1ml BD FACS Lysing solution (BD Biosciences) for 5 min at RT, washed with 2 ml FACS washing buffer, re-suspended in 100 μl FACS washing buffer and analyzed using an LSRII machine. Analysis was done by collecting a maximum of 100,000 total cells depending on cell availability. Live lymphocytes were defined by size and granularity in FSC and SSC and after exclusion of dead cells by manual gating in FSC/SSC. Detection thresholds were set according to isotype-matched negative controls. Results were expressed as Mean Fluorescent Intensity (MFI) and percent (%) of lymphocytes, NK cells (Lin3-CD56+CD16+), CD3+CD4+ or CD3+CD8+ T cells. Data analysis was performed using FloJo software (Tree Star).
Statistical analysis
The primary endpoint of this study was the fraction of subjects surviving 3 mo without disease progression (3 mo PFS rate). The distributions of PFS and OS were estimated by Kaplan-Meier curves. Cytofluorometric data were described as medians with 25th and 75th interquantiles. Cytofluorometric and biochemical parameters obtained from baseline and treatment values were compared by the paired Wilcoxon test. Cytofluorometric data obtained from treated patients (at baseline) and controls were compared by the two-sample Mann-Whitney test. We compared OS in subjects with and without humoral antibody responses by the logrank test.
| Abbreviations: | ||
| Ad.IFN | = | adenovirus expressing type 1 interferon |
| ECOG PS | = | Eastern Cooperative Oncology Group Performance Status |
| FSC | = | forward scatter |
| IV | = | intravenously |
| MMP | = | matrix metalloprotease |
| MPM | = | malignant pleural mesothelioma |
| NK | = | natural killer |
| OS | = | overall survival |
| PAI-1 | = | plasminogen activator inhibitor-1 |
| PBMC | = | peripheral blood mononuclear cells |
| PD-1 | = | Programmed Cell Death-1 protein |
| PD | = | progressive disease |
| PFS | = | progression-free survival |
| SD | = | stable disease |
| SSC | = | side scatter |
| SMRP | = | serum mesothelin reactive protein |
| TGFβ | = | transforming growth factor β |
| Tregs | = | T regulatory cells |
| VEGF | = | vascular endothelial growth factor |
| WT-1 | = | Wilms tumor -1 gene |
Additional material
Download Zip (15.2 MB)Acknowledgments
The study was primarily funded by the Richard Schulze Family Foundation; additional support was provided by grants P30-CA016520 (DFH, SMA, MJ-S), the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health and Cancer Center Grant (P30 CA10815) (LJM and EP) and an NIEHS funded Environmental Health Sciences Core Center grant P30-ES013508 (SMA). The authors wish to thank: (1) Dr. Frank Hsu, Dr. Scott Lonning, Joan Mannick, Eileen Budri, Cliff Hendrick, and Michael Menconi of Genzyme Corporation for administrative, regulatory, and biomarker analysis support; (2) Dr. Guanjun Cheng at Penn for technical support; (3) Jocelin Joseph, Charity Calloway, Brian Ross, Agnieszka Mackiewicz at the Wistar Institute for technical support; (4) Dr. Anil Vachani at Penn for providing control serum; (5) Drs. Carlos Paz, Brian Pucevich, Vesna Petronic-Rosic, and Maria Tsoukas at the University of Chicago for assistance in interpreting the skin pathology; and Fujirebio Inc. for the SMRP ELISA kits.
Disclosure of Potential Conflicts of Interest
John McPherson is an employee of Sanofi-Genzyme
Citation: Stevenson JP, Kindler HL, Papasavvas E, Sun J, Jacobs-Small M, Hull J, Schwed D, Ranganathan A, Newick K, Heitjan DF, et al. Immunological effects of the TGFβ-blocking antibody GC1008 in malignant pleural mesothelioma patients. OncoImmunology 2013; 2:e26218; 10.4161/onci.26218
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/oncoimmunology/article/26218
References
- Massagué J. TGFbeta in Cancer. Cell 2008; 134:215 - 30; http://dx.doi.org/10.1016/j.cell.2008.07.001; PMID: 18662538
- Tian M, Schiemann WP. The TGF-beta paradox in human cancer: an update. Future Oncol 2009; 5:259 - 71; http://dx.doi.org/10.2217/14796694.5.2.259; PMID: 19284383
- Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov 2012; 11:790 - 811; http://dx.doi.org/10.1038/nrd3810; PMID: 23000686
- Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet 2001; 29:117 - 29; http://dx.doi.org/10.1038/ng1001-117; PMID: 11586292
- Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol 2010; 10:554 - 67; http://dx.doi.org/10.1038/nri2808; PMID: 20616810
- Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell 2008; 134:392 - 404; http://dx.doi.org/10.1016/j.cell.2008.07.025; PMID: 18692464
- Yoshimura A, Wakabayashi Y, Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J Biochem 2010; 147:781 - 92; http://dx.doi.org/10.1093/jb/mvq043; PMID: 20410014
- Tran DQ. TGF-β: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol 2012; 4:29 - 37; http://dx.doi.org/10.1093/jmcb/mjr033; PMID: 22158907
- Rook AH, Kehrl JH, Wakefield LM, Roberts AB, Sporn MB, Burlington DB, Lane HC, Fauci AS. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol 1986; 136:3916 - 20; PMID: 2871107
- Bellone G, Aste-Amezaga M, Trinchieri G, Rodeck U. Regulation of NK cell functions by TGF-beta 1. J Immunol 1995; 155:1066 - 73; PMID: 7636180
- Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, Biassoni R, Bottino C, Moretta L, Moretta A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A 2003; 100:4120 - 5; http://dx.doi.org/10.1073/pnas.0730640100; PMID: 12646700
- Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol 2004; 172:7335 - 40; PMID: 15187109
- Park YP, Choi SC, Kiesler P, Gil-Krzewska A, Borrego F, Weck J, Krzewski K, Coligan JE. Complex regulation of human NKG2D-DAP10 cell surface expression: opposing roles of the γc cytokines and TGF-β1. Blood 2011; 118:3019 - 27; http://dx.doi.org/10.1182/blood-2011-04-346825; PMID: 21816829
- Wilson EB, El-Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL, Cook GP. Human tumour immune evasion via TGF-β blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS One 2011; 6:e22842; http://dx.doi.org/10.1371/journal.pone.0022842; PMID: 21909397
- Biswas S, Criswell TL, Wang SE, Arteaga CL. Inhibition of transforming growth factor-beta signaling in human cancer: targeting a tumor suppressor network as a therapeutic strategy. Clin Cancer Res 2006; 12:4142 - 6; http://dx.doi.org/10.1158/1078-0432.CCR-06-0952; PMID: 16857784
- Connolly EC, Freimuth J, Akhurst RJ. Complexities of TGF-β targeted cancer therapy. Int J Biol Sci 2012; 8:964 - 78; http://dx.doi.org/10.7150/ijbs.4564; PMID: 22811618
- Trachtman H, Fervenza FC, Gipson DS, Heering P, Jayne DR, Peters H, Rota S, Remuzzi G, Rump LC, Sellin LK, et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-β antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int 2011; 79:1236 - 43; http://dx.doi.org/10.1038/ki.2011.33; PMID: 21368745
- Lonning S, Mannick J, McPherson JM. Antibody targeting of TGF-β in cancer patients. Curr Pharm Biotechnol 2011; 12:2176 - 89; http://dx.doi.org/10.2174/138920111798808392; PMID: 21619535
- Suzuki E, Kapoor V, Cheung HK, Ling L, DeLong P, Kaiser LR, Albelda SM. Soluble Type II Transforming Growth Factor-β Receptor inhibits both small and large established murine malignant mesothelioma tumor growth by augmenting host anti-tumor Immunity. Clin Ca Res 2004; 10:5907 - 18; http://dx.doi.org/10.1158/1078-0432.CCR-03-0611
- Fitzpatrick DR, Peroni DJ, Bielefeldt-Ohmann H. The role of growth factors and cytokines in the tumorigenesis and immunobiology of malignant mesothelioma. Am J Respir Cell Mol Biol 1995; 12:455 - 60; http://dx.doi.org/10.1165/ajrcmb.12.5.7742009; PMID: 7742009
- Marzo AL, Fitzpatrick DR, Robinson BWS, Scott B. Antisense oligonucleotides specific for transforming growth factor β2 inhibit the growth of malignant mesothelioma both in vitro and in vivo.. Cancer Res 1997; 57:3200 - 7; PMID: 9242450
- Suzuki E, Kim S, Cheung HK, Corbley M, Zhang X, Sun L, Shan F, Singh J, et al. A novel small molecule inhibitor of TGF-beta type I Receptor Kinase (SM16) inhibits murine mesothelioma tumor growth in vivo and prevents the extent of tumor recurrence after surgical resection. Cancer Res 2007; 67:2351 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-06-2389; PMID: 17332368
- Sterman DH, Albelda SM. Advances in the diagnosis, evaluation, and management of malignant pleural mesothelioma. Respirology 2005; 10:266 - 83; http://dx.doi.org/10.1111/j.1440-1843.2005.00714.x; PMID: 15955137
- Fitzpatrick DR, Peroni DJ, Bielefeldt-Ohmann H. The role of growth factors and cytokines in the tumorigenesis and immunobiology of malignant mesothelioma. Am J Respir Cell Mol Biol 1995; 12:455 - 60; http://dx.doi.org/10.1165/ajrcmb.12.5.7742009; PMID: 7742009
- Kuwahara M, Takeda M, Takeuchi Y, Kuwahara M, Harada T, Maita K. Transforming growth factor β production by spontaneous malignant mesothelioma cell lines derived from Fisher 344 rats. Virchows Arch 2001; 438:492 - 7; http://dx.doi.org/10.1007/s004280000363; PMID: 11407478
- Gerwin BI, Lechner JF, Reddel RR, Roberts AB, Robbins KC, Gabrielson EW, Harris CC. Comparison of production of transforming growth factor-beta and platelet-derived growth factor by normal human mesothelial cells and mesothelioma cell lines. Cancer Res 1987; 47:6180 - 4; PMID: 3479241
- Lauber B, Leuthold M, Schmitter D, Cano-Santos J, Waibel R, Stahel RA. An autocrine mitogenic activity produced by a pleural human mesothelioma cell line. Int J Cancer 1992; 50:943 - 50; http://dx.doi.org/10.1002/ijc.2910500621; PMID: 1555893
- Kumar-Singh S, Weyler J, Martin MJH, Vermeulen PB, Van Marck E. Angiogenic cytokines in mesothelioma: a study of VEGF, FGF-1 and -2, and TGF beta expression. J Pathol 1999; 189:72 - 8; http://dx.doi.org/10.1002/(SICI)1096-9896(199909)189:1<72::AID-PATH401>3.0.CO;2-0; PMID: 10451491
- Jagirdar J, Lee TC, Reibman J, Gold LI, Aston C, Bégin R, Rom WN. Immunohistochemical localization of transforming growth factor beta isoforms in asbestos-related diseases. Environ Health Perspect 1997; 105:Suppl 5 1197 - 203; PMID: 9400723
- DeLong P, Carroll RG, Henry AC, Tanaka T, Ahmad S, Leibowitz MS, Sterman DH, June CH, Albelda SM, Vonderheide RH. Regulatory T cells and cytokines in malignant pleural effusions secondary to mesothelioma and carcinoma. Cancer Biol Ther 2005; 4:342 - 6; http://dx.doi.org/10.4161/cbt.4.3.1644; PMID: 15846066
- Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol 2013; 31:227 - 58; http://dx.doi.org/10.1146/annurev-immunol-020711-075005; PMID: 23516982
- Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol 2010; 12:7 - 13; http://dx.doi.org/10.1093/neuonc/nop009; PMID: 20150362
- Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ, Dudley ME, Liu F, Schrump DS, Steinberg SM, Rosenberg SA, Robbins PF. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood 2012; 119:5688 - 96; http://dx.doi.org/10.1182/blood-2011-10-386482; PMID: 22555974
- Morris JC, Shapiro AR, Tan DP et al. Phase I/II study of GC1008: A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody (MAb) in patients with advanced malignant melanoma (MM) or renal cell carcinoma (RCC). J Clin Oncol 26: 2008 (May 20 suppl; abstr 9028).
- Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, Akhurst RJ. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 1996; 86:531 - 42; http://dx.doi.org/10.1016/S0092-8674(00)80127-0; PMID: 8752208
- Kaira K, Dobashi K, Yanagitani N, Sunaga N, Shimizu Y, Hisada T, Ishizuka T, Nakajima T, Mori M. Rapid growth of malignant pleural mesothelioma. J Thorac Oncol 2007; 2:966 - 7; http://dx.doi.org/10.1097/JTO.0b013e31811f474e; PMID: 17909361
- Zucali PA, Simonelli M, Michetti G, Tiseo M, Ceresoli GL, Collovà E, Follador A, Lo Dico M, Moretti A, De Vincenzo F, et al. Second-line chemotherapy in malignant pleural mesothelioma: results of a retrospective multicenter survey. Lung Cancer 2012; 75:360 - 7; http://dx.doi.org/10.1016/j.lungcan.2011.08.011; PMID: 21937142
- Dubey S, Jänne PA, Krug L, Pang H, Wang X, Heinze R, Watt C, Crawford J, Kratzke R, Vokes E, et al. A phase II study of sorafenib in malignant mesothelioma: results of Cancer and Leukemia Group B 30307. J Thorac Oncol 2010; 5:1655 - 61; http://dx.doi.org/10.1097/JTO.0b013e3181ec18db; PMID: 20736856
- Fennell DA, McDowell CM, Busacca S, Webb G, Moulton B, Cakana A, O’Byrne KJ, Meerbeeck JV, Donnellan P, McCaffrey J, et al. Phase II clinical trial of first or second-line treatment with bortezomib in patients with malignant pleural mesothelioma. J Thorac Oncol 2012; 7:1466 - 70; http://dx.doi.org/10.1097/JTO.0b013e318260dfb9; PMID: 22895144
- Stebbing J, Powles T, McPherson K, Shamash J, Wells P, Sheaff MT, Slater S, Rudd RM, Fennell D, Steele JP. The efficacy and safety of weekly vinorelbine in relapsed malignant pleural mesothelioma. Lung Cancer 2009; 63:94 - 7; http://dx.doi.org/10.1016/j.lungcan.2008.04.001; PMID: 18486273
- Nowak AK, Millward MJ, Creaney J, Francis RJ, Dick IM, Hasani A, van der Schaaf A, Segal A, Musk AW, Byrne MJ. A phase II study of intermittent sunitinib malate as second-line therapy in progressive malignant pleural mesothelioma. J Thorac Oncol 2012; 7:1449 - 56; http://dx.doi.org/10.1097/JTO.0b013e31825f22ee; PMID: 22895142
- Sun C, Fu B, Gao Y, Liao X, Sun R, Tian Z, Wei H. TGF-β1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PLoS Pathog 2012; 8:e1002594; http://dx.doi.org/10.1371/journal.ppat.1002594; PMID: 22438812
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003; 198:1875 - 86; http://dx.doi.org/10.1084/jem.20030152; PMID: 14676299
- Petrausch U, Jensen SM, Twitty C, Poehlein CH, Haley DP, Walker EB, Fox BA. Disruption of TGF-beta signaling prevents the generation of tumor-sensitized regulatory T cells and facilitates therapeutic antitumor immunity. J Immunol 2009; 183:3682 - 9; http://dx.doi.org/10.4049/jimmunol.0900560; PMID: 19692636
- Meloni F, Morosini M, Solari N, Passadore I, Nascimbene C, Novo M, Ferrari M, Cosentino M, Marino F, Pozzi E, et al. Foxp3 expressing CD4+ CD25+ and CD8+CD28- T regulatory cells in the peripheral blood of patients with lung cancer and pleural mesothelioma. Hum Immunol 2006; 67:1 - 12; http://dx.doi.org/10.1016/j.humimm.2005.11.005; PMID: 16698419
- Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbé C, Maio M, Binder M, Bohnsack O, Nichol G, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009; 15:7412 - 20; http://dx.doi.org/10.1158/1078-0432.CCR-09-1624; PMID: 19934295
- Madan RA, Gulley JL, Fojo T, Dahut WL. Therapeutic cancer vaccines in prostate cancer: the paradox of improved survival without changes in time to progression. Oncologist 2010; 15:969 - 75; http://dx.doi.org/10.1634/theoncologist.2010-0129; PMID: 20798195
- Disis ML. Immunologic biomarkers as correlates of clinical response to cancer immunotherapy. Cancer Immunol Immunother 2011; 60:433 - 42; http://dx.doi.org/10.1007/s00262-010-0960-8; PMID: 21221967
- Sterman DH, Recio AR, Carroll RG, Gillespie CT, Haas A, Vachani A, Kapoor V, Sun J, Hodinka R, Brown JL, et al. A phase I clinical trial of single-dose intrapleural IFN-beta gene transfer for malignant pleural mesothelioma and metastatic pleural effusions: high rate of antitumor immune responses. Clin Cancer Res 2007; 13:4456 - 66; http://dx.doi.org/10.1158/1078-0432.CCR-07-0403; PMID: 17671130
- Marina O, Hainz U, Biernacki MA, Zhang W, Cai A, Duke-Cohan JS, Liu F, Brusic V, Neuberg D, Kutok JL, et al. Serologic markers of effective tumor immunity against chronic lymphocytic leukemia include nonmutated B-cell antigens. Cancer Res 2010; 70:1344 - 55; http://dx.doi.org/10.1158/0008-5472.CAN-09-3143; PMID: 20124481
- Fonseca C, Soiffer R, Ho V, Vanneman M, Jinushi M, Ritz J, Neuberg D, Stone R, DeAngelo D, Dranoff G. Protein disulfide isomerases are antibody targets during immune-mediated tumor destruction. Blood 2009; 113:1681 - 8; http://dx.doi.org/10.1182/blood-2007-09-114157; PMID: 19008459
- Nemunaitis J, Jahan T, Ross H, Sterman D, Richards D, Fox B, Jablons D, Aimi J, Lin A, Hege K. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther 2006; 13:555 - 62; http://dx.doi.org/10.1038/sj.cgt.7700922; PMID: 16410826
- Ohmori T, Yang JL, Price JO, Arteaga CL. Blockade of tumor cell transforming growth factor-betas enhances cell cycle progression and sensitizes human breast carcinoma cells to cytotoxic chemotherapy. Exp Cell Res 1998; 245:350 - 9; http://dx.doi.org/10.1006/excr.1998.4261; PMID: 9851876
- Taniguchi Y, Kawano K, Minowa T, Sugino T, Shimojo Y, Maitani Y. Enhanced antitumor efficacy of folate-linked liposomal doxorubicin with TGF-β type I receptor inhibitor. Cancer Sci 2010; 101:2207 - 13; http://dx.doi.org/10.1111/j.1349-7006.2010.01646.x; PMID: 20608940
- Kim S, Buchlis G, Fridlender ZG, Sun J, Kapoor V, Cheng G, Haas A, Cheung HK, Zhang X, Corbley M, et al. Systemic blockade of transforming growth factor-β signaling augments the efficacy of immunogene therapy. Cancer Res 2008; 68:10247 - 56; http://dx.doi.org/10.1158/0008-5472.CAN-08-1494; PMID: 19074893
- Wallace A, Kapoor V, Sun J, Mrass P, Weninger W, Heitjan DF, et al. TGF-β receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid tumors. Clin Cancer Res 2008; 14:3966 - 74; http://dx.doi.org/10.1158/1078-0432.CCR-08-0356; PMID: 18559619
- Terabe M, Ambrosino E, Takaku S, O’Konek JJ, Venzon D, Lonning S, McPherson JM, Berzofsky JA. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin Cancer Res 2009; 15:6560 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-09-1066; PMID: 19861451
- Takaku S, Terabe M, Ambrosino E, Peng J, Lonning S, McPherson JM, Berzofsky JA. Blockade of TGF-beta enhances tumor vaccine efficacy mediated by CD8(+) T cells. Int J Cancer 2010; 126:1666 - 74; PMID: 19830696
- Llopiz D, Dotor J, Casares N, Bezunartea J, Díaz-Valdés N, Ruiz M, Aranda F, Berraondo P, Prieto J, Lasarte JJ, et al. Peptide inhibitors of transforming growth factor-beta enhance the efficacy of antitumor immunotherapy. Int J Cancer 2009; 125:2614 - 23; http://dx.doi.org/10.1002/ijc.24656; PMID: 19530254
- Byrne MJ, Nowak AK. Modified RECIST criteria for assessment of response in malignant pleural mesothelioma. Ann Oncol 2004; 15:257 - 60; http://dx.doi.org/10.1093/annonc/mdh059; PMID: 14760119
- O’Brien PJ, Ramanathan R, Yingling JM, Baselga J, Rothenberg ML, Carducci M, Daly T, Adcock D, Lahn M. Analysis and variability of TGFbeta measurements in cancer patients with skeletal metastases. Biologics 2008; 2:563 - 9; PMID: 19707386