
Abstract
Dasatinib (DAS) is a potent inhibitor of the BCR-ABL, SRC, c-KIT, PDGFR, and ephrin tyrosine kinases that has demonstrated only modest clinical efficacy in melanoma patients. Given reports suggesting that DAS enhances T cell infiltration into the tumor microenvironment, we analyzed whether therapy employing the combination of DAS plus dendritic cell (DC) vaccination would promote superior immunotherapeutic benefit against melanoma. Using a M05 (B16.OVA) melanoma mouse model, we observed that a 7-day course of orally-administered DAS (0.1 mg/day) combined with a DC-based vaccine (VAC) against the OVA257–264 peptide epitope more potently inhibited tumor growth and extended overall survival as compared with treatment with either single modality. The superior efficacy of the combinatorial treatment regimen included a reduction in hypoxic-signaling associated with reduced levels of immunosuppressive CD11b+Gr1+ myeloid-derived suppressor cells (MDSC) and CD4+Foxp3+ regulatory T (Treg) populations in the melanoma microenvironment. Furthermore, DAS + VAC combined therapy upregulated expression of Type-1 T cell recruiting CXCR3 ligand chemokines in the tumor stroma correlating with activation and recruitment of Type-1, vaccine-induced CXCR3+CD8+ tumor-infiltrating lymphocytes (TILs) and CD11c+ DC into the tumor microenvironment. The culmination of this bimodal approach was a profound “spreading” in the repertoire of tumor-associated antigens recognized by CD8+ TILs, in support of the therapeutic superiority of combined DAS + VAC immunotherapy in the melanoma setting.
Introduction
A broad range of small molecule kinase inhibitors have been developed as anticancer therapies due to their abilities to antagonize signaling pathways associated with tumor growth, survival, and metastasis, as well as those regulating tumor-associated neo-angiogenesis.Citation1–Citation5 More recently, a number of these inhibitors have also been observed to modulate stromal cell populations, including immune cells, present within the tumor microenvironment, hence expanding appreciation of the clinical utility of these drugs as immune modulators.Citation6–Citation16
Dasatinib (DAS) is a broad-spectrum multi-kinase inhibitor that exhibits modest single-agent clinical efficacy in patients afflicted with diverse forms of cancer, including melanoma.Citation3–Citation5,Citation17,Citation18 Interestingly, although DAS has been reported to inhibit T-cell activation via blockade of lymphocyte-specific protein tyrosine kinase (Lck)-mediated proximal T-cell receptor (TCR) signaling in vitro,Citation19,Citation20 upon administration in vivo, DAS can profoundly enhance T effector cell activation, expansion, and function.Citation21–Citation25 Indeed, pre-clinical modeling suggests that T cells play a dominant role in the antitumor activity of DAS treatment in vivo,Citation21 providing rationale for the development of potential immunotherapeutic applications. In this particular context, Yang et al.Citation21 have also recently reported that the treatment of BALB/c mice bearing P815 mastocytomas with DAS improved recruitment of T cells into the tumor and surrounding stroma, an immunological response also required to achieve the clinical benefit associated with therapeutic vaccines.Citation11,Citation12,Citation26 Consequently, we hypothesized a combinatorial protocol, comprising specific vaccination to activate and expand tumoricidal CD8+ T cells plus systemic administration of DAS to facilitate the refined targeting of vaccine-induced T effector cells to the tumor microenvironment, would be therapeutically superior to monotherapy.
In the current study, we utilized mice bearing established sub-cutaneous M05 (B16.OVA) melanoma to experimentally test this hypothesis. We report here that bimodal therapy with the combination of tumor-specific (OVA peptide) DC-based vaccine (VAC) + DAS provide superior antitumor benefit as compared with treatment using either single-agent modality alone. Our results provide an experimental basis for the clinical translation of combined DAS + VAC immunotherapies for the treatment of cancer, particularly melanoma.
Results
DAS monotherapy mediates substantial anti-melanoma activity in association with enhanced CD8+ T cell infiltration into the tumor microenvironment
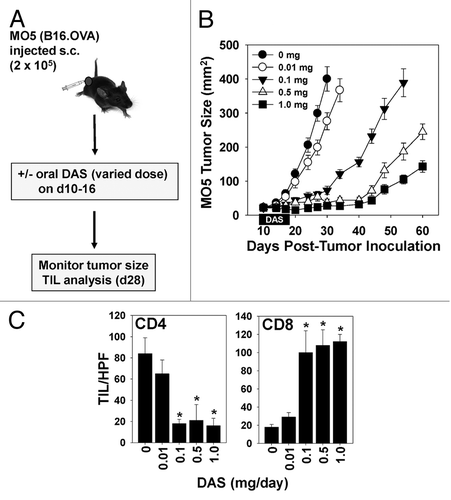
Before assessing the potential immunologic benefits of combining DAS with vaccine-based immunotherapy, we first established an optimal dose of DAS monotherapy based on tumor growth suppression and the preferred immunologic endpoint of tumor-infiltrating lymphocyte (TIL) recruitment. C57BL/6 mice bearing established (implanted 10 d prior) subcutaneous M05 melanomas were left either untreated (controls) or were treated daily with DAS at doses ranging from 0.01–1.0 mg/day for 1 wk by oral gavage (). We observed significant inhibition of melanoma growth at doses of DAS in excess of 0.01 mg/day (, P < 0.05 for all time points past 17d post tumor-inoculation vs. the untreated control, ANOVA), which was associated with a dramatic rise in CD8+ (but not CD4+) TILs (). Based on these data, we selected a DAS dose of 0.1 mg/day for our combinational therapies, as this was the minimal dose of single-agent drug yielding discernable, yet sub-optimal antitumor efficacy and a modest elevation in CD8+ TIL numbers, thereby permitting assessment of improved treatment outcome upon co-administering DAS together with a cancer-specific vaccine.
Figure 1. Therapeutic administration of dasatinib monotherapy elevates the numbers of tumor-infiltrating CD8+ T cells and exhibits dose-dependent anti-melanoma efficacy. (A–C) M05 melanoma cells were injected sub-cutaneously into syngenic C57BL/6 mice and allowed to establish for 10 d, at which time animals were randomized by tumor size and cohorts (n = 7 mice/group) were left untreated (controls) or were treated with orally administered dasatinib (DAS, dose ranging from 0.01–1 mg/day as indicated) for 7 consecutive days. (B) Animals were monitored for tumor growth every 3–4 d using external calipers and estimated tumor volume calculated as the product of the orthogonal measurements. Data are the mean ± SD tumor volume per group. (C) On day 28, 2 mice/group were sacrificed and cryo-preserved tumor sections analyzed by immunofluorescence staining and fluorescence microscopy to assess the numbers of CD4+ and CD8+ tumor-infiltrating lymphocytes (TILs). The mean ± SD number of cells over 10 high-power fields (HPF) are reported. Representative data from 1 of 3 independent experiments are depicted. Statistical analysis was performed by 1-way ANOVA; *P < 0.05 vs. untreated or DAS 0.01 mg/day.
DAS potentiates the immunogenicity and therapeutic efficacy of peptide-based dendritic cell vaccine in vivo
We next sought to test the impact of DAS on vaccine efficacy in vivo. C57BL/6 mice bearing subcutaneous M05 melanomas established 10 d prior were left untreated or were treated with genetically modified dendritic cell (DC) VAC comprising OVA257–264 peptide-pulsed DC overexpressing murine interkeukin-12 (IL-12) that we have previously shown to promote robust T-helper independent anti-OVA Type1 cytotoxic T (Tc) cell responses in C57BL/6 mice.Citation12 Experimental animals were administered either s.c. contralateral VAC on days 10 and 17, DAS (0.1 mg/day via oral gavage on days 10–16) alone, or a combination of the s.c. VAC and oral DAS (). While untreated animals displayed rapidly progressive disease that required euthanasia in accordance with IACUC guidelines by 34 d post-tumor inoculation, M05-bearing mice treated with either single modality (i.e., DAS or VAC) harbored tumors with a slower growth rate and exhibited an extended survival period of approximately 15–25 d relative to untreated control animals (). In contrast, animals treated with combined DAS + VAC therapy exhibited profoundly reduced melanoma growth (, P < 0.05 vs. all other cohorts after day 20).
Figure 2. Combination dasatinib + OVA peptide-based dendritic cell vaccination therapy yields superior antitumor efficacy and immune cell recruitment into the tumor microenvironment vs. either monotherapy. (A–D) C57BL/6 mice bearing subcutaneous M05 melanomas (n = 7 mice/group) established 10 d prior were left untreated or were treated with either 0.1 mg/day dasatinib (DAS) administered by oral gavage for 7 consecutive days, contralateral s.c. vaccination (VAC) consisting of 106 OVA257–264 peptide-pulsed dendritic cells (DCs) genetically modified to overexpress IL-12, or a combination of DAS + VAC. (B) Animals were monitored for tumor growth every 3–4 d using external calipers and estimated tumor volume calculated as the product of the orthogonal measurements. Data are the mean ± SD tumor volume per group. (C) On day 34, 2 mice/group were sacrificed and cryo-preserved tumor sections analyzed by immunofluorescence staining and fluorescence microscopy for CD4+ T cell, CD8+ T cell, and CD11c+ DC content. The numbers of CD4+ and CD8+ tumor-infiltrating lymphocytes (TILs) or DC are reported as the mean ± SD of the indicated cells over 10 high-power fields (HPF). (D) Total tumor mRNA purified from representative tumors from the indicated treatment group was analyzed by RT-PCR for expression of the indicated Type-1-associated cytokine, chemokine, or chemokine receptor transcript. Representative data from 1 of 3 independent experiments are depicted. Statistical analyses were performed by Student’s t test or 1-way ANOVA; *P < 0.05 vs. untreated (t test) and **P < 0.05 vs. all other groups (ANOVA).
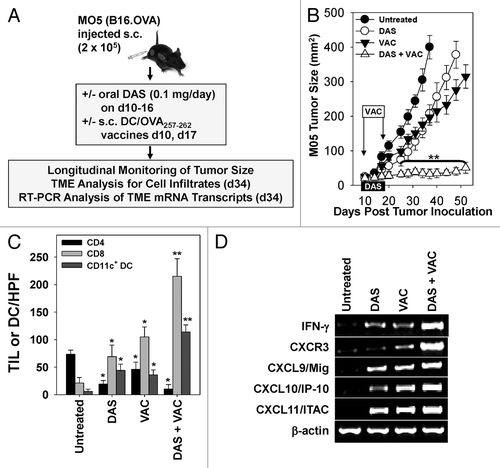
Analyses of tumor-infiltrating immune cells on day 34 revealed significantly increased numbers of CD8+ T lymphocytes and CD11c+ DC in the tumors of mice treated with DAS, VAC, or DAS + VAC, with a statistically elevated level of CD8+ T effector cells in mice receiving the combination therapy (, P < 0.05 for DAS + VAC) in comparison to all other cohorts. In contrast, the levels of CD4+ T cells in all treatment groups were found to be significantly decreased relative to those in untreated controls. As shown in a corresponding RT-PCR analysis of total tumor mRNA extracted from representative tumors revealed that the combination therapy appeared to stimulate the highest expression of transcripts encoding pro-inflammatory cytokine and chemokines. These include interferon-γ (IFNγ) and leukocyte trafficking regulatory proteins chemokine (C-X-C motif) ligand variants 9–11 (CXCL9–11), as well as their corresponding chemokine receptor CXCR3, immunoregulatory molecules known to be expressed by Type1 effector T cells (). In addition, purified CD11c+ DC isolated from tumor digests subsequently challenged with lipopolysaccharide ex vivo also exhibited higher production of IL-12p70 and reduced production of IL-10 in cell preparations from the DAS + VAC cohort vs. all other cohorts (Fig. S1, P < 0.05, ANOVA). Taken together, these data support the notion that combined treatment with DAS + VAC fosters superior pro-inflammatory Type1 CD8+ effector T cell and DC infiltration into the therapeutic tumor microenvironment. Furthermore, based on antibody-mediated T-cell subset depletion studies in vivo, we demonstrate that the superior antitumor efficacy associated with combined DAS + VAC therapy is largely CD8+ T-cell dependent (Fig. S2), whereas depletion of CD4+ T cells has no effect.
Combinatorial DAS + VAC therapy reduces immunoregulatory cell populations and alters hypoxia-mediated signaling in the tumor microenvironment
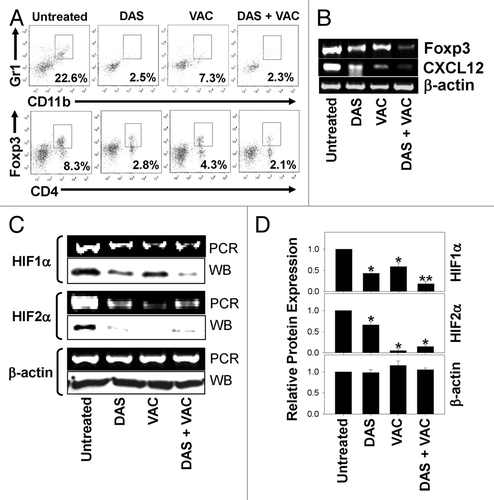
Considering that protective Type1 CD8+ effector T cells would likely mediate more robust anticancer function under conditions that are unopposed by regulatory cell populations,Citation12 we next examined tumors for therapy-associated changes in suppressor cell subsets, i.e., CD11b+Gr1+ myeloid-derived suppressor cell (MDSC) and CD4+Foxp3+ regulatory T (Treg) cells. As shown in , cytofluorometric analysis revealed that progressive and rapidly growing, untreated day 34 M05 tumors accrue high frequencies of immune cells bearing regulatory phenotypes. In contrast, levels of these suppressor cell populations were dramatically reduced in melanomas harvested from animals treated with either single agent modality or with the combination DAS + VAC immunotherapy (). Notably, among CD4+ TILs, CD4+Foxp3+ Tregs appeared to be preferentially reduced relative to untreated control levels, whereas the frequency of CD4+Foxp3- T cells seemed comparable in all treatment cohorts (). Corresponding RT-PCR analyses revealed loss of the Foxp3 and CXCL12, a hypoxia-responsive chemokine known to recruit MDSC and Treg cells,Citation27,Citation28 encoding transcripts in the tumors of treated mice, particularly those receiving the DAS + VAC combined therapy (). Furthermore the canonical markers of hypoxia, hypoxia-inducible factor 1 α (HIF-1α) and hypoxia-inducible factor 2 α (HIF-2α) were found to be reduced in the treated tumors at both the RNA transcript and protein level as a consequence of therapeutic intervention (, P < 0.05 vs. untreated controls, Student’s t test). Densitometry revealed that the greatest reduction of HIF1α, specific protein expression occurred in response to combined DAS + VAC treatment (, P < 0.05 vs. all other cohorts, ANOVA). When taken together, these data suggest that the DAS + VAC combination immunotherapy may recondition the melanoma microenvironment in a manner that reduces hypoxia-driven recruitment and accumulation of regulatory cell populations which may allow for the improved antitumor functionality mediated by vaccine-induced tumor-infiltrating Type-1 CD8+ T cells.
Figure 3. Combinatorial DAS + VAC therapy reduces immunoregulatory cell populations and alters hypoxia-mediated signaling in the tumor microenvironment. (A–D) Day 34 M05 melanomas were isolated from host mice that were left untreated or were treated (starting on d10 post tumor cell s.c. injection) with either 0.1 mg/day dasatinib (DAS) orally administered for 7 consecutive days, or contralateral s.c. vaccination (VAC) consisting of 106 OVA257–264 peptide-pulsed dendritic cells (DCs) genetically modified to overexpress IL-12, or a combination of DAS + VAC. (A) Dissociated tumors from the various treatment groups were analyzed for their content of myeloid-derived suppressor cells (MDSC; CD11b+Gr1+) and regulatory T cells (Tregs, CD4+Foxp3+) by immunofluorescence staining and flow cytometry. The percentage of cells bearing the specified phenotype is reported in panel insets. (B and C) RT-PCR analysis of the levels of Foxp3, CXCL12, HIF1α, HIF2α, and β-actin mRNA transcripts (panels B and C) and western blot analysis for expression of HIF1α, HIF2α, and β-actin protein (panel C). (D) Densitometric analysis of western blots (shown in C) scanned to determine comparative protein levels in each tumor cohort relative to the untreated control that was assigned an arbitrary value of 1.0. Representative data from 1 of 3 independent experiments is depicted. Statistical analyses were performed by Student’s t test or 1-way ANOVA; *P < 0.05 vs. untreated (t test), **P < 0.05 vs. all other groups (ANOVA).
Combination DAS + VAC therapy promotes a broader therapeutic CD8+ T cell repertoire in the tumor-draining lymph node and the tumor microenvironment
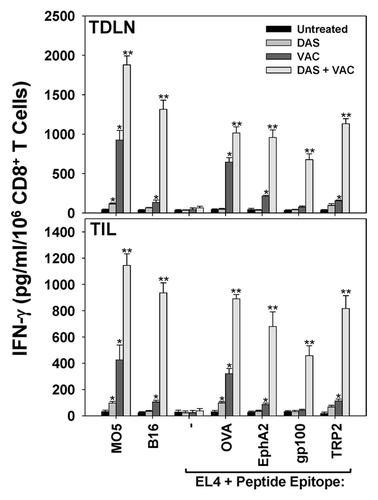
Given the superior enrichment of both Type-1 CD8+ T cells and CD11c+ DC within melanomas treated with bimodal DAS + VAC therapy, we hypothesized that this treatment may reinforce continuous longitudinal cross-priming of anticancer CD8+ T-cell responses in the tumor-draining lymph node (TDLN) against tumor-associated antigens unrelated to the vaccine OVA peptide. To test the possibility that a broader spectrum of cancer cell-specific CD8+ T cells are present in the TDLN or tumor microenvironment in DAS + VAC treated mice, CD8+ T cells were isolated from day 34 TDLN and dissociated tumors and analyzed for their ability to produce IFNγ when stimulated in vitro with intact M05 (OVA+) or B16 (OVA−) melanomas or EL4 thymoma cells +/− H-2b class I-presented peptides derived from OVA, the melanoma-associated antigens gp100 or tyrosinase-related protein 2 (TRP2),Citation29 or the melanoma stromal cell-associated antigen ephrin A2 (EphA2).Citation30 As shown in , CD8+ T cells from the TDLN as well as those infiltrating the tumor isolated from either untreated or DAS alone treated mice displayed weak or no reactivity against any targets evaluated, except for the DAS-treated cohort that exhibited a significant degree of OVA-specific CD8+ T cell reactivity in comparison to T cells from untreated M05 tumors. In marked contrast, CD8+ T cells from the TDLN and TIL population isolated from mice treated with VAC alone or DAS + VAC registered strong anti-OVA responses (i.e., differential reactivity against M05 vs. B16 and EL4 + OVA peptide vs. EL4 alone, P < 0.05, Student’s t test). Notably, the DAS + VAC treatment cohort was unique in developing robust Type1 CD8+ T cell responses among T cells present in both TDLN and TIL against non-vaccine tumor-associated antigens, such as EphA2, gp100 and TRP2 ().
Figure 4. Combined DAS + VAC therapy promotes a broader therapeutic CD8+ T cell repertoire in the tumor-draining lymph node and the tumor microenvironment. M05 melanoma bearing mice were left untreated or were treated (starting on d10 post tumor cell s.c. injection) with either 0.1 mg/day dasatinib (DAS) orally administered for 7 consecutive days, or contralateral s.c. vaccination (VAC) consisting of 106 OVA257–264 peptide-pulsed dendritic cells (DCs) genetically modified to overexpress IL-12, or a combination of DAS + VAC. Tumors and tumor-draining lymph nodes (TDLN) were harvested on day 34 and CD8+ T cells were isolated via anti-CD8 antibody labeling and magnetic beads. CD8+ effector cells were stimulated for 48h with M05 (OVA+) melanoma cells, B16 (OVAneg) melanoma cells, EL4 thymoma control cells or EL4 cells pre-pulsed (for 4h at 37 °C) with CD8+ T cell peptide epitopes derived from other tumor-associated antigens (at an effector-to-target cell ratio of 10:1) as indicated. Cell-free supernatants were then recovered from each culture well and analyzed for IFNγ content by specific ELISA. Data are reported as the mean ± SD from triplicate ELISA determinations. Representative data from 1 of 3 independent experiments is depicted, Statistical analyses were performed by Student’s t test or 1-way ANOVA; *P < 0.05 vs. untreated (t test), **P < 0.05 vs. all other groups (ANOVA).
Discussion
Our major finding is that the multi-kinase small molecule inhibitor DAS serves as an effective adjuvant to peptide-based DC vaccination in the treatment of subcutaneous M05 murine melanoma in vivo. DAS appears to mediate this vaccine-potentiating function via multiple immunological mechanisms (). These include the treatment-associated erosion of immunosuppressive cells populations such as MDSCs and Tregs within the tumor microenvironment. DAS also enhanced the stimulation of anticancer CD8+ T cells in the tumor periphery and promoted the recruitment of therapeutic Type-1 CXCR3+CD8+ T cells via locoregional production of the CXCR3 ligand chemokines CXCL9, CXCL10, and CXCL11. Furthermore, we found that CD8+ T cells are critical for the enhanced efficacy of combinatorial DAS + VAC treatment, as evinced by antibody-based depletion studies.
Figure 5. Proposed mechanism for the superior antitumor action of combined DAS + VAC immunotherapy. We propose that DAS monotherapy exerts an antagonistic effect on tumor-associated myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) manifesting in reduced numbers of such regulatory cells in the melanoma microenvironment. This may be the result of reduced recruitment of MDSCs or Tregs due to reduced levels of relevant recruiting chemokines such as CXCL12, or due to dasitinib (DAS)-mediated inhibition of MDSC or Treg expansion and differentiation within the tumor microenvironment. Removal of such suppression may allow for improved recruitment and function of CD11c+ dendritic cells (DCs) and the effector T (Teff) cells that are cross-primed (by DC-XP) in the tumor-draining lymph node (TDLN). Vaccination (VAC) monotherapy drives anti-OVA CD8+ T cell responses in the vaccine-site draining lymph node (VDLN) and the subsequent trafficking of such effector cells into the tumor area results in locoregional production of IFNγ that stimulates production of CXCR3+ chemokines (such as CXCL9–11) allowing for the corollary recruitment of additional Type-1 CXCR3+ tumor-infiltrating lymphocytes (TILs), a portion of which are the result of cross-priming events in the TDLN. Treatment with DAS + VAC provides an optimal combination therapy that stimulates Type-1 immunity (both T cell and DC) and mitigates immunosuppression associated with MDSCs and Tregs. Re-iterated cycles of IL-12hiIL-10lowCD11c+ DC cross-priming of CD8+ T cells in TDLN results in the rapid broadening of the antitumor T cell repertoire, allowing for effective immune-mediated control of both melanoma cells and tumor-associated stromal cell populations, such as EphA2+ vascular endothelial cells, and superior therapeutic efficacy. Note: Larger symbol sizes and bolder lines infer greater numbers or strength of responses.
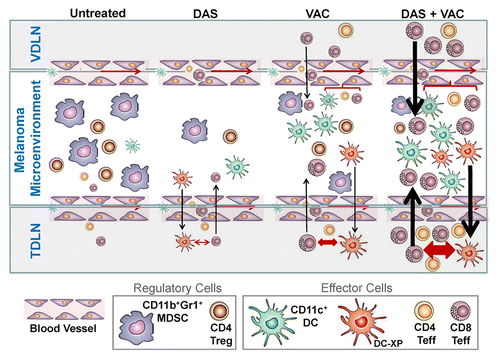
Some of the adjuvant-like qualities associated with DAS are consistent with a recent report by Yang et al.Citation21 and similar to our prior findings regarding alternative tyrosine kinase inhibitors such as axitinibCitation11 and sunitinib.Citation12 These data suggest that DAS treatment benefits may be associated with its potent inhibitory action on the receptor tyrosine kinase c-KIT known to play a supportive role in MDSC immunobiology.Citation13,Citation14,Citation21 It is also conceptually possible that the normalizing of immunologic responses (toward protection) stems from the ability of DAS to inhibit SRC family kinases that act to reinforce STAT3 signaling implicated in Treg immunosuppressive function, effector T cell and DC dysfunction and the pro-angiogenic tumor milieu.Citation31–Citation33 Our findings suggesting that DAS co-treatment attenuates hypoxic signaling in the tumor micrenvironment may also be salient in this context as hypoxia reinforces immune dysfunction and promotes a state of cancer cell resistance to T cell-mediated cytotoxicity.Citation34–Citation38 In the latter scenario, malignant cell escape of cytotoxic immunosurveillance is believed to be regulated by hypoxia-responsive microRNAsCitation34 or the process of autophagy.Citation36
Interestingly, combined DAS + VAC therapy also stimulated elevated IL-12p70hiIL-10lowCD11c+ DC recruitment and accumulation into the tumor microenvironment. This influx of antigen-presenting CD11c+ DCs may coordinate an increase in effector T cell cross priming, thus giving rise to a broader repertoire of CD8+ T cells in the TDLN. Of note, this immunologic manifestation was also reflected within the tumor-infiltrating effector T cell population. This “spreading” in the CD8+ T cell repertoire is presumed to occur secondarily to the initial wave of vaccine-induced TIL-mediated anticancer cytotoxicity. The TIL cancer cell killing would permit the uptake of cellular debris, including tumor-associated antigens, by the recruited CD11c+ DC. These tumor-antigen presenting DCs may then migrate to the TDLN where they may in turn activate antigen-specific CD8+ T cells that may subsequently circulate via the peripheral blood into melanoma sites (, and ref. Citation39). Notably, in our experiment we observed therapeutic cross-priming against both melanoma tumor-associated antigens (i.e., gp100, TRP2), as well as, the EphA2 protein expressed by tumor-associated vascular endothelial cells but not B16 melanoma cells themselves.Citation27,Citation40 This provides an operational paradigm in which DAS co-therapy promotes the expansion of a qualitatively distinct Type-1 CD8+ T cell repertoire that is initiated against a limited set of vaccine-associated antigens (i.e., OVA) but ultimately culminates in reactivity against both tumor and tumor stromal cell populations, thereby providing optimal therapeutic benefit. Although our studies did not address the functional avidity of the various antigen-specific CD8+ T cell populations harvested from each treatment cohort, Wŏlfl et al. have recently reported that at clinically-relevant dosages (i.e., up to 90 nM), DAS pretreatment in vitro enhances DC immunostimulatory activity leading to higher avidity T cells such as those targeting the melanoma antigen MART1.Citation41 The development of complex antigen-specific T cell responses would further aid in immune recognition of heterogeneous populations of melanoma cells that may contain subpopulations with defects in antigen processing and presentation,Citation42 such that only high avidity anti-melanoma T cells may be capable of curative therapeutic impact.Citation41,Citation43
There are theoretical treatment concerns for combinatorial therapies employing DAS and cancer vaccines comprising antigens that are non-mutated gene products overexpressed by the tumor relative to the normal counterpart. By removing regulatory immunity while enhancing specific Type-1 T effector cell function using DAS, one may potentially promote autoimmune pathological responses (colitis, diabetes mellitus, rheumatoid arthritis, among others) as previously observed in clinical trials evaluating immunotherapeutic antibodies such as those targeting cytotoxic T-lymphocyte antigen 4 (CTLA4) or programmed cell death 1 (PD-1).Citation44,Citation45 Although there have been rare case reports of DAS-associated colitis in the setting of leukemia,Citation46,Citation47 in extensive clinical trials so far DAS appears to be a reasonably well-tolerated agent for the treatment of solid tumors, including melanoma.Citation17,Citation48 Indeed, to date, we have not observed any evidence of autoimmune pathology (including vitiligo) in our M05 murine melanoma model.
Further clinically relevant concerns include the known activity of DAS as a reversible inhibitor of Lck, an important mediator of proximal TCR-mediated signaling. This pharmacological activity can transiently inhibit T cell proliferation and IFNγ production (without affecting T cell viability) in vitro.Citation49 While at face value this is potentially problematic, in the cancer setting, chronic Lck activation occurs in antitumor TIL, a molecular phenotype that may lead to phosphorylation of anergy-associated diacylglycerol kinase α (DGKα) in T cells.Citation50 As a consequence, transient interruption of TCR signaling by DAS might actually prevent T effector cell tolerance, potentially re-awakening dysfunctional TILs for therapeutic benefit. We are currently investigating such possibilities in ongoing murine modeling studies that integrate longitudinal monitoring of therapy-associated alterations in T-cell and DC immune function. This should allow for the development of a more detailed mechanistic paradigm underlying the acute vs. sustained antitumor benefits associated with therapeutic administration of DAS monotherapy or combinatorial treatment in conjunction with VAC.
When taken together, our results provide rationale for the clinical translation of combined DAS + VAC protocols for the treatment of melanoma patients. Indeed, we have recently begun evaluating this combination treatment modality in advanced stage melanoma patients at the University of Pittsburgh Cancer Institute (UPCI 12–048, NCT01876212, “A Randomized Phase II Pilot Study of Type I-Polarized Autologous Dendritic Cell Vaccines Incorporating Tumor Blood Vessel Antigen (TBVA)-Derived Peptides in Combination with Dasatinib in Patients with Metastatic Melanoma.”)
Materials and Methods
Mice
Female 6–8 wk old C57BL/6 (H-2b) mice were purchased from the Jackson Laboratory. Animals were maintained in micro-isolator cages and handled under aseptic conditions per an Institutional Animal Care and Use Committee (IACUC)-approved protocol.
Cell lines and culture
The M05 (B16.OVA, H-2b) and wild-type B16 (H-2b) melanoma cell linesCitation12 and the EL4 thymoma (H-2b, American Type Culture Collection, ATCC) were free of Mycoplasma contamination and cultured in complete media (CM) consisting of RPMI 1640 media supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, 100 µg/mL streptomycin, and 10 μM L-glutamine (all from Life Technologies). Cells were cultured at 37 °C with 5% CO2. Expression of the OVA transgene in M05 cells was maintained under culture selection with G418 (Life Technologies).
Peptide
The H-2b class I-presented OVA257–264 (SIINFEKL),Citation12 murine EphA2682–689 (VVSKYKPM),Citation30 murine gp10025–33 (EGSRNQDWL)Citation29 and murine TRP2181–188 (VYDFFVWL)Citation29 peptides were synthesized to > 96% purity by 9-fluorenylmethoxycarbonyl (Fmoc) chemistry by the University of Pittsburgh Cancer Institute’s Peptide Synthesis Facility (a Shared Resource).
VAC generation
DC were generated from the bone marrow of tibias/femurs from naïve C57BL/6 mice as previously described.Citation11,Citation12 Briefly, bone-marrow-derived cells were cultured in CM over a period of 5 d with 1000 U/mL of recombinant murine (rm)GM-CSF and rmIL-4 (both from Peprotech). CD11c+ cells were subsequently purified using magnetic-activated cell sorting (MACS) bead positive selection (Miltenyi Biotec) and infected at a multiplicity of infection = 50 with an adenoviral vector encoding murine IL-12p70 (Ad.mIL-12p70) as previously described.Citation11 After a 48h culture in the presence of rmGM-CSF and rmIL-4, the IL-12 gene-modified DC (DC.IL12) were loaded with 10 μM OVA257–264 peptide for 4 h at 37 °C. After washing with phosphate-buffered saline (PBS), these antigen-loaded cells constituted our VAC formulation.
Tumor therapy
Mice were challenged subcutaneously (s.c., right flank) with 2 × 105 M05 melanoma cells, and tumors were allowed to progress 10 d before randomization into groups of 5 animals each with comparable mean tumor sizes/group. Animals then were left untreated, or received VAC (s.c., left flank) with 1 × 106 OVA peptide-pulsed DC.IL12 cells on days 10 and 17 post-M05 challenge. DAS (0.01–1 mg, BMS-354825/Sprycel, Bristol-Myers Squibb) was dissolved in 50 µl Labrasol (Gattefossé Canada) and delivered via oral gavage daily, alone or in combination with VAC, for one week beginning on day 10. Tumor growth was monitored every 3–4 d and tumor dimensions measured using Vernier calipers. M05 tumor size was recorded in mm2 (mean ± SD) based on the product of orthogonal measurements.
Fluorescence Microscopy
Tumor samples were prepared and sectioned as previously reported.Citation12,Citation40 Briefly, tumor tissues were harvested and fixed in 2% paraformaldehyde (Sigma-Aldrich) at 4 °C for 1h, then cryoprotected in 30% sucrose for 24 h. Tumor tissues were then frozen in liquid nitrogen and 6 μm cryosections prepared. For analysis of T cell subsets, sections were first stained with purified rat anti-mouse CD8α, purified rat anti-mouse CD4 or purified hamster anti-mouse CD11c (all from BD-PharMingen) mAbs for 1h. After washing, sections were stained with PE-conjugated goat anti-rat or anti-hamster secondary antibody (Jackson ImmunoResearch). After washing, sections were then covered in Gelvatol (Monsanto) and a coverslip applied. Slide images were acquired using an Olympus 500 scanning confocal microscope (Olympus America). The positively stained cells were quantified by analyzing the images at a final magnification of × 20. The number of cells in sections with a given fluorescence marked immunophenotype was quantitated using Metamorph Imaging software (Molecular Devices) and data are reported as the mean ± SD number of cells over 10 high-power fields.
Analyses of Type-1 OVA-specific CD8+ T cell responses in TIL and TDLN
At time points ranging from 28–34 d post M05 tumor inoculation, the constituent CD8+ T cells among the TILs and present in the TDLN were harvested from euthanized mice. Single cell suspensions were generated from tumors by enzymatic digestion using DNase I, collagenase, and hyaluronidase (all from Sigma-Aldrich), and from TDLNs by mechanical disruption, with CD8+ T cells subsequently isolated from each tissue preparation using anti-CD8-MACS beads (Miltenyi Biotec), according to the manufacturer’s protocol. In all cases, CD8+ T cells were pooled between 3 animals per group and stimulated for 5 d in vitro with irradiated M05 tumor cells at an effector-to-target (E:T) ratio of 10:1. M05 tumor cells were irradiated prior to the experiment with 100 Gy at room temperature from a 137Cs irradiator (Gammacell40, Atomic Energy of Canada Limited) at a dose rate of 0.87 Gy/min. To assess antigen-specific responses, T cells were then transferred to 96-well round bottom plates (Corning) and incubated with syngenic antigen-loaded EL4 cells (pre-pulsed for 4h at 37 °C with the OVA257–264, EphA2682–689, gp10025–33 or TRP2181–188 peptides or no peptide) at a 10:1 effector-to-target cell ratio for 48 h. Cell-free supernatants were then harvested and assessed for mIFNγ content using a specific ELISA kit (BD Biosciences, lower detection limit = 31.3 pg/mL). Data are reported as mean IFNγ levels (pg/mL) over control based on triplicate determinations ± SD.
Flow cytometry
Single cell suspensions from tumor digests were stained with the following directly-conjugated fluorescein isothiocyanate (FITC) and phycoerythrin (PE) labeled anti-mouse antibodies (all from BD Biosciences): FITC-anti-CD8, FITC-anti-CD4, FITC-anti-CD11b, FITC-anti-CD11c, PE-anti-Gr1, and PE-anti-Foxp3. For Foxp3-specific staining, cells were first labeled with the FITC-anti-CD4 antibody before incubation with the PE-anti-Foxp3 antibody using an Intracellular Fixation and Permeabilization Kit as recommended by the manufacturer (eBioscience). Fluorescence cytometric analysis was performed using Cell Quest software and a FACscan flow cytometer (Becton Dickinson), with FlowJo software (Tree Star) used for data analysis.
RT-PCR
Total RNA was extracted from tumor tissues on day 34 post M05 inoculation using TRIzol reagent (Life Technologies). cDNA was generated using the MuLV reverse transcriptase with random hexamers (both from Applied Biosystems), and gene-specific PCR was performed with AmpliTaq DNA polymerase (Applied Biosystems) and primer pairs for IFNγ, CXCR3, CXCL9, CXCL10, CXCL11, CXCL12, HIF-1α, HIF-2α, Foxp3, and β-actin as previously described.Citation12 Cycling conditions were as followed: initial denaturation at 94 °C for 2 min, denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s, and elongation at 72 °C for 1 min a total of 35–40 cycles followed by a final elongation at 72 °C for 5 min. RT-PCR products were resolved on 2% ethidium bromide stained agarose gels (Sigma–Aldrich), and gel images were captured and analyzed using a GDS 8000 bio-imaging system and Labworks software (UVP, LLC).
Western blotting
Single cell digests derived from tumors harvested on d34 were incubated in cell lysis buffer, with cell-free lysate protein preparations resolved by sodium-dodecyl sulfate PAGE (SDS-PAGE) prior to electro-transfer onto polyvinylidene difluoride membranes, that were subsequently probed with polyclonal rabbit anti-mHIF-1α, anti-mHIF-2α, or β-actin primary antibodies and horseradish peroxidase-conjugated goat anti-rabbit secondary antibody reagents (all from Santa Cruz Biotechnology). Probed blots were visualized using the Western Lighting chemiluminescence detection kit (Perkin–Elmer) and exposed to X-Omat film (Eastman Kodak).
Statistical analysis
Statistical analyses between groups were performed using a 2-tailed Student’s t test or 1-way ANOVA with post hoc analysis, as indicated. All data were analyzed using SigmaStat software, version 3.5 (Systat Software). Differences with a P value < 0.05 were considered significant.
| Abbreviations: | ||
| CTLA-4 | = | cytotoxic T-lymphocyte antigen 4 |
| DAS | = | dasatinib |
| DC | = | dendritic cell |
| DGKα | = | diacylglycerol kinase α |
| Fmoc | = | 9-fluorenylmethyloxycarbonyl |
| GM-CSF | = | granulocyte-macrophage colony-stimulating factor, IACUC, Institutional Animal Care and Use Committee |
| IL | = | interleukin |
| Lck | = | lymphocyte-specific protein tyrosine kinase |
| MACS | = | magnetic-activated cell sorting |
| MART1 | = | melanoma antigen recognized by T cells 1 |
| MDSC | = | myeloid-derived suppressor cells |
| OVA | = | ovalbumin |
| PD-1 | = | programmed cell death protein 1 |
| PDGFR | = | platelet-derived growth factor receptor |
| STAT | = | signal transducer and activator of transcription |
| TCR | = | T cell receptor |
| TDLN | = | tumor-draining lymph node |
| TIL | = | tumor-infiltrating lymphocyte |
| Treg | = | regulatory T cell |
| VAC | = | DC vaccine |
Additional material
Download Zip (105.7 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by NIH R01s CA143075 and CA169118 (to W.J.S.) and P50 CA121973 (to J.M.K.). These studies employed recombinant adenoviruses produced by the UPCI Vector Core Facility supported by the University of Pittsburgh Cancer Center Support Grant (CCSG) P30 CA047904. D.B.L. was supported by a Postdoctoral Fellowship (PF-11–151–01-LIB) from the American Cancer Society.
Supplemental materials may be found here
http://www.landesbioscience.com/journals/oncoimmunology/article/27589
Citation: Lowe DB, Bose A, Taylor JL, Tawbi H, Lin Y, Kirkwood JM, Storkus WJ. Dasatinib promotes the expansion of a therapeutically superior T cell repertoire in response to dendritic cell vaccination against melanoma. OncoImmunology 2014; 3:e27589; 10.4161/onci.27589
Related Research Data
References
- Fabbro D, Cowan-Jacob SW, Möbitz H, Martiny-Baron G. Targeting cancer with small-molecular-weight kinase inhibitors. Methods Mol Biol 2012; 795:1 - 34; http://dx.doi.org/10.1007/978-1-61779-337-0_1; PMID: 21960212
- Natarajan N, Telang S, Miller D, Chesney J. Novel immunotherapeutic agents and small molecule antagonists of signalling kinases for the treatment of metastatic melanoma. Drugs 2011; 71:1233 - 50; http://dx.doi.org/10.2165/11591380-000000000-00000; PMID: 21770473
- Montero JC, Seoane S, Ocaña A, Pandiella A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res 2011; 17:5546 - 52; http://dx.doi.org/10.1158/1078-0432.CCR-10-2616; PMID: 21670084
- Puls LN, Eadens M, Messersmith W. Current status of SRC inhibitors in solid tumor malignancies. Oncologist 2011; 16:566 - 78; http://dx.doi.org/10.1634/theoncologist.2010-0408; PMID: 21521831
- Kim LC, Rix U, Haura EB. Dasatinib in solid tumors. Expert Opin Investig Drugs 2010; 19:415 - 25; http://dx.doi.org/10.1517/13543781003592097; PMID: 20113198
- Matsumoto S, Batra S, Saito K, Yasui H, Choudhuri R, Gadisetti C, Subramanian S, Devasahayam N, Munasinghe JP, Mitchell JB, et al. Antiangiogenic agent sunitinib transiently increases tumor oxygenation and suppresses cycling hypoxia. Cancer Res 2011; 71:6350 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-11-2025; PMID: 21878530
- Tailor TD, Hanna G, Yarmolenko PS, Dreher MR, Betof AS, Nixon AB, Spasojevic I, Dewhirst MW. Effect of pazopanib on tumor microenvironment and liposome delivery. Mol Cancer Ther 2010; 9:1798 - 808; http://dx.doi.org/10.1158/1535-7163.MCT-09-0856; PMID: 20515941
- Qayum N, Muschel RJ, Im JH, Balathasan L, Koch CJ, Patel S, McKenna WG, Bernhard EJ. Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Res 2009; 69:6347 - 54; http://dx.doi.org/10.1158/0008-5472.CAN-09-0657; PMID: 19622766
- Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res 2009; 15:2148 - 57; http://dx.doi.org/10.1158/1078-0432.CCR-08-1332; PMID: 19276286
- Kao J, Ko EC, Eisenstein S, Sikora AG, Fu S, Chen SH. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit Rev Oncol Hematol 2011; 77:12 - 9; http://dx.doi.org/10.1016/j.critrevonc.2010.02.004; PMID: 20304669
- Bose A, Lowe DB, Rao A, Storkus WJ. Combined vaccine+axitinib therapy yields superior antitumor efficacy in a murine melanoma model. Melanoma Res 2012; 22:236 - 43; http://dx.doi.org/10.1097/CMR.0b013e3283538293; PMID: 22504156
- Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, Ko JS, Cohen PA, Finke JH, Storkus WJ. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer 2011; 129:2158 - 70; http://dx.doi.org/10.1002/ijc.25863; PMID: 21170961
- Pan PY, Wang GX, Yin B, Ozao J, Ku T, Divino CM, Chen SH. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood 2008; 111:219 - 28; http://dx.doi.org/10.1182/blood-2007-04-086835; PMID: 17885078
- Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res 2009; 69:2514 - 22; http://dx.doi.org/10.1158/0008-5472.CAN-08-4709; PMID: 19276342
- Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res 2009; 69:2506 - 13; http://dx.doi.org/10.1158/0008-5472.CAN-08-4323; PMID: 19244102
- Coluccia AM, Cirulli T, Neri P, Mangieri D, Colanardi MC, Gnoni A, Di Renzo N, Dammacco F, Tassone P, Ribatti D, et al. Validation of PDGFRbeta and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood 2008; 112:1346 - 56; http://dx.doi.org/10.1182/blood-2007-10-116590; PMID: 18524994
- Kluger HM, Dudek AZ, McCann C, Ritacco J, Southard N, Jilaveanu LB, Molinaro A, Sznol M. A phase 2 trial of dasatinib in advanced melanoma. Cancer 2011; 117:2202 - 8; http://dx.doi.org/10.1002/cncr.25766; PMID: 21523734
- Algazi AP, Weber JS, Andrews SC, Urbas P, Munster PN, DeConti RC, Hwang J, Sondak VK, Messina JL, McCalmont T, et al. Phase I clinical trial of the Src inhibitor dasatinib with dacarbazine in metastatic melanoma. Br J Cancer 2012; 106:85 - 91; http://dx.doi.org/10.1038/bjc.2011.514; PMID: 22127285
- Lee KC, Ouwehand I, Giannini AL, Thomas NS, Dibb NJ, Bijlmakers MJ. Lck is a key target of imatinib and dasatinib in T-cell activation. Leukemia 2010; 24:896 - 900; http://dx.doi.org/10.1038/leu.2010.11; PMID: 20147973
- Weichsel R, Dix C, Wooldridge L, Clement M, Fenton-May A, Sewell AK, Zezula J, Greiner E, Gostick E, Price DA, et al. Profound inhibition of antigen-specific T-cell effector functions by dasatinib. Clin Cancer Res 2008; 14:2484 - 91; http://dx.doi.org/10.1158/1078-0432.CCR-07-4393; PMID: 18413841
- Yang Y, Liu C, Peng W, Lizée G, Overwijk WW, Liu Y, Woodman SE, Hwu P. Antitumor T-cell responses contribute to the effects of dasatinib on c-KIT mutant murine mastocytoma and are potentiated by anti-OX40. Blood 2012; 120:4533 - 43; http://dx.doi.org/10.1182/blood-2012-02-407163; PMID: 22936666
- Rohon P, Porkka K, Mustjoki S. Immunoprofiling of patients with chronic myeloid leukemia at diagnosis and during tyrosine kinase inhibitor therapy. Eur J Haematol 2010; 85:387 - 98; http://dx.doi.org/10.1111/j.1600-0609.2010.01501.x; PMID: 20662899
- Mustjoki S, Auvinen K, Kreutzman A, Rousselot P, Hernesniemi S, Melo T, Lahesmaa-Korpinen AM, Hautaniemi S, Bouchet S, Molimard M, et al. Rapid mobilization of cytotoxic lymphocytes induced by dasatinib therapy. Leukemia 2013; 27:914 - 24; http://dx.doi.org/10.1038/leu.2012.348; PMID: 23192016
- Mustjoki S, Ekblom M, Arstila TP, Dybedal I, Epling-Burnette PK, Guilhot F, Hjorth-Hansen H, Höglund M, Kovanen P, Laurinolli T, et al. Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia 2009; 23:1398 - 405; http://dx.doi.org/10.1038/leu.2009.46; PMID: 19295545
- Fei F, Yu Y, Schmitt A, Rojewski MT, Chen B, Götz M, Döhner H, Bunjes D, Schmitt M. Dasatinib inhibits the proliferation and function of CD4+CD25+ regulatory T cells. Br J Haematol 2009; 144:195 - 205; http://dx.doi.org/10.1111/j.1365-2141.2008.07433.x; PMID: 19016717
- Quezada SA, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med 2008; 205:2125 - 38; http://dx.doi.org/10.1084/jem.20080099; PMID: 18725522
- Yan M, Jene N, Byrne D, Millar EK, O’Toole SA, McNeil CM, Bates GJ, Harris AL, Banham AH, Sutherland RL, et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res 2011; 13:R47; http://dx.doi.org/10.1186/bcr2869; PMID: 21521526
- Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res 2011; 71:7463 - 70; http://dx.doi.org/10.1158/0008-5472.CAN-11-2449; PMID: 22025564
- Xiang R, Lode HN, Chao TH, Ruehlmann JM, Dolman CS, Rodriguez F, Whitton JL, Overwijk WW, Restifo NP, Reisfeld RA. An autologous oral DNA vaccine protects against murine melanoma. Proc Natl Acad Sci U S A 2000; 97:5492 - 7; http://dx.doi.org/10.1073/pnas.090097697; PMID: 10779556
- Hatano M, Kuwashima N, Tatsumi T, Dusak JE, Nishimura F, Reilly KM, Storkus WJ, Okada H. Vaccination with EphA2-derived T cell-epitopes promotes immunity against both EphA2-expressing and EphA2-negative tumors. J Transl Med 2004; 2:40; http://dx.doi.org/10.1186/1479-5876-2-40; PMID: 15563374
- Johnson FM, Saigal B, Tran H, Donato NJ. Abrogation of signal transducer and activator of transcription 3 reactivation after Src kinase inhibition results in synergistic antitumor effects. Clin Cancer Res 2007; 13:4233 - 44; http://dx.doi.org/10.1158/1078-0432.CCR-06-2981; PMID: 17634553
- Kujawski M, Zhang C, Herrmann A, Reckamp K, Scuto A, Jensen M, Deng J, Forman S, Figlin R, Yu H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res 2010; 70:9599 - 610; http://dx.doi.org/10.1158/0008-5472.CAN-10-1293; PMID: 21118964
- Cohen PA, Ko JS, Storkus WJ, Spencer CD, Bradley JM, Gorman JE, McCurry DB, Zorro-Manrique S, Dominguez AL, Pathangey LB, et al. Myeloid-derived suppressor cells adhere to physiologic STAT3- vs STAT5-dependent hematopoietic programming, establishing diverse tumor-mediated mechanisms of immunologic escape. Immunol Invest 2012; 41:680 - 710; http://dx.doi.org/10.3109/08820139.2012.703745; PMID: 23017141
- Noman MZ, Buart S, Romero P, Ketari S, Janji B, Mari B, Mami-Chouaib F, Chouaib S. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res 2012; 72:4629 - 41; http://dx.doi.org/10.1158/0008-5472.CAN-12-1383; PMID: 22962263
- Hasmim M, Noman MZ, Lauriol J, Benlalam H, Mallavialle A, Rosselli F, Mami-Chouaib F, Alcaide-Loridan C, Chouaib S. Hypoxia-dependent inhibition of tumor cell susceptibility to CTL-mediated lysis involves NANOG induction in target cells. J Immunol 2011; 187:4031 - 9; http://dx.doi.org/10.4049/jimmunol.1101011; PMID: 21911602
- Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib F, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 2011; 71:5976 - 86; http://dx.doi.org/10.1158/0008-5472.CAN-11-1094; PMID: 21810913
- Noman MZ, Buart S, Van Pelt J, Richon C, Hasmim M, Leleu N, Suchorska WM, Jalil A, Lecluse Y, El Hage F, et al. The cooperative induction of hypoxia-inducible factor-1 α and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J Immunol 2009; 182:3510 - 21; http://dx.doi.org/10.4049/jimmunol.0800854; PMID: 19265129
- Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res 2008; 14:5947 - 52; http://dx.doi.org/10.1158/1078-0432.CCR-08-0229; PMID: 18829471
- van der Bruggen P, Van den Eynde BJ. Processing and presentation of tumor antigens and vaccination strategies. Curr Opin Immunol 2006; 18:98 - 104; http://dx.doi.org/10.1016/j.coi.2005.11.013; PMID: 16343880
- Zhao X, Bose A, Komita H, Taylor JL, Kawabe M, Chi N, Spokas L, Lowe DB, Goldbach C, Alber S, et al. Intratumoral IL-12 gene therapy results in the crosspriming of Tc1 cells reactive against tumor-associated stromal antigens. Mol Ther 2011; 19:805 - 14; http://dx.doi.org/10.1038/mt.2010.295; PMID: 21189473
- Wölfl M, Schwinn S, Yoo YE, Reß ML, Braun M, Chopra M, Schreiber SC, Ayala VI, Ohlen C, Eyrich M, et al. Src-kinase inhibitors sensitize human cells of myeloid origin to Toll-like-receptor-induced interleukin 12 synthesis. Blood 2013; 122:1203 - 13; http://dx.doi.org/10.1182/blood-2013-03-488072; PMID: 23836556
- Seliger B, Ritz U, Abele R, Bock M, Tampé R, Sutter G, Drexler I, Huber C, Ferrone S. Immune escape of melanoma: first evidence of structural alterations in two distinct components of the MHC class I antigen processing pathway. Cancer Res 2001; 61:8647 - 50; PMID: 11751378
- Zeh HJ 3rd, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol 1999; 162:989 - 94; PMID: 9916724
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252 - 64; http://dx.doi.org/10.1038/nrc3239; PMID: 22437870
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443 - 54; http://dx.doi.org/10.1056/NEJMoa1200690; PMID: 22658127
- Shimokaze T, Mitsui T, Takeda H, Kawakami T, Arai T, Ito M, Iwaba A, Izumino H, Takahashi N, Kanno M, et al. Severe hemorrhagic colitis caused by dasatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia. Pediatr Hematol Oncol 2009; 26:448 - 53; PMID: 19657995
- Erkut M, Erkut N, Ersoz S, Arslan M, Sonmez M. A case of acute colitis with severe rectal bleeding in a patient with chronic myeloid leukemia after dasatinib use. Acta Haematol 2010; 123:205 - 6; http://dx.doi.org/10.1159/000306070; PMID: 20375493
- Breccia M, Alimena G. Activity and safety of dasatinib as second-line treatment or in newly diagnosed chronic phase chronic myeloid leukemia patients. BioDrugs 2011; 25:147 - 57; http://dx.doi.org/10.2165/11591840-000000000-00000; PMID: 21528941
- Tinsley SM. Safety profiles of second-line tyrosine kinase inhibitors in patients with chronic myeloid leukaemia. J Clin Nurs 2010; 19:1207 - 18; http://dx.doi.org/10.1111/j.1365-2702.2009.03167.x; PMID: 20345830
- Kim LC, Rix U, Haura EB. Dasatinib in solid tumors. Expert Opin Investig Drugs 2010; 19:415 - 25; http://dx.doi.org/10.1517/13543781003592097; PMID: 20113198