
Abstract
Cis-diamminedichloridoplatinum(II) (CDDP), commonly referred to as cisplatin, is a chemotherapeutic drug used for the treatment of a wide range of solid cancers. CDDP is a relatively poor inducer of immunogenic cell death (ICD), a cell death modality that converts dying cells into a tumor vaccine, stimulating an immune response against residual cancer cells that permits long-lasting immunity and a corresponding reduction in tumor growth. The incapacity of CDDP to trigger ICD is at least partially due to its failure to stimulate the premortem endoplasmic reticulum (ER)-stress response required for the externalization of the “eat-me” signal calreticulin (CRT) on the surface of dying cancer cells. Here, we developed a murine cancer cell line genetically modified to express the ER resident protein reticulon-1c (Rtn-1c) by virtue of tetracycline induction and showed that enforced Rtn-1c expression combined with CDDP treatment promoted CRT externalization to the surface of cancer cells. In contrast to single agent treatments, the tetracycline-mediated Rtn-1c induction combined with CDDP chemotherapy stimulated ICD as measured by the capacity of dying tumor cells, inoculated into syngenic immunocompetent mice, to mount an immune response to tumor re-challenge 1 week later. More importantly, established tumors, forced to constitutively express Rtn-1c in vivo by continuous treatment with tetracycline, became responsive to CDDP and exhibited a corresponding reduction in the rate of tumor growth. The combined therapeutic effects of Rtn-1c induction with CDDP treatment was only detected in the context of an intact immune system and not in nu/nu mice lacking thymus-dependent T lymphocytes. Altogether, these results indicate that the artificial or “synthetic” induction of immunogenic cell death by genetic manipulation of the ER-stress response can improve the efficacy of chemotherapy with CDDP by stimulating anticancer immunity.
Introduction
Anticancer chemotherapies do not only act on tumor cells to induce growth arrest (cytostasis) and death (cytotoxicity) to mediate their therapeutic effects. A significant portion of their efficacy involves the elicitation of anticancer immune responses that appear particularly important for extending the lifespan of patients and for mediating long-term effects on tumor growth. Thus, successful chemotherapeutics initiate, or reinstate efficient immunosurveillance mechanisms, thereby allowing the host’s immune system to eliminate cancer cells or at least attenuate their proliferation.Citation1-Citation3
There are multiple pathways through which antineoplastic agents can stimulate anticancer immune responses.Citation4,Citation5 On one hand, chemotherapeutics can act on immune cells by stimulating effector mechanisms, for instance by activating dendritic cells (DCs) and cytotoxic T lymphocytes (CTLs). Alternatively, such immunomodulatory moieties could act by repressing or depleting inhibitory elements, such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs). On the other hand, cytotoxic anticancer agents may kill tumor cells in a way that their death is preceded and accompanied by “danger” signals that induce the local recruitment and subsequent activation of innate and acquired immune effectors, hence favoring a cellular immune response against tumor-associated antigens.Citation6,Citation7 Distinct from physiological apoptosis that fails to elicit inflammatory and immune reactions, “immunogenic cell death” (ICD) is presaged by multiple stress response-associated cellular and molecular changes. As an example, ICD is preceded by autophagy, which then allows the tumor cells to liberate significant amounts of ATP as they succumb to caspase-dependent apoptosis.Citation8-Citation12 Cancer cell-derived extracellular ATP subsequently acts as a potent chemoattractant for myeloid cells, including the precursors of inflammatory DCs that then act as antigen-presenting cells (APCs) within the tumor bed.Citation13,Citation14 Moreover, ICD follows endoplasmic reticulum (ER)-stress responses. One particular arm of the ER-stress response, that involves the phosphorylation of eukaryotic initiation factor 2α (eIF2α), stimulates a complex molecular cascade that culminates in the translocation of the most abundant ER-resident soluble protein, calreticulin (CRT), to the cell surface.Citation15-Citation19 Once it has been exposed at the cell surface, CRT acts as an “eat-me” signal for phagocytes,Citation20,Citation21 and hence favors the transfer of tumor antigens into antigen-presenting DCs.Citation13,Citation17
Extensive screening programs for the identification of ICD inducers have been launched. However, to date only a limited number of anticancer agents have been found to possess ICD provoking potential. Thus, anthracyclines,Citation17,Citation22 oxaliplatin,Citation19,Citation23 histone deacetylase inhibitors (such as vorinostat),Citation24,Citation25 taxanes,Citation26,Citation27 zinc,Citation28 irradiation,Citation17,Citation29-Citation31 and photodynamic therapyCitation15,Citation32 were found to potently induce ICD. In contrast, many other anticancer agents are comparably inefficient ICD inducers, including cis-diamminedichloridoplatinum(II) (CDDP also known in the clinic as “cisplatin”) which is widely used for the treatment of testicular, germinal, bronchial, gastrointestinal, and triple-negative breast cancers.Citation17,Citation22 CDDP is an agent that, in spite of its cytotoxic and pro-apoptotic effects, induces rather low ER-stress responses and hence, fails to stimulate CRT exposure.Citation23,Citation33-Citation36 Pharmacological induction of ER stress combined with CDDP could restore CRT externalization on cancer cells and render CDDP an efficient ICD inducer.Citation17,Citation35
Based on these premises, we explored the possibility of specifically restoring the failing ER-stress response in vivo, in the context of CDDP-mediated chemotherapy. To this end, we established a synthetic system for inducing ER stress, namely by the tetracycline (TET)-inducible expression of an ER-restricted protein, reticulon-1, splice variant 1c (Rtn-1c),Citation37,Citation38 in murine cancer cells. In a panel of human cancer cell lines, we have previously shown that CDDP alone fails to induce CRT exposure, correlating with its inability to induce ER stress.Citation23,Citation33-Citation36 However, when combined with a tetracycline-inducible system of Rtn-1c expression in mice, CDDP was capable of inducing CRT externalization,Citation37,Citation38 signifying that induction of the missing ER-stress response compensated for the deficient induction of this hallmark of immunogenicity. Herein, we show that the synthetic induction of ER stress in tumor-bearing immunocompetent mice that express Rtn-1C upon treatment with TET can improve the efficacy of CDDP-based chemotherapy in vivo.
Results and Discussion
An inducible system that promotes CRT surface exposure on fibroblasts in response to Rtn-1 expression and CDDP treatment in vitro
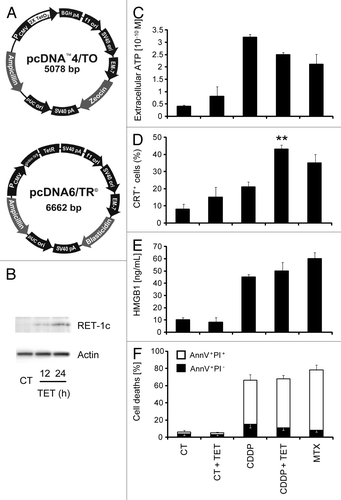
MCA205, a C57BL/6 mouse-derived fibrosarcoma cell line that has been extensively used for the characterization of ICD,Citation17,Citation22,Citation29,Citation39 was engineered to express a tetracycline-inducible (Tet-On) reticulon-1c (Rtn-1c) transgene (). The resulting iRTN-1c MCA205 cells were able to express significant amounts of immunoblot-detectable Rtn-1c protein after 24 h of treatment with TET (). TET-induced expression of Rtn-1c for 48 h alone failed to elicit any of the hallmarks of ICD, such as ATP release, (), CRT surface exposure (), release of high-mobility group box-1 (HMGB1) (), or apoptosis and associated secondary necrosis (). Moreover, the TET-mediated induction of Rtn-1c in iRTN-1c MCA205 cells co-stimulated by simultaneous addition of CDDP did not alter cell death (), or the release of ATP () or HMGB1 (). In contrast, the combination of TET and CDDP induced higher CRT exposure than either of the 2 compounds alone. In fact, the addition of TET plus CDDP-induced translocation of CRT to the plasma membrane surface with equal efficiency as that observed after exposure of cells to the positive control, the quintessential ICD inducer mitoxantrone (MTX; ). We refer to this genetic manipulation facilitating CRT exposure as a “synthetic” procedure.
Figure 1. Establishment and characterization of an MCA205 cell line expressing tetracycline-inducible Rtn-1c. (A) Vectors used to stably transduce murine fibrosarcoma MCA205 cells to generate tetracycline (TET)-inducible iRTN-1c MCA 205 cells. (B) Western-blot analysis of the expression of reticulon-1 (Rtn-1C) in control conditions (CT) or upon TET treatment for 12 or 24 h. β-actin was used as loading control. A representative experiment is shown. (C‒F) Immunogenic cell death markers of iRTN-1c MCA 205 cells analyzed in response to 0.3 µM TET, 150 µM cisplatin (CDDP), CDDP + TET, 1 µM MTX, or no treatment. (C) Extracellular ATP as measured by luciferin-luciferase assay, (D) Immunofluorescence staining of calreticulin (CRT) exposure to the surface, (E) HMGB1 release from cells as detected by ELISA. (F) Apoptosis assayed by Annexin V detection of externalized phosphatidylserine and secondary necrosis as detected by staining with the vital dye propidium iodide. MTX treatment was used as a positive control. Tetracycline treated samples were compared with their untreated counterpart. Results are reported means ± SEM of triplicates. *P < 0.05, **P < 0.01 (unpaired Student’s t test).
Synthetic induction of ER stress by Rtn-1c facilitates the induction of ICD, as measured in vaccination assays
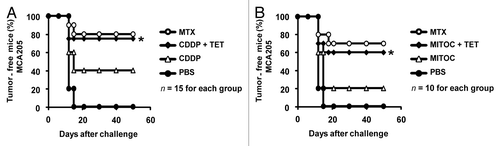
The aforementioned results recapitulate prior experimental evidence obtained in human cell lines showing that RTN-1c can stimulate CRT exposure in cells responding to CDDP.Citation38 However, our synthetic mouse model has a technical advantage over prior experiments in human cells in that iRTN-1c MCA205 cells can be used in immunological experiments performed in histocompatible, immunocompetent C57BL/6 mice in vivo.Citation17,Citation22,Citation29,Citation39 In a first round of experiments, we determined the capacity of TET-inducible Rtn-1c to elicit the immunogenicity of cancer cell death mediated by CDDP, an agent that alone is unable to induce ICD due to its intrinsic incapacity to stimulate a relevant ER-stress response.Citation23,Citation35 For this, iRTN-1c MCA205 cells were pre-incubated with TET to upregulate Rtn-1c (vs. untreated controls) and then subjected to subsequent CDDP treatment in vitro. The CDDP-killed cells were then washed and injected subcutaneously into the left flank of C57Bl/6 mice. One week later, the animals were rechallenged with live, untreated MCA205 cells, and the incidence of tumor development was monitored. While CDDP-only treated cells were relatively poor anticancer vaccines, TET (i.e., Rtn-1c overexpressing) plus CDDP-treated iRTN-1c MCA205 cells effectively protected most of the mice against rechallenge with live tumor cells. This protective effect was as high as that observed for the MTX-treated iRTN-1c MCA205 positive control cells (). In a subsequent round of experiments, we confirmed that pre-treatment with TET also greatly improved the immunogenicity of cells treated with mitomycin C. In line with previous reports, mitomycin C alone was a poor inducer of ICD,Citation17,Citation22 whereas cells that had been treated with TET prior to mitomycin C exposure induced a potent anticancer immune response (). Altogether, these results indicate that TET-induced Rtn-1c expression can cause cells to undergo ICD, even in response to agents like CDDP and mitomycin C that are innately poor ICD initiators when used alone.
Figure 2. Rtn-1c expression triggers immunogenicity of cell death mediated by CDDP or mitomycin C. (A and B) iRTN-1c MCA205 cells were treated with a single agent or combined with the tetracycline (TET)-induced expression of reticulon-1c (Rtn-1c) and assayed as anticancer vaccines by tumor re-challenge. iRTN-1c MCA205 cells were treated with 150 µM cisplatin (CDDP) or CDDP+ 0.3µM TET (A) or 200 µM mitomycin C (MITOC) or MITOC+ 0.3µM TET (B). 3 x 105 of these dying iRTN-1c MCA205 cells were inoculated s.c. into the left flank of C57BL/6 mice. PBS was injected as a negative control, and mitoxantrone (MTX) treated cells were used as a positive control. One week later, mice were re-challenged with 3 x 104 living MCA205 cells injected s.c. in the contralateral flank, and the absence of tumor growth was scored 60 d later as an indication of an anticancer immune response. The total number of mice for each experiment is indicated (n = 10–15/group) and was obtained by adding independent experiments. Differences between TET-treated groups and their untreated counterparts were analyzed using χ2 test (*P < 0.05).
Synthetic induction of ER stress induced by Rtn-1c improves cisplatin/ chemotherapy in vivo
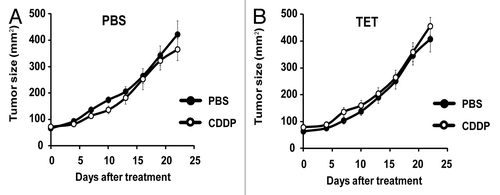
CDDP-based chemotherapy of MCA205 cells is inefficient unless CDDP is combined with agents capable of inducing ER stress.Citation40 Accordingly, we observed that the rate of tumor growth of either parental MCA205 cells or iRTN-1c MCA205 cultured in the absence of TET (i.e., without exogenous Rtn-1c) was not inhibited by systemic administration of CDDP (). Similar results were achieved with TET-treated parental MCA205 cells, such that both PBS- and CDDP-exposed cells gave rise to tumors with similar growth rates (). However, when the mice bearing iRTN-1c MCA205 tumor received continuous treatment with TET in the drinking water, and when TET was injected concomitantly with the treatment, such tumors did respond to CDDP chemotherapy by reducing their growth rate (). This TET effect was “on-target” (i.e., dictated by the expression of the Rtn-1c transgene) because parental MCA205 cells did not respond to a similar combination regimen of TET + CDDP (). Moreover, the combination of TET and CDDP failed to reduce the growth of iRTN-1c MCA205 cancers in immunodeficient nu/nu mice lacking thymus-dependent T lymphocytes evincing the requirement for an intact immune system in the implementation of the Rtn-1c-mediated protective effect (). Taken together, these results support the notion that synthetic induction of ER-stress response via Rtn-1c can stimulate anticancer immunity that significantly increases the efficacy of cisplatin/CDDP-based chemotherapy.
Figure 3. Tumor growth modulation by CDDP and tetracycline treatment in parental and iRTN-1c MCA205 tumors. (A‒D) Tetracycline (TET) mediated reticulon-1c (Rtn-1c) expression combined with cisplatin/CDDP chemotherapy effects the growth rate of established tumors. 2 × 105 parental MCA205 (A and B) or iRTN-1c MCA205 cells (C and D) were injected s.c. into the flank of C57Bl/6 mice. Once palpable, tumors were treated by a single i.p. injection of 0.25 mg/kg CDDP or vehicle (PBS) (day 0) and tumor growth was monitored with a caliper for 25 d. Normal water (A and C) or 100 µM dose TET (B and D) were added in bibber waters for the duration, starting 7 d prior to the cell injection. Moreover, PBS (A and C) or 0.3 µM tetracycline (B and D) were injected intratumorally on day 0. Experiments were done on groups of n = 5 mice and repeated at least twice. Results are reported as means ± SEM *P < 0.05 (unpaired Student’s t test).
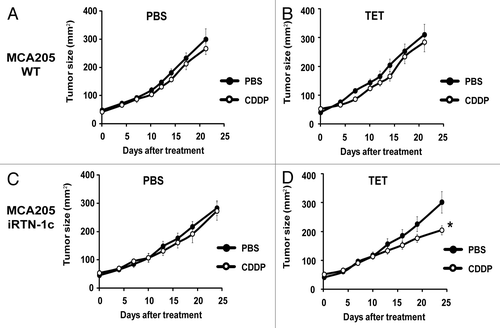
Figure 4. CDDP and tetracycline fail to reduce tumor growth in immunodeficient hosts. (A and B) Tetracycline (TET) mediated reticulon-1c (Rtn-1c) expression combined with cisplatin/CDDP chemotherapy is ineffective against established tumors in athymic nu/nu mice. 2 x 105 iRTN-1c MCA205 cells were injected s.c. into the flank of athymic nu/nu mice. Once palpable, tumors were treated by a single i.p. injection of 0.25 mg/kg cisplatin (CDDP) or vehicle (PBS) (day 0) and tumor growth was monitored with a caliper for 25 d. Normal water (A) or 100 µM dose TET-enriched water (B) were added in bibber bottles for the duration of the experiment, starting 7 d prior to the injection. Moreover, PBS (A) or 0.3 µM tetracycline (B) were injected intratumorally on day 0. Experiments were done on n = 5 mice per group and repeated at least twice. Results are reported as means ± SEM *P < 0.05 (unpaired Student’s t test).
Materials and Methods
Chemical, cell lines, and culture conditions
Unless otherwise indicated, media, antibiotics, and supplements for cell culture were purchased from Gibco-Invitrogen, chemicals were obtained from Sigma‒Aldrich, and plastic ware from Corning B.V. Life Sciences. Blasticidin and zeocin were obtained from Invivogen.
Murine fibrosarcoma MCA205 cells (Class I MHC haplotype H-2b, syngenic for C57BL/6 mice) cells were cultured in RPMI 1640 medium. iRTN-1c MCA cells were generated by transducing MCA205 cells with plasmids from the T-Rex system (Invitrogen). Briefly, Rtn-1, splice variant c (Rtn-1c) cDNA was inserted into the pcDNA4/TO vector, then co-transfected into MCA205 cells with the pcDNA6/TR plasmid expressing the TET repressor. MCA cells were further selected by blasticidin and zeocin for 1 week, and then cloned. All media were supplemented with 10% heat-inactivated fetal bovine serum (FBS), 10 mM HEPES, 10 U/mL penicillin sodium and 10 μg/mL streptomycin sulfate.
Immunoblotting
For immunoblotting, approximately 1 × 106 iRTN-1c MCA cells that had been incubated (or not, as control) with 0.3 µM tetracycline for 12–24 h, were washed with cold PBS, and lysed following standard procedures. Forty µg of proteins were separated according to molecular weight on NuPAGE® Novex® Bis-Tris 4–12% pre-cast gels (Invitrogen) and then electrotransferred to nitrocellulose membranes (Bio-Rad). Unspecific binding sites were blocked by incubating the membranes for 1 h in 0.05% Tween 20 (v/v in TBS) and supplemented with 5% bovine serum albumin, followed by overnight incubation at 4 °C with primary antibodies specific for β-actin (MAB1501) or reticulon-1c (sc-71982, Santa Cruz Biotechnology). Primary antibodies were detected with the appropriate horseradish peroxidase-labeled secondary antibodies (Southern Biotechnologies Associates) and were revealed with the ECL Plus Western Blotting Detection System (GE Healthcare). The abundance of β-actin was monitored to ensure equal lane loading.
Cell death assays
iRTN-1c MCA205 cells were treated (or not, as control) with 1 µM MTX, 150 µM CDDP, 0.3 µM TET, or CDDP+TET, for 18 h followed by the cytofluorometric assessment of propidium iodide uptake (PI) and phosphatidylserine (PS) externalization. Briefly, cells were washed twice with PBS and resuspended in 1X binding buffer supplemented with FITC-conjugated AnnexinV (BD Biosciences) and 0.5 μg/mL PI, following the manufacturer’s instructions. Cytofluorometric determinations were performed using a FACSCalibur cytofluorometer (Becton Dickinson) equipped with a 70 µm nozzle. First line statistical analysis of cytofluorometric results was performed using CellQuest™ software (BD Biosciences) by gating on the events characterized by normal forward scatter and side scatter parameters.
Determination of cell surface-exposed CRT by flow cytometry
iRTN-1c MCA205 cells were treated (or not) by 1 µM MTX, 150 µM CDDP, 0.3 µM TET, or CDDP+TET, for 24 h, prior to cell harvest and subsequent wash with ice cold PBS. After 30 min incubation with an anti-CRT antibody (Abcam) in cold blocking buffer (5% FBS, v/v in PBS) on ice, cells were further washed and incubated with AlexaFluor®488-conjugated secondary antibody (Life Technologies) in blocking buffer (for 30 min). Cells were finally washed and maintained in cold PBS with 1 μg/mL PI and samples were analyzed by means of a FACS Calibur cytofluorometer (BD Biosciences). Isotype-matched IgG antibodies (Cell Signaling Technology) were used a negative staining control, and the analysis was performed exclusively on non-permeabilized (PI-) cells. Data were statistically evaluated by means of the Cell Quest Software package (BD Biosciences).
ATP release assays
iRTN-1c MCA205 cells were treated (or not) by 1 µM MTX, 150 µM CDDP, 0.3 µM TET, or CDDP+TET for 24 h, then extracellular ATP levels were measured by the luciferin-based ENLITEN® ATP Assay (Promega), in excess of luciferin and luciferase, as indicated by the manufacturer. ATP-driven chemoluminescence was recorded on a Fluostar Multiwell Plate Luminometer (BMG Labtech).
Detection of HMGB1 release
2.5 × 105 iRTN-1c MCA205 cells were seeded in 6-well plate, permitted to adhere overnight and then treated (or not) with 1 µM MTX, 150 µM CDDP, 0.3 µM TET, or CDDP+TET for 48 h. Supernatants were collected and cleared of dying tumor cells by centrifugation (800 g, 5 min), then frozen at −80 °C or immediately analyzed for HMGB1 abundance. HMGB1 quantification was performed by an enzyme-linked immunosorbent assay (ELISA) kit (HMBG1 ELISA kit II, Gentaur Europe), according to the manufacturer's instructions.
Animal experimentations
Animals were maintained in specific pathogen-free conditions, and experiments followed the Federation of European Laboratory Animal Science Association (FELASA) guidelines. Animal experiments were approved by the local Ethics Committee (CEEA IRCIV / IGR n°26, registered with the French Ministry of Research) and were in compliance with EU 63/2010 directive. Animals were used between 6 and 20 wk of age and those bearing tumors exceeding 20–25% body mass were euthanized. C57BL/6 (H-2b) mice were obtained from Harlan, and athymic nu/nu mice were obtained from IGR local breeding.
Antitumor vaccination experiments
MCA205 or iRTN-1c MCA205 cells were incubated with 150 µM cisplatin or 200 µM mitomycin C for 24 h, alone or in combination with 0.3 µM tetracycline, and then subcutaneously inoculated (3 × 105 in 200 μL PBS, pH 7.4) into the lower flank of 6-wk-old C57BL/6 female mice. Seven days later, 3 × 104 untreated MCA205 cells were inoculated into the contralateral flank. Mice were then monitored for the appearance of tumors for 60 d.
Chemotherapy of established tumors in mice
C57BL/6 mice or athymic nu/nu mice were inoculated with 2 × 105 SCR MCA205 or iRTN-1c MCA205 cells. When tumor size reached 40–80 mm2, mice were assigned into homogenous groups (5 mice per group) and then treated intratumorally with 0.25 mg/kg CDDP (in 100 μL sterile water), alone or in combination with tetracycline (one 0.3 µM intratumoral dose (in 100 μL PBS pH 7.4) preceded by 100 µM dose added in Bibber water bottles 7 d prior to cell injection. The tumor surface was then monitored with a common caliper every 2/3 d for up to 25 d.
Statistical analyses
Data were compared by the χ2, or Student’s t tests, as appropriate. All P values were 2 tailed. P values < 0.05 were considered as statistically significant for all experiments. GraphPadPrism version 5 and Microsoft Excel 2007 software were used in data analyses.
Concluding Remarks
The efficacy of anticancer chemotherapeutics greatly depends on their capacity to stimulate anticancer immune responses.Citation2,Citation14,Citation28,Citation41-Citation43 One particular pathway allowing for the elicitation of immunotherapeutic activity against neoplastic lesions is via the induction of immunogenic cell death.Citation7 In this scenario, a selected panel of chemotherapeutic agents (such as anthracylines, including MTX) induces particular cellular stress and death pathways stimulating dying cancer cells to emit signals that activate pattern recognition receptors in innate immune effectors, thereby converting the tumor into a therapeutic vaccine. It appears that one particularly important limiting factor for the induction of ICD centers upon the induction of an ER-stress response culminating in the translocation of the ER chaperone CRT to the cell surface. Chemotherapeutic agents such as CDDP that fail to induce an ER-stress responseCitation17,Citation23 are unable to induce ICD unless they are combined with pharmacological ER-stress inducers. These include inhibitors of the protein phosphatase 1 (PP1)/GADD34 complex, e.g., tautomycin, calyculin A, salubrinal, or peptide antagonists,Citation16,Citation17 inhibitors of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), e.g., thapsigargin,Citation35 or inhibitors of the Na+/K+-ATPase, e.g., cardiac glycosides.Citation40 Here, we show that a precise genetic manipulation to induce ER stress, namely the TET-inducible expression of Rtn-1c, was capable of improving the chemotherapeutic efficacy of CDDP treatment in mice by enhancing anticancer immune responses mediated by thymus-dependent T lymphocytes. In contrast to the addition of chemical ER-stress inducers (such as inhibitors of the protein phosphatase 1/GADD34 complex, SERCA, or Na+/K+-ATPase), all of which may have a significant level of off-target effects, the expression of Rtn-1c can be expected to yield a relatively specific, on-target ER-stress response,Citation38 likely through an increase in the Ca2+ influx into the ER.Citation37 Hence, the outcome of our studies using the TET-inducible system to drive Rtn-1c mediated CRT surface exposure supports the conclusion that artificial enforcement of ER stress has a major therapeutic impact on the dialog between dying tumor cells and the immune system.
At present, the precise molecular mechanisms by which Rtn-1c overexpression combined with CDDP induces CRT exposure (relative to either agent alone that fails to do so) remains a conundrum. CDDP induces a major degree of redox stress,Citation33 which may be an important contributing factor in the induction of CRT surface exposure.Citation19 Furthermore, it is possible that the resultant changes in reactive oxygen species and altered luminal ER ion homeostasisCitation37 facilitate the synergistic interaction between CCDP and Rtn-1c. Elucidating a more detailed molecular comprehension of these mechanisms may open avenues for the design of pharmacological ICD inducers.
| Abbreviations: | ||
| APCs | = | antigen-presenting cells |
| CDDP/cisplatin | = | Cis-diamminedichloridoplatinum(II) |
| CRT | = | calreticulin |
| CTL | = | cytotoxic T lymphocyte |
| DC | = | dendritic cell |
| eIF2α | = | eukaryotic initiation factor 2α |
| ER | = | endoplasmic reticulum |
| ICD | = | immunogenic cell death |
| MDSC | = | myeloid-derived suppressor cell |
| MTX | = | mitoxantrone |
| Rtn1c | = | reticulon-1, splice variant c |
| TET | = | tetracycline |
| Tregs | = | regulatory T cells |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work is supported by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR); Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
Citation: Michaud M, Sukkurwala AQ, Di Sano F, Zitvogel L, Kepp O, Kroemer G. Synthetic induction of immunogenic cell death by genetic stimulation of endoplasmic reticulum stress. OncoImmunology 2014; 3:e28276; 10.4161/onci.28276
Related Research Data
References
- Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success?. J Clin Invest 2008; 118:1991 - 2001; http://dx.doi.org/10.1172/JCI35180; PMID: 18523649
- Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity 2013; 39:74 - 88; http://dx.doi.org/10.1016/j.immuni.2013.06.014; PMID: 23890065
- Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol 2011; 8:151 - 60; http://dx.doi.org/10.1038/nrclinonc.2010.223; PMID: 21364688
- Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol 2008; 8:59 - 73; http://dx.doi.org/10.1038/nri2216; PMID: 18097448
- Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol 2006; 6:715 - 27; http://dx.doi.org/10.1038/nri1936; PMID: 16977338
- Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009; 9:353 - 63; http://dx.doi.org/10.1038/nri2545; PMID: 19365408
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol 2013; 31:51 - 72; http://dx.doi.org/10.1146/annurev-immunol-032712-100008; PMID: 23157435
- Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Séror C, Métivier D, Perfettini JL, Zitvogel L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle 2009; 8:3723 - 8; http://dx.doi.org/10.4161/cc.8.22.10026; PMID: 19855167
- Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Métivier D, Galluzzi L, Perfettini JL, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ 2014; 21:79 - 91; http://dx.doi.org/10.1038/cdd.2013.75; PMID: 23852373
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334:1573 - 7; http://dx.doi.org/10.1126/science.1208347; PMID: 22174255
- Michaud M, Sukkurwala AQ, Martins I, Shen S, Zitvogel L, Kroemer G. Subversion of the chemotherapy-induced anticancer immune response by the ecto-ATPase CD39. Oncoimmunology 2012; 1:393 - 5; http://dx.doi.org/10.4161/onci.19070; PMID: 22737627
- Wang Y, Martins I, Ma Y, Kepp O, Galluzzi L, Kroemer G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy 2013; Forthcoming http://dx.doi.org/10.4161/auto.25873; PMID: 23989612
- Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013; 38:729 - 41; http://dx.doi.org/10.1016/j.immuni.2013.03.003; PMID: 23562161
- Ma Y, Adjemian S, Yang H, Catani JP, Hannani D, Martins I, Michaud M, Kepp O, Sukkurwala AQ, Vacchelli E, et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013; 2:e24568; http://dx.doi.org/10.4161/onci.24568; PMID: 23894718
- Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012; 31:1062 - 79; http://dx.doi.org/10.1038/emboj.2011.497; PMID: 22252128
- Kepp O, Gdoura A, Martins I, Panaretakis T, Schlemmer F, Tesniere A, Fimia GM, Ciccosanti F, Burgevin A, Piacentini M, et al. Lysyl tRNA synthetase is required for the translocation of calreticulin to the cell surface in immunogenic death. Cell Cycle 2010; 9:3072 - 7; http://dx.doi.org/10.4161/cc.9.15.12459; PMID: 20699648
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13:54 - 61; http://dx.doi.org/10.1038/nm1523; PMID: 17187072
- Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, Fimia GM, Kepp O, Piacentini M, Froehlich KU, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ 2008; 15:1499 - 509; http://dx.doi.org/10.1038/cdd.2008.67; PMID: 18464797
- Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 2009; 28:578 - 90; http://dx.doi.org/10.1038/emboj.2009.1; PMID: 19165151
- Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, Weiskopf K, Willingham SB, Raveh T, Park CY, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med 2010; 2:63ra94; http://dx.doi.org/10.1126/scitranslmed.3001375; PMID: 21178137
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005; 123:321 - 34; http://dx.doi.org/10.1016/j.cell.2005.08.032; PMID: 16239148
- Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 2005; 202:1691 - 701; http://dx.doi.org/10.1084/jem.20050915; PMID: 16365148
- Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010; 29:482 - 91; http://dx.doi.org/10.1038/onc.2009.356; PMID: 19881547
- West AC, Christiansen AJ, Smyth MJ, Johnstone RW. The combination of histone deacetylase inhibitors with immune-stimulating antibodies has potent anti-cancer effects. Oncoimmunology 2012; 1:377 - 9; http://dx.doi.org/10.4161/onci.18804; PMID: 22737621
- West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, Johnstone RW. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res 2013; 73:7265 - 76; http://dx.doi.org/10.1158/0008-5472.CAN-13-0890; PMID: 24158093
- Senovilla L, Vacchelli E, Galon J, Adjemian S, Eggermont A, Fridman WH, Sautès-Fridman C, Ma Y, Tartour E, Zitvogel L, et al. Trial watch: Prognostic and predictive value of the immune infiltrate in cancer. Oncoimmunology 2012; 1:1323 - 43; http://dx.doi.org/10.4161/onci.22009; PMID: 23243596
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science 2012; 337:1678 - 84; http://dx.doi.org/10.1126/science.1224922; PMID: 23019653
- Cirone M, Garufi A, Di Renzo L, Granato M, Faggioni A, D’Orazi G. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. Oncoimmunology 2013; 2:e26198; http://dx.doi.org/10.4161/onci.26198; PMID: 24228232
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007; 13:1050 - 9; http://dx.doi.org/10.1038/nm1622; PMID: 17704786
- Galluzzi L, Kepp O, Kroemer G. Immunogenic cell death in radiation therapy. Oncoimmunology 2013; 2:e26536; http://dx.doi.org/10.4161/onci.26536; PMID: 24404424
- Vacchelli E, Vitale I, Tartour E, Eggermont A, Sautès-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Anticancer radioimmunotherapy. Oncoimmunology 2013; 2:e25595; http://dx.doi.org/10.4161/onci.25595; PMID: 24319634
- Garg AD, Krysko DV, Vandenabeele P, Agostinis P. The emergence of phox-ER stress induced immunogenic apoptosis. Oncoimmunology 2012; 1:786 - 8; http://dx.doi.org/10.4161/onci.19750; PMID: 22934283
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene 2012; 31:1869 - 83; http://dx.doi.org/10.1038/onc.2011.384; PMID: 21892204
- Galluzzi L, Vitale I, Senovilla L, Olaussen KA, Pinna G, Eisenberg T, Goubar A, Martins I, Michels J, Kratassiouk G, et al. Prognostic impact of vitamin B6 metabolism in lung cancer. Cell Rep 2012; 2:257 - 69; http://dx.doi.org/10.1016/j.celrep.2012.06.017; PMID: 22854025
- Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011; 30:1147 - 58; http://dx.doi.org/10.1038/onc.2010.500; PMID: 21151176
- Tajeddine N, Galluzzi L, Kepp O, Hangen E, Morselli E, Senovilla L, Araujo N, Pinna G, Larochette N, Zamzami N, et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 2008; 27:4221 - 32; http://dx.doi.org/10.1038/onc.2008.63; PMID: 18362892
- Fazi B, Melino S, Di Sano F, Cicero DO, Piacentini M, Paci M. Cloning, expression, and preliminary structural characterization of RTN-1C. Biochem Biophys Res Commun 2006; 342:881 - 6; http://dx.doi.org/10.1016/j.bbrc.2006.02.036; PMID: 16500619
- Tufi R, Panaretakis T, Bianchi K, Criollo A, Fazi B, Di Sano F, Tesniere A, Kepp O, Paterlini-Brechot P, Zitvogel L, et al. Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ 2008; 15:274 - 82; http://dx.doi.org/10.1038/sj.cdd.4402275; PMID: 18034188
- Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 2009; 15:1170 - 8; http://dx.doi.org/10.1038/nm.2028; PMID: 19767732
- Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, Yamazaki T, Sukkurwala AQ, Michaud M, Mignot G, Schlemmer F, Sulpice E, Locher C, Gidrol X, Ghiringhelli F, Modjtahedi N, Galluzzi L, Andre F, Zitvogel L, Kepp O, Kroemer G. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med 2012; 4:143ra99; http://dx.doi.org/10.1126/scitranslmed.3003807
- Ma Y, Yamazaki T, Yang H, Kepp O, Galluzzi L, Zitvogel L, Smyth MJ, Kroemer G. Tumor necrosis factor is dispensable for the success of immunogenic anticancer chemotherapy. Oncoimmunology 2013; 2:e24786; http://dx.doi.org/10.4161/onci.24786; PMID: 23894723
- Riganti C, Massaia M. Inhibition of the mevalonate pathway to override chemoresistance and promote the immunogenic demise of cancer cells: Killing two birds with one stone. Oncoimmunology 2013; 2:e25770; http://dx.doi.org/10.4161/onci.25770; PMID: 24327936
- Vacchelli E, Galluzzi L, Fridman WH, Galon J, Sautès-Fridman C, Tartour E, Kroemer G. Trial watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2012; 1:179 - 88; http://dx.doi.org/10.4161/onci.1.2.19026; PMID: 22720239