Abstract
Renal biopsies commonly display tissue remodeling with a combination of many different findings. In contrast to trauma, kidney remodeling largely results from intrinsic responses, but why? Distinct danger response programs were positively selected throughout evolution to survive traumatic injuries and to regenerate tissue defects. These are: (1) clotting to avoid major bleeding, (2) immunity to control infection, (3) epithelial repair and (4) mesenchymal repair. Collateral damages are acceptable for the sake of host survival but causes for kidney injury commonly affect the kidneys in a diffuse manner. This way, coagulation, inflammation, deregulated epithelial healing or fibrosis contribute to kidney remodeling. Here, I focus on how these ancient danger response programs determine renal pathology mainly because they develop in a deregulated manner, either as insufficient or overshooting processes that modulate each other. From a therapeutic point of view, immunopathology can be prevented by suppressing sterile renal inflammation, a useless atavism with devastating consequences. In addition, it appears as an important goal for the future to promote podocyte and tubular epithelial cell repair, potentially by stimulating the differentiation of their newly discovered intrarenal progenitor cells. By contrast, it is still unclear whether selectively targeting renal fibrogenesis can preserve or bring back lost renal parenchyma, which would be required to maintain or improve kidney function. Thus, renal pathology results from ancient danger responses that evolved because of their evolutional benefits upon trauma. Understanding these causalities may help to shape the search for novel treatments for kidney disease patients.
Introduction
In Greek mythology Perseus used his sword to strike off Medusa's head, probably because he anticipated that this would kill her immediately. The causal relationship between this injurious trigger and its immediate tissue consequences fostered the culture of executions in its various forms. So far only Victual Brother captain Klaus Störtebeker on (October 20th, 1401 in Hamburg) was reported to contradict that scenario after his beheading, when his body arose and he walked upright past 11 of his men to save them from being executed along with him.Citation1
Most non-communicable diseases, however, arise from a mix of injury and the host's responses to it. Renal pathology, especially, is rather dominated by a complex process of tissue remodeling to which the injurious trigger accounts only partially and the kidneys get rather destroyed by the body's response to this trigger. But why did evolution develop processes that destroy our organs? Decisions during evolution are generally based on a careful risk-benefit assessment which implies that the processes that finally destroy kidneys provide some other significant survival benefits that outweigh renal damage. I will address this question by elaborating a unifying concept of (renal) pathology which is based on the following assumptions: (1) evolution has positively selected four major response programs to address the potentially life threatening dangers of traumatic injury from the state of primitive multicellular organisms, i.e., clotting, inflammation, epithelial and mesenchymal healing, (2) kidney remodeling results from deregulated danger response programs and (3) suppressing inflammation and promoting epithelial regeneration are the most promising approaches to prevent the loss of renal tissue, kidney remodeling and chronic kidney disease (CKD).
Danger Control after Trauma
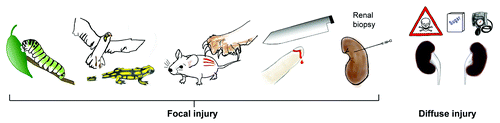
Multicellular organisms need to maintain tissue integrity and any traumatic injury creates an immediate and potential fatal threat.Citation2 As such there is a need for sealing the wound and for tissue regeneration, a concept that applies to plants and animals.Citation3,Citation4 Upon focal injury, danger responses control the problem locally, which is an important benefit to assure the survival of the entire organism. Survival outweighs the risk of any focal collateral damage that may be associated with these danger responses, e.g., focal immunopathology and scarring (). The same concept applies to focal injuries to the kidney, such as biopsy injuries, infective pyelonephritis or even unilateral renal artery stenosis. However, nowadays many clinically common entities of kidney injury are of toxic, hemodynamic or metabolic nature, which hit both of the kidneys in all of their compartments. Nature has not yet developed (and probably will not in the future) different danger response programs to avoid the loss of solid organs that are absolutely necessary for survival. The reason for this is simple, even though it may be hard to accept for the renal scientist: renal failure was not (and probably never will be) a significant selection pressure for the survival of a species as a whole. Therefore, kidney tissue remodeling and renal failure in humans is an—from an evolutionary point of view—acceptable side effect of the otherwise very effective danger response programs that effectively assured the survival of multicellular organisms after traumatic tissue injuries.
Figure 1. Traumatic injuries across species and kidney disease. Coordinated danger response programs are of benefit for multicellular organisms, even if they cause some collateral tissue damage or persisting defects. However, metabolic, hemodynamic or toxic factors rather hit the kidney in a diffuse manner which is why collateral damage affects the entire renal tissue and progresses to end-stage renal disease.
The Four Elementary Danger Response Programs in Skin Trauma
Wound healing after skin injury is a useful example to illustrate the four danger response programs in a temporal manner.Citation5-Citation7
Clotting
Skin trauma usually induces bleeding which involves the potential risk of significant blood loss and hemorrhagic shock. Clotting, a complex interplay of the injured endothelial cells, the coagulation factors and platelet aggregation, represents the first danger response program that, within minutes, often perfectly controls this risk.Citation8 Insufficient clotting implies the risk for major bleedings, while overshooting clotting causes tissue ischemia via intravascular coagulation or thromboembolism.
Inflammation
Eukarionts compete with prokarionts for nutritients and space of their biotope. This battle between eukarionts and prokarionts started from monocellular organisms and represents the underlying concept behind the evolution of the immune system and the clinical concepts of infectious diseases and host defense.Citation9 Traumatic injury of metazoan, such as skin wounding, involves the risk of pathogen entry and potentially fatal sepsis, a danger that has been addressed by rapid induction of a local inflammatory state that intends to control pathogen entry and spreading. Sponges and other primitive organisms use several hundreds of Toll-like receptors to recognize their microbial environment and to initiate inflammation and appropriate host defense mechanisms.Citation10-Citation12 It is important to note that extrinsic pathogen-associated molecular patterns (PAMPs), as well as intrinsic damage-associated molecular patterns (DAMPs), can activate the same innate immunity pattern recognition receptor platforms which explains why infectious and sterile forms of inflammation initiate identical inflammatory responses.Citation13,Citation14 Inflammation is already activated by clotting, as aggregating platelets release chemokines that trigger the recruitment of neutrophils and subsequently other leukocytes that attack, and ideally neutralize, pathogens.Citation15 Local inflammation and abscess formation support host survival while systemic inflammation during sepsis, a deregulated version of the same response program, implicates life-threating consequences.Citation16 Insufficient host defense implies the risk of potentially fatal infection while overshooting local inflammation causes unnecessary immunopathology and loss of parenchyma, e.g., in pyoderma gangraenosum.Citation17
Epithelial healing
In wounds of non-sterile outer and inner surfaces the risk of infection can only be sufficiently controlled when the external barrier defect is rapidly repaired.Citation7,Citation18 This is achieved by immediate signals that trigger re-epitheliazation from the borders of the wound.Citation4,Citation7,Citation19 Superficial erosions of the epidermis or the corneal and conjunctival epithelium heal rapidly also in adults.Citation4,Citation7,Citation19 Coagulation factors like fibrinogen and platelet-derived growth factors are the first mediators within the wound with direct mitogenic effects on surviving cells in the epithelial compartment.Citation4,Citation8,Citation15,Citation19,Citation20 During the inflammatory phase, epithelial growth factors, as well as certain interleukins like IL-6, IL-17 and IL-22, stimulate epithelial repair.Citation19,Citation21-Citation27 Epithelial healing depends on the presence of local progenitor cells that are committed to the specific epithelial lineage phenotype.Citation18,Citation28,Citation29 Progenitor-mediated re-epitheliazation should be a highly controlled and coordinated process requiring appropriate activation, proliferation, migration and finally differentiation into the final phenotype. Insufficient re-epitheliazation implies a persistent risk for infections and creates chronic wounds while overshooting or uncoordinated re-epitheliazation which causes hyperplastic wounds.Citation4
Mesenchymal healing/fibrosis
Insufficient re-epitheliazation or injury to mesenchymal tissues activates the wound healing program of mesenchymal healing. This process stabilizes the organ structure, regrows vasculature or fills the gap of lost parenchymal cells that cannot regrow. After a bone fracture or a tear of a ligament, but also insufficient epithelial repair stimulates mesenchymal healing, e.g., through the process of epithelial-mesenchymal transition (EMT) of epithelial cells that get arrested in the G2/M phase.Citation30 The number of collagen-producing cells increases either by EMT,Citation31,Citation32 by the influx of bone marrow-derived fibrocytes,Citation33,Citation34 by the transition of pericytes or endothelial cellsCitation34,Citation35 or simply by proliferating resident fibroblasts and their transition to myofibroblasts.Citation35 The accumulation of extracellular matrix, i.e., fibrosis, leads to stiffening of the tissue, i.e., sclerosis. Insufficient mesenchymal healing may destabilize tissues and result in chronic wounds, while overshooting mesenchymal healing produces keloid and scleroderma.
The sequential activation of these danger response programs has been, and still is, essential to assure the survival of multicellular organisms so that they can reach the age of reproduction. Children and adolescents have to survive many skin wounds until the next generation is born; wound healing is also necessary to survive a baby’s delivery. Given the enigmatic importance of these danger response programs it comes as no surprise that injuries to internal organs involve the activation of the identical series of mechanisms.
Clotting in Renal Pathology
There are several renal disorders that directly relate to clotting disorders such as the thrombotic microangiopathies or phospholipid antibody syndrome, which originate from intense activation of the endothelia.Citation36,Citation37 Microvascular clotting causes tissue ischemia and eventually necrosis (). Vascular damage with GBM perforations and plasma leakage also occurs in intense tissue inflammation, e.g., in crescentic glomerulonephritis.Citation38 Vascular damage implies bleeding, which manifests as hematuria in the kidney, a process that should activate clotting. It is well documented that crescentic glomerulonephritis is associated with glomerular fibrinogen deposition especially in the area of loop necrosis.Citation39,Citation40 The same applies to vascular disintegration in Alport nephropathy which lacks glomerular inflammation but where the genetic GBM abnormality promotes vascular injury.Citation41 Activated platelets release coagulation factors as well as proinflammatory and mitogenic factors which activate inflammation and epithelial healing, e.g., CC-chemokines, platelet-derived growth factors (PDGF) and others.Citation8,Citation15,Citation19
Table 1. Danger response programs and typical histopathologic abnormalities of the kidney
Insufficient or Overshooting Inflammation in Renal Pathology
Renal inflammation is a major determinant of AKI, as well as the progression of CKD. The network of resident dendritic cells inside the kidney and infiltrating mononuclear phagocytes are fully equipped with the entire spectrum of pattern recognition receptors that can activate innate immunity.Citation42,Citation43 In addition, the renal parenchymal cells express a limited spectrum of such receptors.Citation44,Citation45 Mesangial cells, endothelial cells, podocytes, tubular cells and renal fibroblasts lack some of the endosomal nucleic acid-specific Toll-like receptors and the NLRP3 inflammasome but they readily get activated by exposure to bacterial endotoxin and other PAMPs and DAMPs.Citation46,Citation47 For example, infections cause a transient exposure to PAMPs that have the potential to activate cells and trigger inflammatory responses inside and outside the kidney. We have demonstrated this concept by transiently injecting agonists into mice with experimental immune complex glomerulonephritis or lupus nephritis and could demonstrate that they activate the intrarenal production of cytokines, type I interferons which increases inflammation and tissue damage.Citation48-Citation58 By contrast, the NLRP3 inflammasome contributes to interstitial, but not to glomerular, disease.Citation59-Citation61 The type of response depended on the cell-type specific expression of the involved receptors which explains the heterogeneity of infection-associated flares of, e.g, IgA nephropathy, lupus nephritis and renal vasculitis.
Another important aspect of PAMP-mediated renal inflammation is the loss of renal parenchymal cells, especially podocytes, because these cannot be easily regenerated. When mice with Alport nephropathy were transiently injected with bacterial CpG-DNA, resident dendritic cells and infiltrating Ly6Chigh+ macrophages were stimulated to produce more proinflammatory mediators including the proapoptotic cytokine tumor necrosis factor-α which induced podocyte apoptosis and accelerated progressive glomerulosclerosis.Citation62
Renal inflammation does not only result from extrinsic stimuli. Any kind of tissue damage that causes renal cell necrosis can cause the release of DAMPs from intracellular compartments, which signals “danger” to the innate immune system.Citation44,Citation63,Citation64 For example, renal ischemia-reperfusion injury does not involve PAMP exposure but tubular cell necrosis induces a TLR-dependent acute intrarenal inflammatory response which largely contributes to the extent of AKI and loss of renal function.Citation65-Citation68 Surprisingly, TLR signaling occurs mostly in renal parenchymal cellsCitation44 because TLR signaling in the intrarenal network of dendritic cells is efficiently suppressed by the constitutive and induced expression of TLR signaling inhibitors thatare absent or dysfunctional in tubular epithelial cells.Citation69-Citation72 As another example, lupus nephritis is a state of abnormal immune recognition of endogenous nucleic acids.Citation73,Citation74 Nuclear particles that contain immunostimulatory endogenous RNA and/or DNA activate antigen-presenting dendritic cells, macrophages, and B cells, to mature and to release numerous proinflammatory mediators including type I interferon.Citation74,Citation75 The latter set off a coordinated antiviral immune response explaining the similarities between the clinical manifestations of viral infections and systemic lupus.Citation76 This process also occurs inside the kidney as documented by the antiviral gene expression signature in renal biopsies.Citation77,Citation78 For example, mesangial cells and glomerular endothelial cells use their cytosolic nucleic acid sensors to translate nucleic acid recognition into the release of type I interferons which contribute to renal inflammation and tissue damage.Citation79-Citation84
Innate immune activation triggers the local release of cytokines and chemokines which attracts various subsets of leukocytes into the kidney.Citation85-Citation88 Macrophages, T cells and B cells occur in different subsets that all differently contribute to the regulation of inflammation and renal immunopathology.Citation89,Citation90 Activating intrarenal macrophages, e.g., by transient infections or certain drugs, can turn non-activated into activated leukocytes which strongly contribute to renal pathology.Citation91-Citation93 Blocking the recruitment of pro-inflammatory macrophages by interfering with the CC-chemokine CCL2 or its receptor CCR2 probably reduces renal immunopathology in the glomerular as well as the tubulointerstitial compartment in those diseases that are associated with renal inflammation, but not in non-inflammatory forms like Alport nephropathy.Citation94-Citation99 However, TLR-mediated renal inflammation remains an important element of pathogen control in renal infections. For example, BK virus infection of the allograft requires TLRs and cytosolic nucleic acid receptors to translate pathogen recognition into appropriate antiviral host defense.Citation100 The enigmatic importance of renal inflammation also becomes evident during bacterial pyelonephritis.Citation101 Insufficient pathogen recognition and renal inflammation will allow pathogen overgrowth and pathogen spreading, which holds the risk of systemic infection. For example, mice that lack a functional LPS receptor, i.e., TLR4, and that are inoculated with uropathogenic E. coli into the urinary bladder, are protected from renal abscess formation because they can no longer recognize E. coli LPS and lack the appropriate chemokine signaling that would be required to recruit neutrophils into the infected kidneys.Citation102 This apparent protection from immunopathology occurs at the price of insufficient pathogen control at the entry site and could cause fatal bacterial spreading across the entire organism.
Together, the kidney is mostly a sterile organ in which pathogen control, the evolutionary rationale for inducing innate immunity, remains a rare event. As such, the kidney is mostly affected by renal inflammation that is triggered by remote infections that release immunostimulatory elements into the circulation or by intrarenal release of DAMPs that promote a sterile inflammatory response which predominately promotes unnecessary (collateral) damage to renal cells, a useless atavism of wound healing (). Thus, suppressing renal inflammation should be a valid strategy to preserve renal tissue, especially renal epithelial cells and vasculature. Drugs with anti-inflammatory properties without systemic immunosuppressive effects may be sufficient for that.Citation103 The protection of podocytes from unnecessary inflammatory stress is of major importance because lost podocytes can hardly be regenerated and the subsequent glomerulosclerosis still accounts for the majority of CKD cases.
Insufficient or Overshooting Epithelial Healing in Renal Pathology
In the glomerulus as well as within the tubules the epithelial cells are of enigmatic importance to the specific function of the compartment. After a transient and short-term ischemic or toxic tubular injury sufficient epithelial repair can rapidly restore renal function.Citation104,Citation105 Numerous growth factors drive the repair of the epithelial monolayers after injury.Citation19 For example, PDGF is already released by platelets or injured epithelia during the early phase of injury.Citation8,Citation15 BMP-7 and its receptor activin-like kinase-3 also contribute to epithelial healing.Citation106 Furthermore, cell cycle regulators like murine double minute (MDM)-2 assure the proliferative response during AKI by inhibiting p53-dependent cell cycle arrest.Citation107 Epithelial healing becomes more evident after the down-modulation of intrarenal inflammation.Citation108 The removal of necrotic cells and their DAMPs by infiltrating phagocytes changes the renal microenvironment which promotes a phenotype shift of the intrarenal mononuclear cells toward anti-inflammatory and pro-regeneratory phenotypes.Citation90 This process is associated with the release of additional growth factors that drive epithelial repair in the kidney (and the liver).Citation108-Citation111 A persistent activation of intrarenal mononuclear cells toward proinflammatory phenotypes, e.g., due to aberrant genetic macrophage priming such as IRAK-M deficiency (own unpublished data) or repetitive/persistent triggers of kidney injury impair this epithelial healing response. In addition, severe forms of kidney injury may also kill the epithelial progenitor cells which are located within the renal epithelial monolayer and that have a higher capacity to survive stress.Citation112 Even though it was clearly demonstrated that renal epithelial cells repair without a cellular contribution from bone marrow stem cells, the contribution of local progenitor cells vs. epithelial renewal from differentiated tubular epithelial cells to tubular repair from inside the tubular compartment remains under debate.Citation113-Citation117 However, it is obvious that an insufficient repair of the tubular epithelial monolayer will lead to tubular atrophy and nephron loss, a typical characteristic of progressive CKD ().
Insufficient epithelial repair in the glomerular compartment is the predominant cause for CKD. The particular structure of the differentiated podocyte which is required to fulfill its important function at the glomerular filtration barrier remains a major obstacle for rapid repair.Citation118-Citation120 There has been a controversial debate regarding whether bone marrow-derived progenitors are able to replace lost podocytes.Citation121-Citation123 It has now been convincingly demonstrated that podocytes originate from local epithelial progenitors at the urinary pole of the glomerulus that can migrate to the vascular pole and differentiate into terminally differentiated podocytes on the glomerular tuft.Citation124 This process clearly operates during renal development and early childhoodCitation101 but its capacity to replace injured podocytes seems to be limited in adult mice.Citation125,Citation126 The mechanisms that regulate podocyte renewal within the epithelial monolayer of glomerular visceral and parietal epithelial cells are still poorly understood. There is increasing evidence that those factors that regulate other stem cell compartments, such as Notch and Wnt signaling, EGF and SDF-1/CXCL12 also regulate podocyte renewal from their local progenitors.Citation127-Citation130 Particularly, heavy proteinuria seems to suppress podocyte repair because glomerular diseases that are associated with nephrotic syndrome show the highest tendency to progressive glomerular scarring, i.e., glomerulosclerosis.Citation131,Citation132 This process is associated with specific epigenetic imprinting at histone H3K9, H3K23 (acetylation), H3K4 (dimethylation) and H3K4 phosphorylation at serine 10 which alters gene expression and cell growth.Citation133,Citation134
Overshooting epithelial repair also exists in the kidney, especially when the epithelial cells at the progenitor cell location get heavily activated in the absence of the necessary signals for differentiation.Citation112,Citation135,Citation136 For example, rapid progressive glomerulonephritis combines glomerular vascular damage and fibrinogen activation with intense intraglomerular inflammation.Citation128,Citation137,Citation138 All these factors heavily activate parietal epithelial cells to proliferate without any apparent differentiation into podoctes.Citation41,Citation136 Upon rupture of Bowman’s capsule infiltrating macrophages and T cells provide additional stimuli for the proliferation of epithelial cells.Citation137,Citation138 We used the model of Col4A3-deficient mice in the 129/X1/SvJ genetic background to study crescent formation in the absence of glomerular inflammation. Disruption of glomerular capillaries causes plasma leakage which is sufficient to trigger the uncoordinated proliferation of parietal epithelial cells, a cell type that normally resides, devoid of serum contact, in the urinary space.Citation41
Overshooting epithelial healing in the tubular compartment is less evident. The tubular progenitor cells are located at the junction of glomeruli and proximal tubules while single progenitor cells are scattered in the proximal and distal tubules of the cortex.Citation139 Chevalier, et al. have recently demonstrated that the phenomenon of atubular glomeruli originates from an obstruction of the tubular lumen by epithelial cells.Citation140,Citation141
Together, epithelial repair is an important element to regain homeostasis after injury. Insufficient epithelial repair is the central element of glomerulosclerosis and tubular atrophy. Overactive epithelial repair can cause other forms of renal pathology, i.e., glomerular crescents. It should be desirable to stimulate a coordinated proliferation and differentiation of surviving epithelial cells of their local progenitor cells.
Mesenchymal Healing in Renal Pathology
Tissue healing is not limited to epithelia. Bleeding (skin) wounds imply damage to mesenchymal structures. Neo-angiogenesis, bone fractures, ligament ruptures or muscle damage require mesenchymal healing. In addition, healing in general implies the need for mechanical stabilization also of parenchymal tissues like the kidney, i.e., epithelial growth factors are secreted together with pro-fibrotic growth factors that stimulate the mesenchymal cells of the tissues to mature and to increase the secretion of extracellular matrix.Citation19 For example, in skin wounds resident tissue fibroblasts are activated to mature to myofibroblasts that produce type I and other collagens and to promote wound contraction which reduces the wound area, a process that limits infections and that supports re-epithelisation.Citation4 In areas of irreversible tissue loss, e.g., after ligament rupture or severe burning of the skin, fibrous tissue fills the gap of the lost tissue. In the long run, fibrolysis, the process of removing fibrous tissue after the disease process has ended, leads to a reduction of scar size to the minimum of what is absolutely required to stabilize the tissue.Citation4 These outstanding beneficial effects of mesenchymal healing should not be forgotten when questioning why many different renal disorders lead to a common scarring pathway. Again, during evolution the benefits of fibrosis outweigh the problems which, however, can become the predominant problem of few individuals. For example, the contraction of the fibrous skin wounds of badly injured skin can reduce joint mobility which, still, remains a focal problem. In the kidney, again, the diffuse nature of metabolic, hemodynamic, and toxic injuries implies that the process of fibrosis affects all of the kidney tissue to the same extent (). In this context, wound contraction implies the development of shrunken kidneys. However, it is likely that fibrosis itself does not destroy the kidney. The extracellular matrix mainly replaces lost renal epithelia. As such, reducing fibrosis may result in even smaller kidneys, if not accompanied by regeneration of intact nephrons. For example, repetitive mesenchymal stem cell injection into Col4A3-deficient mice with progressive Alport nephropathy significantly reduced interstitial fibrosis of the kidney but did not improve renal function and overall survival.Citation142 Renal interstitial fibrosis, even though an extremely interesting process with multiple fascinating aspects to study with the latest technologies,Citation32,Citation35,Citation143 rather seems to be a marker of loss of renal parenchyma rather than a pathogenic factor of renal failure by itself. It is of note that the driving factor of interstitial fibrosis seems to be an insufficient epithelial repair, e.g., when proliferating epithelial cells get arrested in the G2/M phase and start to produce tumor growth factor-β,30 a process, e.g., also triggered by aristocholic acid,Citation30,Citation144 the pathogenic element of Chinese herb nephropathy.Citation145 This concept implies that in the kidney mesenchymal healing could be a second-line healing program that predominates whenever epithelial healing is too slow or of low capacity.
This certainly applies for the glomerular compartment because podocyte renewal from local progenitors is mostly insufficient, especially in proteinuric disorders of the adult.Citation112,Citation132 Smeets et al. have recently demonstrated that in FSGS parietal epithelial cells do not sufficiently differentiate into mature podocytes but rather increase the production extracellular matrix.Citation146 Ideally, this scar formation stabilizes the glomerular pathology by forming a stable synechia or focal scar, like it may happen in some forms of secondary focal-segmental glomerulosclerosis. However, when a higher percentage of glomerular podocyte gets lost, the scarring process has its own dynamic, and rapidly progresses to global glomerulosclerosis,Citation120 mostly because the hyperfiltration of the remaining glomerular loops puts additional dynamic stress on the initially unaffected podocytes.Citation118,Citation147 A mesenchymal transition of parietal epithelial cells also contributes to the formation of fibrocellular crescents.Citation148 We recently documented that crescent formation in Col4A3-deficient mice with Alport nephropathy is associated with a phenotype switch of the proliferating parietal epithelial cells.Citation41 Parietal cells in cellular crescents lose their epithelial polarity and start to produce extracellular matrix into all directions like mesenchymal cells which leads to the honeycomb-like transformation of the Bowman's space into fibrocellular crescents.Citation41 This process is conceptually similar to the epithelial-mesenchymal transition of tubular epithelial cells which is thought to contribute to the heterogeneity of interstitial fibroblasts.Citation35 The significance of this phenomenon for human kidney disease remains under debate because clear evidence is still lacking that the transformed epithelial cells actually leave the tubular compartment by passing through their tubular basement membrane in vivo.Citation32,Citation149-Citation151 In the glomerulus, however, parietal epithelial cells that do not adequately differentiate into podocytes clearly undergo this mesenchymal transition and cause scarring without leaving their compartment.Citation41,Citation148
Infiltrating leukocytes and other bone marrow progenitors contribute to renal fibrosis, as reducing leukocyte recruitment markedly reduces interstitial fibrosis in various models of glomerular or tubulointerstitial kidney disease.Citation85,Citation152-Citation155 Whether this is a direct or an indirect effect is less clear.Citation99,Citation156,Citation157 For example, blocking or deletion of the chemokine receptor CCR1 prevents the recruitment of alternatively activated macrophages with a “wound healing” phenotype which can modulate the fibrotic process directly.Citation90,Citation158-Citation165
Together, mesenchymal repair is an important element to stabilize tissues after traumatic injury, especially after loss of parenchymal tissue. In the kidney, insufficient scarring does not seem to be a problem. Glomerular scarring, glomerulosclerosis and the formation of fibrocellular crescents, is the greatest concern because of their irreversible nature and the missing capacity to regrow lost nephrons. Why scarring does not occur inside the tubular compartment remains a miracle, but the interstitial compartment is commonly affected. Fibroblast activation and matrix deposition seem to fill the gaps left by dying nephrons. However, because nephrons do not grow back, this mechanical stabilization of the renal tissue is another useless atavism and the contraction of the fibrous tissue further contributes to kidney shrinking. Whether reducing renal fibrosis alone can regain renal function without replacing the lost nephrons remains under debate.
Summary
Traumatic tissue injury endangers the survival of multicellular organisms, hence, a series of danger response mechanisms were positively selected throughout evolution. Clotting and inflammation prevent bleeding and infection and are followed by epithelial and mesenchymal healing for tissue repair. During focal injuries these responses assure survival even when collateral damage leads to persistent defects such as focal immunopathology and scarring. The kidneys, however, are mostly affected by toxic, metabolic, hemodynamic or immune-mediated triggers of injury which affect all parts of both kidneys at the same time. Therefore, clotting and inflammation, as well as abnormal healing responses, often trigger remodeling of the renal tissue and promote additional and potentially progressive kidney injury. From a therapeutic perspective, the inhibition of renal inflammation together with the stimulation of epithelial healing without promoting mesenchymal healing should be the most promising strategy.
Dr Jeffrey Miner, Professor of Medicine, Washington University School of Medicine:
Do you know when in the Alport disease the proliferative response begins? When can you first detect Ki67 positive podocytes or parietal cells?
Dr Hans-Joachim Anders, Professor of Medicine, Klinikum der Universitat Munchen:
We noticed crescents already at early stages of the disease. Within weeks there are global crescents. Then later crescents become more prominent. We have not really tried to find out what is the earliest time point when that starts. There is quite some variability also on that point. In the end as you saw, the mortality curve is quite tight. The mice more or less died between 8 and 10 weeks of age.
Dr Miner:
My immunologist colleagues tell me that LPS induces TNF α expression by the liver such that it can explain all of the downstream effects. Are you saying that it must be produced within the kidney to have the effects that you are seeing?
Dr Anders:
It was an unexpected finding for us to see that repetitive LPS injections did not do anything to impact the progression of Alport nephropathy. Of course, then we focused more on the positive finding on the CpG-DNA, so we did not really follow-up on the question, why didn’t we see anything with LPS. The easiest explanation would be that the repetitive injections were more or less turning down the system. Which should also work somehow with CpG-DNA but it is far less dominant because the LPS receptor TLR4 is expressed in all cells, also inside the kidney. Whereas the receptor for CpG-DNA is only expressed on the immune cells, on B cells and macrophages of which we mainly find the macrophages inside the kidney of Alport mice. So we think with that we mainly modulated the inflammatory component that comes from the macrophages. Whereas, one would have expected that, because the receptors are everywhere, the effect of LPS would be much stronger, so the only explanation I have is that it is a downregulatory effect that starts from the second injection.
Dr Miner:
Did you try combining the LPS with the CpG-DNA?
Dr Anders:
No.
Dr Helen Liapis, Professor of Pathology and Immunology and Medicine, Washington University School of Medicine:
I would like you to expand on your concept about environmental dangers. In the Alport case, what do you think may be an environmental danger response in humans who carry an inherited mutation and how that will work with your concept.
Dr Anders:
I mean, we used agonists for bacteria and injected them systemically. So from that point of view one would conclude that at least systemic infections should provide that kind of stimulus to promote the progression of the disease. We have done a lot like that in lupus models where you clinically already have the experience that infections could cause a flare. We did the same kind of studies in lupus mice and were injecting all different kinds of Toll-like receptor agonists and then carefully described how does the flare develop and that may be related to the cell-type specific expression of the Toll-like receptors in the end.Citation14,Citation93 From there we knew that it is really the cells inside or outside the kidney get stimulated by that particular agonist and then you get more inflammation. In Alport's you anyway have a low grade of inflammation and the inflammation increases only upon a particular stimuli. I would raise the hypothesis, that infections are no good for Alport kids or any people with CKD, with a smoldering disease process and that any infection that would transiently activate innate immunity could then increase cytokine release and attract more leukocytes, and cause more kidney damage. I think the clinical experience from IgA nephropathy is pretty clear, that you have even vascular ruptures causing macrohematuria in association with an infection which then goes away after a while. Those of you who see lupus patients or patients with renal ANCA vasculitis know very well that each flare takes away some part of the renal function and that flares are indicators of bad outcomes.
Dr Liapis:
Are there any data on TNF in humans or in the serum in patients with Alport?
Dr Anders:
Not that I know. No one has looked at it.
Dr Feng Chen, Associate Professor of Medicine, Washington University School of Medicine:
You mentioned that scars occur in tubular injury because the repair may come too slowly. That certainly is the case. In the glomerulus, proliferation at a high rate may also cause problems like the loss of polarity. For the parietal cells to replace the damaged podocytes, they have to migrate a long distance in order to reach the correct location for repair. Is it possible that for optimal repair of the glomerulus, it might be better to slow down the proliferation? Maybe slower is better in this case.
Dr Anders:
Yes, we actually do that in renal vasculitis by treating the patient with cyclophosphamide so we reduce the proliferation. Repair mechanisms have their good and bad sides. It is well known that an overgrowth of bone marrow stem cells can lead to myeloproliferative syndromes and if bone marrow stem cells don’t proliferate enough you get myelodysplastic syndromes. That means you need a coordinated proliferation of those cells to maintain homeostasis. If it is too few it can cause problems. If it is too much it can also cause problems. Now that the cells have been identified, I think it is pretty clear that, for example FSGS is a disease, where the podocyte repair is insufficient. There is loss of podocytes and somehow the podocyte progenitors within the parietal epithelial cell compartment cannot compensate that and that is why you end up with a scar. If you have crescentic disease you get dramatic activation of this regenerative response. That is why you get a crescent. What we need is that repair happens in a coordinated manner and I think you can imagine that if you have a progenitor which is quiescent it first needs to be activated. We saw that WT-1 gets induced, which means these cells get activated toward a podocyte phenotype. Then they should proliferate first and then migrate and when it arrives at the glomerular tuft it should differentiate into a podocyte. You need those four steps to happen in a coordinated way. Let’s just imagine that if you have a vascular lesion like in ANCA vasculitis you have a dramatic inflammation of the glomerulus, vascular disruption, you have seen the picture by Steve Bonsib. You get serum leakage, these cells get heavily activated to proliferate but not to differentiate to the same extent. That means you get an overgrowth of undifferentiated progenitors, you get a crescent. That is probably what happens and somehow in FSGS you get podocyte loss. There is proteinuria. The progenitor cells should get activated but somehow they don’t proliferate as they should or a least they don’t differentiate. I think Marcus Moeller has shown, just a few months back, very nicely, that the parietal cell makes the scar in FSGS.Citation146 The cells go there but then they are unable to differentiate so they stay there and they use the second program, they produce matrix and that is why you end up with a scar. This work raises the idea that many of the diseases that we see relate to an over stimulation or blockade of the intrinsic capacity of the kidney to repair itself.
Dr Ying Maggie Chen, Assistant Professor of Medicine, Washington University School of Medicine:
The GBM gaps are caused by matrix metalloproteinase (MMP) digestion, right? Is MMP mainly produced by macrophage cells or by podocytes because podocytes can also upregulate MMP expression?
Dr Anders:
The published data are more in support of that MMP12 is released from macrophages.Citation166
Dr Y.M. Chen:
Regarding the animal model, you are using collagen IV α 3 knockout mice on the genetic background of SV129, which develops Alport much faster than another genetic background. Would you think that the findings on SV129 can be replicated in another genetic background?
Dr Anders:
That is a good question. We thought about this but could not find a reason to do the experiment because we were mainly using that as a model so that means we needed crescents to ask the question: how do they develop? Dr. Liapis had looked at human Alport cases and were able to find only very few crescents. There are a small number of papers describing crescent also in human Alport but, at least, in this cohort it was less than 1%.Citation41 So that means, I think it is not so interesting to find out whether it also happens in another mouse, if it is not a big problem in humans. I would think that certainly the genetic background plays an important role here.
Dr Marc Hammerman, Chromalloy Professor of Medicine, Washington University School of Medicine:
Are there any circumstances in humans or in mammals in which we have evidence that renal repair is accomplished on the basis of stem cell migration and differentiation or does this only apply in fish?
Dr Anders:
Marcus Moelle was able to trace the parietal cells that were stained in blue in his mouse then move on the tuft.Citation125 It was published in the same JASN edition along with the paper of Paola Romagnani demonstrating that parietal cells harbour renal progenitor cells. Her data were all generated in humans.Citation124 Markus data were generated in the mouse.Citation125 He was able to show that this happens mostly in young but no longer in older mice. From that the idea arises that maybe the capacity to regrow podocytes from progenitors is much higher in kids than in adults and from that you can speculate a lot. For example, why FSGS is much more frequent in adults than kids or minimal change diseases almost never ends up in scarring in kids. So that could mean that kids maintain a higher capacity to repair damaged podocytes from their local progenitors. And with aging this capacity gets lost somehow. We still don’t know the reasons and why adults much more frequently end up with FSGS a podocyte injury. There are no data yet to support that. These papers were published 2 years back, I think now many people move into this field and each of them with different questions then hopefully in a few years we will be able to get answers to these questions.
Acknowledgments
The author thanks Jan Hagemann for creating . This work was funded by the Deutsche Forschungsgemeinschaft (AN372/11-1, 12-1, 14-1, 15-1) and the Else Kröner-Fresenius Stiftung (P22/09/A14/09).
Note
Edited transcripts of research conferences sponsored by Organogenesis and the Washington University George M. O’Brien Center for Kidney Disease Research (P30 DK079333) are published in Organogenesis. These conferences cover organogenesis in all multicellular organisms including research into tissue engineering, artificial organs and organ substitutes and are participated in by faculty at Washington University School of Medicine, St. Louis, MO USA.
References
- Puhle M. Die Vitalienbrüder: Klaus Störtebeker und die Seeräuber der Hansezeit. Frankfurt am Main, Campus Verlag. 1992.
- Nichols SA, Dirks W, Pearse JS, King N. Early evolution of animal cell signaling and adhesion genes. Proc Natl Acad Sci U S A 2006; 103:12451 - 6; http://dx.doi.org/10.1073/pnas.0604065103; PMID: 16891419
- Schilmiller AL, Howe GA. Systemic signaling in the wound response. Curr Opin Plant Biol 2005; 8:369 - 77; http://dx.doi.org/10.1016/j.pbi.2005.05.008; PMID: 15939667
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature 2008; 453:314 - 21; http://dx.doi.org/10.1038/nature07039; PMID: 18480812
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med 1999; 341:738 - 46; http://dx.doi.org/10.1056/NEJM199909023411006; PMID: 10471461
- Clark RA. Cutaneous tissue repair: basic biologic considerations. I. J Am Acad Dermatol 1985; 13:701 - 25; http://dx.doi.org/10.1016/S0190-9622(85)70213-7; PMID: 2416789
- Martin P. Wound healing--aiming for perfect skin regeneration. Science 1997; 276:75 - 81; http://dx.doi.org/10.1126/science.276.5309.75; PMID: 9082989
- Nurden AT. Platelets, inflammation and tissue regeneration. Thromb Haemost 2011; 105:Suppl 1 S13 - 33; http://dx.doi.org/10.1160/THS10-11-0720; PMID: 21479340
- Medzhitov R. Origin and physiological roles of inflammation. Nature 2008; 454:428 - 35; http://dx.doi.org/10.1038/nature07201; PMID: 18650913
- Messier-Solek C, Buckley KM, Rast JP. Highly diversified innate receptor systems and new forms of animal immunity. Semin Immunol 2010; 22:39 - 47; http://dx.doi.org/10.1016/j.smim.2009.11.007; PMID: 20022762
- Gauthier ME, Du Pasquier L, Degnan BM. The genome of the sponge Amphimedon queenslandica provides new perspectives into the origin of Toll-like and interleukin 1 receptor pathways. Evol Dev 2010; 12:519 - 33; http://dx.doi.org/10.1111/j.1525-142X.2010.00436.x; PMID: 20883219
- Wiens M, Korzhev M, Perovic-Ottstadt S, Luthringer B, Brandt D, Klein S, et al. Toll-like receptors are part of the innate immune defense system of sponges (demospongiae: Porifera). Mol Biol Evol 2007; 24:792 - 804; http://dx.doi.org/10.1093/molbev/msl208; PMID: 17190971
- Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol 2010; 28:321 - 42; http://dx.doi.org/10.1146/annurev-immunol-030409-101311; PMID: 20307211
- Anders HJ. Toll-like receptors and danger signaling in kidney injury. J Am Soc Nephrol 2010; 21:1270 - 4; http://dx.doi.org/10.1681/ASN.2010030233; PMID: 20651159
- Semple JW, Italiano JE Jr., Freedman J. Platelets and the immune continuum. Nat Rev Immunol 2011; 11:264 - 74; http://dx.doi.org/10.1038/nri2956; PMID: 21436837
- Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol 2011; 6:19 - 48; http://dx.doi.org/10.1146/annurev-pathol-011110-130327; PMID: 20887193
- Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol 2012; 13:191 - 211; http://dx.doi.org/10.2165/11595240-000000000-00000; PMID: 22356259
- Romagnani P. From Proteus to Prometheus: learning from fish to modulate regeneration. J Am Soc Nephrol 2010; 21:726 - 8; http://dx.doi.org/10.1681/ASN.2010020228; PMID: 20378822
- Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev 2003; 83:835 - 70; PMID: 12843410
- Sopova K, Tatsidou P, Stellos K. Platelets and Platelet Interaction with Progenitor Cells in Vascular Homeostasis and Inflammation. Curr Vasc Pharmacol 2012; PMID: 22338570
- Zenewicz LA, Flavell RA. Recent advances in IL-22 biology. Int Immunol 2011; 23:159 - 63; http://dx.doi.org/10.1093/intimm/dxr001; PMID: 21393631
- Sugawara T, Gallucci RM, Simeonova PP, Luster MI. Regulation and role of interleukin 6 in wounded human epithelial keratinocytes. Cytokine 2001; 15:328 - 36; http://dx.doi.org/10.1006/cyto.2001.0946; PMID: 11594800
- Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 2009; 206:1465 - 72; http://dx.doi.org/10.1084/jem.20082683; PMID: 19564350
- Nishida T, Nakamura M, Mishima H, Otori T. Interleukin 6 promotes epithelial migration by a fibronectin-dependent mechanism. J Cell Physiol 1992; 153:1 - 5; http://dx.doi.org/10.1002/jcp.1041530102; PMID: 1522123
- Mizoguchi A. Healing of intestinal inflammation by IL-22. Inflamm Bowel Dis 2012; In press http://dx.doi.org/10.1002/ibd.22929; PMID: 22359410
- Jiang GX, Zhong XY, Cui YF, Liu W, Tai S, Wang ZD, et al. IL-6/STAT3/TFF3 signaling regulates human biliary epithelial cell migration and wound healing in vitro. Mol Biol Rep 2010; 37:3813 - 8; http://dx.doi.org/10.1007/s11033-010-0036-z; PMID: 20229017
- Braun RK, Ferrick C, Neubauer P, Sjoding M, Sterner-Kock A, Kock M, et al. IL-17 producing gammadelta T cells are required for a controlled inflammatory response after bleomycin-induced lung injury. Inflammation 2008; 31:167 - 79; http://dx.doi.org/10.1007/s10753-008-9062-6; PMID: 18338242
- Brockes JP. Amphibian limb regeneration: rebuilding a complex structure. Science 1997; 276:81 - 7; http://dx.doi.org/10.1126/science.276.5309.81; PMID: 9082990
- Sipos F, Valcz G, Molńr B. Physiological and pathological role of local and immigrating colonic stem cells. World J Gastroenterol 2012; 18:295 - 301; http://dx.doi.org/10.3748/wjg.v18.i4.295; PMID: 22294835
- Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 2010; 16:535 - 43, 1p, 143; http://dx.doi.org/10.1038/nm.2144; PMID: 20436483
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol 2010; 21:212 - 22; http://dx.doi.org/10.1681/ASN.2008121226; PMID: 20019167
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 2003; 112:1776 - 84; PMID: 14679171
- Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Göbel N, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A 2009; 106:17892 - 7; http://dx.doi.org/10.1073/pnas.0906070106; PMID: 19815530
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010; 176:85 - 97; http://dx.doi.org/10.2353/ajpath.2010.090517; PMID: 20008127
- Zeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol 2010; 21:1819 - 34; http://dx.doi.org/10.1681/ASN.2010080793; PMID: 20864689
- Chapman K, Seldon M, Richards R. Thrombotic microangiopathies, thrombotic thrombocytopenic purpura, and ADAMTS-13. Semin Thromb Hemost 2012; 38:47 - 54; http://dx.doi.org/10.1055/s-0031-1300951; PMID: 22314603
- Amengual O, Atsumi T, Koike T. Pathophysiology of thrombosis and potential targeted therapies in antiphospholipid syndrome. Curr Vasc Pharmacol 2011; 9:606 - 18; http://dx.doi.org/10.2174/157016111796642715; PMID: 21692741
- Bonsib SM. Glomerular basement membrane discontinuities. Scanning electron microscopic study of acellular glomeruli. Am J Pathol 1985; 119:357 - 60; PMID: 4014431
- Sörensen I, Susnik N, Inhester T, Degen JL, Melk A, Haller H, et al. Fibrinogen, acting as a mitogen for tubulointerstitial fibroblasts, promotes renal fibrosis. Kidney Int 2011; 80:1035 - 44; http://dx.doi.org/10.1038/ki.2011.214; PMID: 21734641
- Drew AF, Tucker HL, Liu H, Witte DP, Degen JL, Tipping PG. Crescentic glomerulonephritis is diminished in fibrinogen-deficient mice. Am J Physiol Renal Physiol 2001; 281:F1157 - 63; PMID: 11704568
- Ryu M, Migliorini M, Miosge N, Gross O, Shankland S, Brinkoetter PT, et al. Plasma leakage through glomerular basement membrane ruptures triggers the proliferation of parietal epithelial cells and crescent formation in non-inflammatory glomerular injury. J Pathol 2012; 226:120 - 31; PMID: 21953121
- Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J. The renal mononuclear phagocytic system. J Am Soc Nephrol 2012; 23:194 - 203; http://dx.doi.org/10.1681/ASN.2011070680; PMID: 22135312
- Lech M, Avila-Ferrufino A, Skuginna V, Susanti HE, Anders HJ. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int Immunol 2010; 22:717 - 28; http://dx.doi.org/10.1093/intimm/dxq058; PMID: 20584763
- Anders HJ, Banas B, Schlöndorff D. Signaling danger: toll-like receptors and their potential roles in kidney disease. J Am Soc Nephrol 2004; 15:854 - 67; http://dx.doi.org/10.1097/01.ASN.0000121781.89599.16; PMID: 15034087
- Patole PS, Pawar RD, Lech M, Zecher D, Schmidt H, Segerer S, et al. Expression and regulation of Toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol Dial Transplant 2006; 21:3062 - 73; http://dx.doi.org/10.1093/ndt/gfl336; PMID: 16954173
- Anders HJ. Innate pathogen recognition in the kidney: toll-like receptors, NOD-like receptors, and RIG-like helicases. Kidney Int 2007; 72:1051 - 6; http://dx.doi.org/10.1038/sj.ki.5002436; PMID: 17653134
- Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol 2011; 22:1007 - 18; http://dx.doi.org/10.1681/ASN.2010080798; PMID: 21566058
- Pawar RD, Castrezana-Lopez L, Allam R, Kulkarni OP, Segerer S, Radomska E, et al. Bacterial lipopeptide triggers massive albuminuria in murine lupus nephritis by activating Toll-like receptor 2 at the glomerular filtration barrier. Immunology 2009; 128:Suppl e206 - 21; http://dx.doi.org/10.1111/j.1365-2567.2008.02948.x; PMID: 19175801
- Patole PS, Gröne HJ, Segerer S, Ciubar R, Belemezova E, Henger A, et al. Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J Am Soc Nephrol 2005; 16:1326 - 38; http://dx.doi.org/10.1681/ASN.2004100820; PMID: 15772251
- Pawar RD, Patole PS, Zecher D, Segerer S, Kretzler M, Schlöndorff D, et al. Toll-like receptor-7 modulates immune complex glomerulonephritis. J Am Soc Nephrol 2006; 17:141 - 9; http://dx.doi.org/10.1681/ASN.2005070714; PMID: 16280469
- Anders HJ, Banas B, Linde Y, Weller L, Cohen CD, Kretzler M, et al. Bacterial CpG-DNA aggravates immune complex glomerulonephritis: role of TLR9-mediated expression of chemokines and chemokine receptors. J Am Soc Nephrol 2003; 14:317 - 26; http://dx.doi.org/10.1097/01.ASN.0000042169.23931.73; PMID: 12538732
- Anders HJ, Vielhauer V, Eis V, Linde Y, Kretzler M, Perez de Lema G, et al. Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB J 2004; 18:534 - 6; PMID: 14734643
- Allam R, Pawar RD, Kulkarni OP, Hornung V, Hartmann G, Segerer S, et al. Viral 5′-triphosphate RNA and non-CpG DNA aggravate autoimmunity and lupus nephritis via distinct TLR-independent immune responses. Eur J Immunol 2008; 38:3487 - 98; http://dx.doi.org/10.1002/eji.200838604; PMID: 19009528
- Brown HJ, Lock HR, Sacks SH, Robson MG. TLR2 stimulation of intrinsic renal cells in the induction of immune-mediated glomerulonephritis. J Immunol 2006; 177:1925 - 31; PMID: 16849506
- Brown HJ, Lock HR, Wolfs TG, Buurman WA, Sacks SH, Robson MG. Toll-like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody-mediated glomerulonephritis via CXCL1 and CXCL2. J Am Soc Nephrol 2007; 18:1732 - 9; http://dx.doi.org/10.1681/ASN.2006060634; PMID: 17460147
- Brown HJ, Sacks SH, Robson MG. Toll-like receptor 2 agonists exacerbate accelerated nephrotoxic nephritis. J Am Soc Nephrol 2006; 17:1931 - 9; http://dx.doi.org/10.1681/ASN.2005111167; PMID: 16738018
- Wörnle M, Schmid H, Banas B, Merkle M, Henger A, Roeder M, et al. Novel role of toll-like receptor 3 in hepatitis C-associated glomerulonephritis. Am J Pathol 2006; 168:370 - 85; http://dx.doi.org/10.2353/ajpath.2006.050491; PMID: 16436653
- Lichtnekert J, Vielhauer V, Zecher D, Kulkarni OP, Clauss S, Segerer S, et al. Trif is not required for immune complex glomerulonephritis: dying cells activate mesangial cells via Tlr2/Myd88 rather than Tlr3/Trif. Am J Physiol Renal Physiol 2009; 296:F867 - 74; http://dx.doi.org/10.1152/ajprenal.90213.2008; PMID: 19158348
- Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol 2010; 21:1732 - 44; http://dx.doi.org/10.1681/ASN.2010020143; PMID: 20688930
- Lichtnekert J, Kulkarni OP, Mulay SR, Rupanagudi KV, Ryu M, Allam R, et al. Anti-GBM glomerulonephritis involves IL-1 but is independent of NLRP3/ASC inflammasome-mediated activation of caspase-1. PLoS One 2011; 6:e26778; http://dx.doi.org/10.1371/journal.pone.0026778; PMID: 22046355
- Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A 2009; 106:20388 - 93; http://dx.doi.org/10.1073/pnas.0908698106; PMID: 19918053
- Ryu M, Kulkarni OP, Radomska E, Miosge N, Gross O, Anders HJ. Bacterial CpG-DNA accelerates Alport glomerulosclerosis by inducing an M1 macrophage phenotype and tumor necrosis factor-α-mediated podocyte loss. Kidney Int 2011; 79:189 - 98; http://dx.doi.org/10.1038/ki.2010.373; PMID: 20962742
- Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol 2007; 18:1721 - 31; http://dx.doi.org/10.1681/ASN.2006101162; PMID: 17460144
- Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol 2010; 21:1878 - 90; http://dx.doi.org/10.1681/ASN.2009101048; PMID: 20847143
- Shigeoka AA, Holscher TD, King AJ, Hall FW, Kiosses WB, Tobias PS, et al. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol 2007; 178:6252 - 8; PMID: 17475853
- Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest 2005; 115:2894 - 903; http://dx.doi.org/10.1172/JCI22832; PMID: 16167081
- Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest 2007; 117:2847 - 59; http://dx.doi.org/10.1172/JCI31008; PMID: 17853945
- Anders HJ, Schlöndorff D. Toll-like receptors: emerging concepts in kidney disease. Curr Opin Nephrol Hypertens 2007; 16:177 - 83; http://dx.doi.org/10.1097/MNH.0b013e32803fb767; PMID: 17420659
- Lech M, Garlanda C, Mantovani A, Kirschning CJ, Schlöndorff D, Anders HJ. Different roles of TiR8/Sigirr on toll-like receptor signaling in intrarenal antigen-presenting cells and tubular epithelial cells. Kidney Int 2007; 72:182 - 92; http://dx.doi.org/10.1038/sj.ki.5002293; PMID: 17495864
- Lassen S, Lech M, Römmele C, Mittruecker HW, Mak TW, Anders HJ. Ischemia reperfusion induces IFN regulatory factor 4 in renal dendritic cells, which suppresses postischemic inflammation and prevents acute renal failure. J Immunol 2010; 185:1976 - 83; http://dx.doi.org/10.4049/jimmunol.0904207; PMID: 20601597
- Lech M, Avila-Ferrufino A, Allam R, Segerer S, Khandoga A, Krombach F, et al. Resident dendritic cells prevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J Immunol 2009; 183:4109 - 18; http://dx.doi.org/10.4049/jimmunol.0900118; PMID: 19692646
- Gong J, Wei T, Stark RW, Jamitzky F, Heckl WM, Anders HJ, et al. Inhibition of Toll-like receptors TLR4 and 7 signaling pathways by SIGIRR: a computational approach. J Struct Biol 2010; 169:323 - 30; http://dx.doi.org/10.1016/j.jsb.2009.12.007; PMID: 20025973
- Pawar RD, Patole PS, Wörnle M, Anders HJ. Microbial nucleic acids pay a Toll in kidney disease. Am J Physiol Renal Physiol 2006; 291:F509 - 16; http://dx.doi.org/10.1152/ajprenal.00453.2005; PMID: 16597607
- Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol 2007; 25:419 - 41; http://dx.doi.org/10.1146/annurev.immunol.22.012703.104514; PMID: 17378763
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 2002; 416:603 - 7; http://dx.doi.org/10.1038/416603a; PMID: 11948342
- Migliorini A, Anders HJ. A novel pathogenetic concept-antiviral immunity in lupus nephritis. Nat Rev Nephrol 2012; 8:183 - 9; http://dx.doi.org/10.1038/nrneph.2011.197; PMID: 22249778
- Anders HJ. Pseudoviral immunity—a novel concept for lupus. Trends Mol Med 2009; 15:553 - 61; http://dx.doi.org/10.1016/j.molmed.2009.10.004; PMID: 19896418
- Anders HJ, Lichtnekert J, Allam R. Interferon-alpha and -beta in kidney inflammation. Kidney Int 2010; 77:848 - 54; http://dx.doi.org/10.1038/ki.2010.71; PMID: 20237459
- Flür K, Allam R, Zecher D, Kulkarni OP, Lichtnekert J, Schwarz M, et al. Viral RNA induces type I interferon-dependent cytokine release and cell death in mesangial cells via melanoma-differentiation-associated gene-5: Implications for viral infection-associated glomerulonephritis. Am J Pathol 2009; 175:2014 - 22; http://dx.doi.org/10.2353/ajpath.2009.080585; PMID: 19850889
- Hägele H, Allam R, Pawar RD, Anders HJ. Double-stranded RNA activates type I interferon secretion in glomerular endothelial cells via retinoic acid-inducible gene (RIG)-1. Nephrol Dial Transplant 2009; 24:3312 - 8; http://dx.doi.org/10.1093/ndt/gfp339; PMID: 19608629
- Hägele H, Allam R, Pawar RD, Reichel CA, Krombach F, Anders HJ. Double-stranded DNA activates glomerular endothelial cells and enhances albumin permeability via a toll-like receptor-independent cytosolic DNA recognition pathway. Am J Pathol 2009; 175:1896 - 904; http://dx.doi.org/10.2353/ajpath.2009.090182; PMID: 19834059
- Allam R, Lichtnekert J, Moll A, Taubitz A, Vielhauer V, Anders HJ. Viral RNA and DNA sense common antiviral responses including type I interferons in mesangial cells. J Am Soc Nephrol 2009; 20:1986 - 96; http://dx.doi.org/10.1681/ASN.2008101067; PMID: 19713315
- Fairhurst AM, Mathian A, Connolly JE, Wang A, Gray HF, George TA, et al. Systemic IFN-alpha drives kidney nephritis in B6.Sle123 mice. Eur J Immunol 2008; 38:1948 - 60; http://dx.doi.org/10.1002/eji.200837925; PMID: 18506882
- Fairhurst AM, Xie C, Fu Y, Wang A, Boudreaux C, Zhou XJ, et al. Type I interferons produced by resident renal cells may promote end-organ disease in autoantibody-mediated glomerulonephritis. J Immunol 2009; 183:6831 - 8; http://dx.doi.org/10.4049/jimmunol.0900742; PMID: 19864599
- Anders HJ, Vielhauer V, Schlöndorff D. Chemokines and chemokine receptors are involved in the resolution or progression of renal disease. Kidney Int 2003; 63:401 - 15; http://dx.doi.org/10.1046/j.1523-1755.2003.00750.x; PMID: 12631106
- Vielhauer V, Kulkarni O, Reichel CA, Anders HJ. Targeting the recruitment of monocytes and macrophages in renal disease. Semin Nephrol 2010; 30:318 - 33; http://dx.doi.org/10.1016/j.semnephrol.2010.03.006; PMID: 20620675
- Heller F, Lindenmeyer MT, Cohen CD, Brandt U, Draganovici D, Fischereder M, et al. The contribution of B cells to renal interstitial inflammation. Am J Pathol 2007; 170:457 - 68; http://dx.doi.org/10.2353/ajpath.2007.060554; PMID: 17255314
- Steinmetz OM, Stahl RA, Panzer U. Chemokines and B cells in renal inflammation and allograft rejection. Front Biosci (Schol Ed) 2009; 1:13 - 22; PMID: 19482678
- Panzer U, Kurts C. T cell cross-talk with kidney dendritic cells in glomerulonephritis. J Mol Med (Berl) 2010; 88:19 - 26; http://dx.doi.org/10.1007/s00109-009-0541-5; PMID: 19798477
- Anders HJ, Ryu M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int 2011; 80:915 - 25; http://dx.doi.org/10.1038/ki.2011.217; PMID: 21814171
- Anders HJ, Frink M, Linde Y, Banas B, Wörnle M, Cohen CD, et al. CC chemokine ligand 5/RANTES chemokine antagonists aggravate glomerulonephritis despite reduction of glomerular leukocyte infiltration. J Immunol 2003; 170:5658 - 66; PMID: 12759447
- Pawar RD, Patole PS, Ellwart A, Lech M, Segerer S, Schlondorff D, et al. Ligands to nucleic acid-specific toll-like receptors and the onset of lupus nephritis. J Am Soc Nephrol 2006; 17:3365 - 73; http://dx.doi.org/10.1681/ASN.2006030263; PMID: 17082246
- Anders HJ, Zecher D, Pawar RD, Patole PS. Molecular mechanisms of autoimmunity triggered by microbial infection. Arthritis Res Ther 2005; 7:215 - 24; http://dx.doi.org/10.1186/ar1818; PMID: 16207351
- Ble A, Mosca M, Di Loreto G, Guglielmotti A, Biondi G, Bombardieri S, et al. Antiproteinuric effect of chemokine C-C motif ligand 2 inhibition in subjects with acute proliferative lupus nephritis. Am J Nephrol 2011; 34:367 - 72; http://dx.doi.org/10.1159/000330685; PMID: 21876349
- Kulkarni O, Pawar RD, Purschke W, Eulberg D, Selve N, Buchner K, et al. Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. J Am Soc Nephrol 2007; 18:2350 - 8; http://dx.doi.org/10.1681/ASN.2006121348; PMID: 17625118
- Ninichuk V, Clauss S, Kulkarni O, Schmid H, Segerer S, Radomska E, et al. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36-3’PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am J Pathol 2008; 172:628 - 37; http://dx.doi.org/10.2353/ajpath.2008.070601; PMID: 18258851
- Kulkarni O, Eulberg D, Selve N, Zöllner S, Allam R, Pawar RD, et al. Anti-Ccl2 Spiegelmer permits 75% dose reduction of cyclophosphamide to control diffuse proliferative lupus nephritis and pneumonitis in MRL-Fas(lpr) mice. J Pharmacol Exp Ther 2009; 328:371 - 7; http://dx.doi.org/10.1124/jpet.108.142711; PMID: 18997060
- Sayyed SG, Ryu M, Kulkarni OP, Schmid H, Lichtnekert J, Grüner S, et al. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int 2011; 80:68 - 78; http://dx.doi.org/10.1038/ki.2011.102; PMID: 21508925
- Clauss S, Gross O, Kulkarni O, Avila-Ferrufino A, Radomska E, Segerer S, et al. Ccl2/Mcp-1 blockade reduces glomerular and interstitial macrophages but does not ameliorate renal pathology in collagen4A3-deficient mice with autosomal recessive Alport nephropathy. J Pathol 2009; 218:40 - 7; http://dx.doi.org/10.1002/path.2505; PMID: 19156777
- Ribeiro A, Wörnle M, Motamedi N, Anders HJ, Gröne EF, Nitschko H, et al. Activation of innate immune defense mechanisms contributes to polyomavirus BK-associated nephropathy. Kidney Int 2012; 81:100 - 11; http://dx.doi.org/10.1038/ki.2011.311; PMID: 21918500
- Anders HJ, Patole PS. Toll-like receptors recognize uropathogenic Escherichia coli and trigger inflammation in the urinary tract. Nephrol Dial Transplant 2005; 20:1529 - 32; http://dx.doi.org/10.1093/ndt/gfh922; PMID: 15941847
- Patole PS, Schubert S, Hildinger K, Khandoga S, Khandoga A, Segerer S, et al. Toll-like receptor-4: renal cells and bone marrow cells signal for neutrophil recruitment during pyelonephritis. Kidney Int 2005; 68:2582 - 7; http://dx.doi.org/10.1111/j.1523-1755.2005.00729.x; PMID: 16316333
- Kulkarni OP, Ryu M, Kantner C, Sárdy M, Naylor D, Lambert D, et al. Recombinant chaperonin 10 suppresses cutaneous lupus and lupus nephritis in MRL-(Fas)lpr mice. Nephrol Dial Transplant 2012; 27:1358 - 67; http://dx.doi.org/10.1093/ndt/gfr544; PMID: 21987536
- Bonventre JV. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J Am Soc Nephrol 2003; 14:Suppl 1 S55 - 61; http://dx.doi.org/10.1097/01.ASN.0000067652.51441.21; PMID: 12761240
- Abuelo JG. Normotensive ischemic acute renal failure. N Engl J Med 2007; 357:797 - 805; http://dx.doi.org/10.1056/NEJMra064398; PMID: 17715412
- Sugimoto H, Lebleu VS, Bosukonda D, Keck P, Taduri G, Bechtel W, et al. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat Med 2012; 18:396 - 404; http://dx.doi.org/10.1038/nm.2629; PMID: 22306733
- Mulay SR, Thomasova D, Ryu M, Anders HJ. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int 2012; 81:1199 - 211; http://dx.doi.org/10.1038/ki.2011.482; PMID: 22297670
- Weidenbusch M, Anders HJ. Tissue microenvironments define and get reinforced by macrophage phenotypes in homeostasis or during inflammation, repair, and fibrosis. J Innate Immun 2012; In press
- Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 2005; 115:56 - 65; PMID: 15630444
- Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 2011; 22:317 - 26; http://dx.doi.org/10.1681/ASN.2009060615; PMID: 21289217
- Lee SB, Kalluri R. Mechanistic connection between inflammation and fibrosis. Kidney Int Suppl 2010; 78:S22 - 6; http://dx.doi.org/10.1038/ki.2010.418; PMID: 21116313
- Lasagni L, Romagnani P. Glomerular epithelial stem cells: the good, the bad, and the ugly. J Am Soc Nephrol 2010; 21:1612 - 9; http://dx.doi.org/10.1681/ASN.2010010048; PMID: 20829409
- Humphreys BD, Czerniak S, DiRocco DP, Hasnain W, Cheema R, Bonventre JV. Repair of injured proximal tubule does not involve specialized progenitors. Proc Natl Acad Sci U S A 2011; 108:9226 - 31; http://dx.doi.org/10.1073/pnas.1100629108; PMID: 21576461
- Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008; 2:284 - 91; http://dx.doi.org/10.1016/j.stem.2008.01.014; PMID: 18371453
- Duffield JS, Park KM, Hsiao LL, Kelley VR, Scadden DT, Ichimura T, et al. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. J Clin Invest 2005; 115:1743 - 55; http://dx.doi.org/10.1172/JCI22593; PMID: 16007251
- Romagnani P. Family portrait: renal progenitor of Bowman’s capsule and its tubular brothers. Am J Pathol 2011; 178:490 - 3; http://dx.doi.org/10.1016/j.ajpath.2010.11.044; PMID: 21281781
- Tögel FE, Westenfelder C. Mesenchymal stem cells: a new therapeutic tool for AKI. Nat Rev Nephrol 2010; 6:179 - 83; http://dx.doi.org/10.1038/nrneph.2009.229; PMID: 20186233
- Kriz W, Lemley KV. The role of the podocyte in glomerulosclerosis. Curr Opin Nephrol Hypertens 1999; 8:489 - 97; http://dx.doi.org/10.1097/00041552-199907000-00014; PMID: 10491745
- Teixeira VdeP, Blattner SM, Li M, Anders HJ, Cohen CD, Edenhofer I, et al. Functional consequences of integrin-linked kinase activation in podocyte damage. Kidney Int 2005; 67:514 - 23; http://dx.doi.org/10.1111/j.1523-1755.2005.67108.x; PMID: 15673299
- Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 2005; 16:2941 - 52; http://dx.doi.org/10.1681/ASN.2005010055; PMID: 16107576
- Sugimoto H, Mundel TM, Sund M, Xie L, Cosgrove D, Kalluri R. Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc Natl Acad Sci U S A 2006; 103:7321 - 6; http://dx.doi.org/10.1073/pnas.0601436103; PMID: 16648256
- LeBleu V, Sugimoto H, Mundel TM, Gerami-Naini B, Finan E, Miller CA, et al. Stem cell therapies benefit Alport syndrome. J Am Soc Nephrol 2009; 20:2359 - 70; http://dx.doi.org/10.1681/ASN.2009010123; PMID: 19833902
- Gross O, Borza DB, Anders HJ, Licht C, Weber M, Segerer S, et al. Stem cell therapy for Alport syndrome: the hope beyond the hype. Nephrol Dial Transplant 2009; 24:731 - 4; http://dx.doi.org/10.1093/ndt/gfn722; PMID: 19110486
- Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, et al. Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 2009; 20:322 - 32; http://dx.doi.org/10.1681/ASN.2008070709; PMID: 19092120
- Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, et al. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 2009; 20:333 - 43; http://dx.doi.org/10.1681/ASN.2008070795; PMID: 19092119
- Lazzeri E, Crescioli C, Ronconi E, Mazzinghi B, Sagrinati C, Netti GS, et al. Regenerative potential of embryonic renal multipotent progenitors in acute renal failure. J Am Soc Nephrol 2007; 18:3128 - 38; http://dx.doi.org/10.1681/ASN.2007020210; PMID: 17978305
- Lasagni L, Ballerini L, Angelotti ML, Parente E, Sagrinati C, Mazzinghi B, et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells 2010; 28:1674 - 85; http://dx.doi.org/10.1002/stem.492; PMID: 20680961
- Bollée G, Flamant M, Schordan S, Fligny C, Rumpel E, Milon M, et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat Med 2011; 17:1242 - 50; http://dx.doi.org/10.1038/nm.2491; PMID: 21946538
- Sayyed SG, Hägele H, Kulkarni OP, Endlich K, Segerer S, Eulberg D, et al. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia 2009; 52:2445 - 54; http://dx.doi.org/10.1007/s00125-009-1493-6; PMID: 19707743
- Darisipudi MN, Kulkarni OP, Sayyed SG, Ryu M, Migliorini A, Sagrinati C, et al. Dual blockade of the homeostatic chemokine CXCL12 and the proinflammatory chemokine CCL2 has additive protective effects on diabetic kidney disease. Am J Pathol 2011; 179:116 - 24; http://dx.doi.org/10.1016/j.ajpath.2011.03.004; PMID: 21703397
- Macconi D, Sangalli F, Bonomelli M, Conti S, Condorelli L, Gagliardini E, et al. Podocyte repopulation contributes to regression of glomerular injury induced by ACE inhibition. Am J Pathol 2009; 174:797 - 807; http://dx.doi.org/10.2353/ajpath.2009.080227; PMID: 19164508
- Remuzzi G, Benigni A, Remuzzi A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J Clin Invest 2006; 116:288 - 96; http://dx.doi.org/10.1172/JCI27699; PMID: 16453013
- Gaikwad AB, Sayyed SG, Lichtnekert J, Tikoo K, Anders HJ. Renal failure increases cardiac histone h3 acetylation, dimethylation, and phosphorylation and the induction of cardiomyopathy-related genes in type 2 diabetes. Am J Pathol 2010; 176:1079 - 83; http://dx.doi.org/10.2353/ajpath.2010.090528; PMID: 20075197
- Sayyed SG, Gaikwad AB, Lichtnekert J, Kulkarni O, Eulberg D, Klussmann S, et al. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol Dial Transplant 2010; 25:1811 - 7; http://dx.doi.org/10.1093/ndt/gfp730; PMID: 20067909
- Smeets B, Angelotti ML, Rizzo P, Dijkman H, Lazzeri E, Mooren F, et al. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J Am Soc Nephrol 2009; 20:2593 - 603; http://dx.doi.org/10.1681/ASN.2009020132; PMID: 19875807
- Smeets B, Uhlig S, Fuss A, Mooren F, Wetzels JF, Floege J, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol 2009; 20:2604 - 15; http://dx.doi.org/10.1681/ASN.2009010122; PMID: 19917779
- Atkins RC, Nikolic-Paterson DJ, Song Q, Lan HY. Modulators of crescentic glomerulonephritis. J Am Soc Nephrol 1996; 7:2271 - 8; PMID: 8959617
- Tipping PG, Kitching PR, Holdsworth SR. The formation of the glomerular crescent. In: Immunologic Renal Diseases. 2. Neilson EG, Couser WG, eds. Philadelphia: Lippincott Williams & Wilkins Publishers, 2001.
- Lindgren D, Boström AK, Nilsson K, Hansson J, Sjölund J, Möller C, et al. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am J Pathol 2011; 178:828 - 37; http://dx.doi.org/10.1016/j.ajpath.2010.10.026; PMID: 21281815
- Forbes MS, Thornhill BA, Chevalier RL. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol 2011; 301:F110 - 7; http://dx.doi.org/10.1152/ajprenal.00022.2011; PMID: 21429968
- Chevalier RL, Forbes MS. Generation and evolution of atubular glomeruli in the progression of renal disorders. J Am Soc Nephrol 2008; 19:197 - 206; http://dx.doi.org/10.1681/ASN.2007080862; PMID: 18199796
- Ninichuk V, Gross O, Segerer S, Hoffmann R, Radomska E, Buchstaller A, et al. Multipotent mesenchymal stem cells reduce interstitial fibrosis but do not delay progression of chronic kidney disease in collagen4A3-deficient mice. Kidney Int 2006; 70:121 - 9; http://dx.doi.org/10.1038/sj.ki.5001521; PMID: 16723981
- Higgins DF, Lappin DW, Kieran NE, Anders HJ, Watson RW, Strutz F, et al. DNA oligonucleotide microarray technology identifies fisp-12 among other potential fibrogenic genes following murine unilateral ureteral obstruction (UUO): modulation during epithelial-mesenchymal transition. Kidney Int 2003; 64:2079 - 91; http://dx.doi.org/10.1046/j.1523-1755.2003.00306.x; PMID: 14633130
- Li Y, Liu Z, Guo X, Shu J, Chen Z, Li L. Aristolochic acid I-induced DNA damage and cell cycle arrest in renal tubular epithelial cells in vitro. Arch Toxicol 2006; 80:524 - 32; http://dx.doi.org/10.1007/s00204-006-0090-4; PMID: 16609888
- Debelle FD, Vanherweghem JL, Nortier JL. Aristolochic acid nephropathy: a worldwide problem. Kidney Int 2008; 74:158 - 69; http://dx.doi.org/10.1038/ki.2008.129; PMID: 18418355
- Smeets B, Kuppe C, Sicking EM, Fuss A, Jirak P, van Kuppevelt TH, et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol 2011; 22:1262 - 74; http://dx.doi.org/10.1681/ASN.2010090970; PMID: 21719782
- Helal I, Fick-Brosnahan GM, Reed-Gitomer B, Schrier RW. Glomerular hyperfiltration: definitions, mechanisms and clinical implications. Nat Rev Nephrol 2012; 8:293 - 300; http://dx.doi.org/10.1038/nrneph.2012.19; PMID: 22349487
- Bariety J, Hill GS, Mandet C, Irinopoulou T, Jacquot C, Meyrier A, et al. Glomerular epithelial-mesenchymal transdifferentiation in pauci-immune crescentic glomerulonephritis. Nephrol Dial Transplant 2003; 18:1777 - 84; http://dx.doi.org/10.1093/ndt/gfg231; PMID: 12937224
- Duffield JS. Epithelial to mesenchymal transition in injury of solid organs: fact or artifact?. Gastroenterology 2010; 139:1081 - 3, 1083, e1-5; http://dx.doi.org/10.1053/j.gastro.2010.08.017; PMID: 20800655
- Zeisberg M, Duffield JS. Resolved: EMT produces fibroblasts in the kidney. J Am Soc Nephrol 2010; 21:1247 - 53; http://dx.doi.org/10.1681/ASN.2010060616; PMID: 20651165
- Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy?. J Clin Invest 2011; 121:468 - 74; http://dx.doi.org/10.1172/JCI44595; PMID: 21370523
- Ninichuk V, Anders HJ. Bone marrow-derived progenitor cells and renal fibrosis. Front Biosci 2008; 13:5163 - 73; http://dx.doi.org/10.2741/3072; PMID: 18508578
- Vielhauer V, Anders HJ, Mack M, Cihak J, Strutz F, Stangassinger M, et al. Obstructive nephropathy in the mouse: progressive fibrosis correlates with tubulointerstitial chemokine expression and accumulation of CC chemokine receptor 2- and 5-positive leukocytes. J Am Soc Nephrol 2001; 12:1173 - 87; PMID: 11373340
- Anders HJ, Vielhauer V, Kretzler M, Cohen CD, Segerer S, Luckow B, et al. Chemokine and chemokine receptor expression during initiation and resolution of immune complex glomerulonephritis. J Am Soc Nephrol 2001; 12:919 - 31; PMID: 11316850
- Mayer V, Hudkins KL, Heller F, Schmid H, Kretzler M, Brandt U, et al. Expression of the chemokine receptor CCR1 in human renal allografts. Nephrol Dial Transplant 2007; 22:1720 - 9; http://dx.doi.org/10.1093/ndt/gfm007; PMID: 17298994
- Vielhauer V, Anders HJ. Chemokines and chemokine receptors as therapeutic targets in chronic kidney disease. Front Biosci (Schol Ed) 2009; 1:1 - 12; PMID: 19482677
- Jedlicka J, Soleiman A, Draganovici D, Mandelbaum J, Ziegler U, Regele H, et al. Interstitial inflammation in Alport syndrome. Hum Pathol 2010; 41:582 - 93; http://dx.doi.org/10.1016/j.humpath.2009.08.024; PMID: 20004949
- Anders HJ, Ninichuk V, Schlöndorff D. Progression of kidney disease: blocking leukocyte recruitment with chemokine receptor CCR1 antagonists. Kidney Int 2006; 69:29 - 32; http://dx.doi.org/10.1038/sj.ki.5000053; PMID: 16374420
- Eis V, Vielhauer V, Anders HJ. Targeting the chemokine network in renal inflammation. Arch Immunol Ther Exp (Warsz) 2004; 52:164 - 72; PMID: 15247883
- Eis V, Luckow B, Vielhauer V, Siveke JT, Linde Y, Segerer S, et al. Chemokine receptor CCR1 but not CCR5 mediates leukocyte recruitment and subsequent renal fibrosis after unilateral ureteral obstruction. J Am Soc Nephrol 2004; 15:337 - 47; http://dx.doi.org/10.1097/01.ASN.0000111246.87175.32; PMID: 14747380
- Anders HJ, Vielhauer V, Frink M, Linde Y, Cohen CD, Blattner SM, et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J Clin Invest 2002; 109:251 - 9; PMID: 11805137
- Anders HJ, Belemezova E, Eis V, Segerer S, Vielhauer V, Perez de Lema G, et al. Late onset of treatment with a chemokine receptor CCR1 antagonist prevents progression of lupus nephritis in MRL-Fas(lpr) mice. J Am Soc Nephrol 2004; 15:1504 - 13; http://dx.doi.org/10.1097/01.ASN.0000130082.67775.60; PMID: 15153561
- Vielhauer V, Berning E, Eis V, Kretzler M, Segerer S, Strutz F, et al. CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int 2004; 66:2264 - 78; http://dx.doi.org/10.1111/j.1523-1755.2004.66038.x; PMID: 15569315
- Ninichuk V, Gross O, Reichel C, Khandoga A, Pawar RD, Ciubar R, et al. Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J Am Soc Nephrol 2005; 16:977 - 85; http://dx.doi.org/10.1681/ASN.2004100871; PMID: 15716328
- Ninichuk V, Khandoga AG, Segerer S, Loetscher P, Schlapbach A, Revesz L, et al. The role of interstitial macrophages in nephropathy of type 2 diabetic db/db mice. Am J Pathol 2007; 170:1267 - 76; http://dx.doi.org/10.2353/ajpath.2007.060937; PMID: 17392166
- Rao VH, Meehan DT, Delimont D, Nakajima M, Wada T, Gratton MA, et al. Role for macrophage metalloelastase in glomerular basement membrane damage associated with alport syndrome. Am J Pathol 2006; 169:32 - 46; http://dx.doi.org/10.2353/ajpath.2006.050896; PMID: 16816359