
Abstract
The normal development and function of photoreceptors is essential for eye health and visual acuity in vertebrates. Mutations in genes encoding proteins involved in photoreceptor development and function are associated with a suite of inherited retinal dystrophies, often as part of complex multi-organ syndromic conditions. In this review, we focus on the role of the photoreceptor outer segment, a highly modified and specialized primary cilium, in retinal health and disease. We discuss the many defects in the structure and function of the photoreceptor primary cilium that can cause a class of inherited conditions known as ciliopathies, often characterized by retinal dystrophy and degeneration, and highlight the recent insights into disease mechanisms.
Introduction: the neurosensory retina and photoreceptor cells
The retina is the internal layer of the eyeball, responsible for converting light signals from the environment (focused by the anterior features of the eye) into neural impulses to be sent to the brain. This thin (0.56 – 0.1mm) membrane can be further divided into two distinct layers: an inner neurosensory layer and an outer pigmented layer, the retinal pigment epithelium (RPE). The most prevalent cell-type of the retina is the neuron, of which there are three main groups responsible for relaying light generated impulses. These are the bipolar cells, the ganglion cells and the photoreceptors. Photoreceptor cells are long and narrow, and are sub-divided into inner segments (IS) and outer segments (OS) connected by a connecting cilium (CC).Citation1 The OS of the photoreceptor develops from a primitive primary cilium, and the OS is widely considered to be a highly modified primary cilium,Citation1-Citation4 with the retinal connecting cilium homologous to the transition zone of the primary cilium.Citation5-Citation7
There are two types of photoreceptor cells, rods () and cones, which are named after the shape of their respective OS. These specialized regions of the cell contain high concentrations of the components of the phototransduction cascade such as the G-protein transducin and visual pigments, and low concentrations of proteins involved in other cellular functions.Citation8 Rod OS contain the visual pigment rhodopsin within membrane-bound discs, which stack together to form the rod shape. Cones contain several visual pigments, known as opsins, inserted into the highly invaginated plasma membranes which form the conical shape of these cells. While rod OS tips are phagocytosed daily by the RPE and new discs formed at the OS base, the cone OS are not phagocytosed in this manner. Rods are shed daily on light onsetCitation9 while cones are shed during light offset.Citation10
Figure 1. Schematic representation of a rod photoreceptor cell and localization of ciliary proteins.(A) The schematic represents the rod photoreceptor cell outer segment, connecting cilium, inner segment, outer fiber, cell body, inner fiber and synaptic terminus. A number of key components of the ciliary apparatus are color coded and indicated. The IFT complex A (blue) and complex B (red) are represented in the magnified inset. A retinal pigmentary epithelial (RPE) cell is shown in gray at the top. (B) Confocal microscopy images of an immunofluorescent stained P20 mouse retinal cryosection showing the stratified layers of the retina. Cilium transition zone and basal body protein MKS1 is stained in green, and a novel interactant of MKS1, RNF34, is stained in red. These proteins localize to the base of the connecting cilium, as shown by the arrowheads in the enlarged insets. (C) Confocal microscopy image of a human adult retinal pigment epithelium (ARPE19) cell overexpressing enhanced-GFP-tagged lebercilin and immunostained with an antibody against acetylated α tubulin, which marks the axoneme of the cilium. Lebercilin can be seen in a punctuate pattern along the axoneme. Scale bar = 10μm.
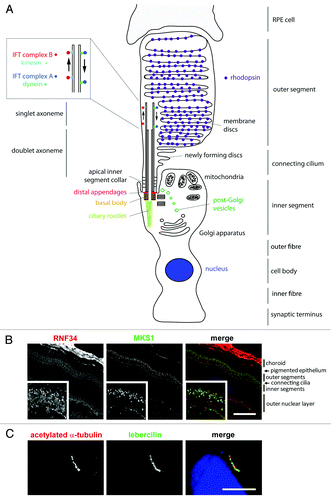
The CC connects the OS to the IS and consists of an axoneme of nine microtubule doublets nucleated by a triplet microtubule basal body. This is derived from the mother centriole, at the apical surface of the IS. This axoneme extends into the photoreceptor OS, converting to singlet microtubules toward the distal end ().Citation11 The axoneme often reaches near the distal tip of cone OS and at least half-way along the rod OS, reaching the distal tip of rod OS in some cases.Citation12 The axoneme is stabilized by post-translational modifications such as glutamylation and acetylation in rods, but is turned over as membranes are replaced at the distal surface of cones.Citation13
The ciliary rootlet extends from the basal side of the basal body and anchors the cilium to the cell, extending deep into IS.Citation14 It is composed of rootletin and is required for the structural stability of the photoreceptor OS (). It is thought to also act as a docking point for transport motor proteins carrying vesicular cargo which may be destined for the CC or OS. The IS of the photoreceptors is where protein synthesis occurs, and where ATP is produced. The IS are split into two regions. The distal or ellipsoid region is packed with mitochondria, reflecting the high metabolic demands of the cells, whereas the proximal or myoid region is predominantly filled with the Golgi apparatus and the endoplasmic reticulum that enable high rates of protein synthesis in these parts of the cell. The remainder of the photoreceptors consist of the outer fiber connecting the IS to the cell body (containing the cell nucleus), which in turn is connected via the inner fiber to the synaptic terminal, forming junctions with the retinal bipolar cells for transmission of nerve impulses. These cells perform essential roles in phototransduction, and ciliary signaling is a crucial component of this activity that, when disrupted, can lead to retinal disease. The role of ciliary signaling in retinal disease has been reviewed recentlyCitation15 and the present review will instead focus on photoreceptor assembly, organization and maintenance.
Model Systems for the Study Of Photoreceptor Development and Function
A range of organisms are used as models of retinal development, each with its own individual benefits and drawbacks (). The interested reader is referred to Table S1 for further details. Diurnal primates are the best model of human retinal development, since they share several important features of the human retina that are absent in other mammals such as the fovea, the region of the central retina populated by cones and with the highest visual acuity.Citation16 Most mammals have dichroic vision, with two opsins (blue-sensitive and green-sensitive) whereas humans and diurnal primates have trichroic vision (with an additional red-sensitive opsin).Citation17 Ethical and practical factors prevent the use of such primates in widespread retinal research, and mice remain the most commonly-used mammalian model. However, mice do not differentiate their photoreceptor OS until P8-P16,Citation18 preventing the study of photoreceptor development and/or degeneration in mouse models of conditions that are embryonic or perinatal lethal, as is the case for many ciliopathies. The use of conditional knockouts by crossing to lines expressing Cre recombinase in the rods and/or cones can help to overcome such lethality. Crossing of conditional knockouts to lines expressing Cre recombinase under the control of a cone-specific gene promoter have been successfully used to develop models of foveal diseases.Citation19
Table 1. The most comprehensively characterized photoreceptor proteins mutated in human disease
Additionally, mice photoreceptors lack calyceal processes (CPs), finger-like structures that protrude from the apical region of the inner segment and surround the base of the outer segment, further compromising their utility as models of retinal ciliopathies. This is particularly true in the case of mouse models of Usher syndrome (USH), an inherited condition involving sensorineural hearing loss and retinal dystrophy. Mouse knockouts of the USH1 proteins (myosin VIIa, harmonin, cadherin-23, protocadherin-15, SANS) often develop hearing loss and vestibular dysfunction but do not display retinal degeneration due to the lack of CPs.Citation20 Amphibians (especially Xenopus tadpoles) or macaques are a better model of USH1 as they possess CPs, but zebrafish are more commonly used for most models of Usher syndrome.
The retina is fully laminated and light responsive by the third day of embryogenesis in this organism, and zebrafish mutants, morphants and TALEN models provide tractable and versatile resources for the study of retinal ciliopathies. Knockouts and knock-ins by genomic editing with TALENs (or CRISPRs) are a recent exciting addition to the techniques developed for the study of photoreceptor degeneration in zebrafish. Morpholino oligonucleotide knockdown of gene transcript levels is only effective for around three days after microinjection, and so is applicable for studies of retinal development but not slower or later-onset forms of retinal degeneration. Medaka fish are also popular model organisms for studying eye development because the embryos are transparent embryos, allowing simple visual assays of mutant phenotypes, with comparatively simple and affordable animal husbandry.Citation21 However, whole genome duplication in fish can complicate genetic studies in this class of organisms due to the presence of multiple orthologs of genes.Citation22 For example, PCDH15 (protocadherin-15) is mutated in humans with Usher syndrome type 1FCitation23 and both humans and other mammals have a single PCDH15 gene that is expressed in both inner ear and retina. In contrast, the homolog has duplicated and diverged in zebrafish. The two copies of the fish gene have evolved independent, tissue-specific functions: pcdh15a is expressed in inner ear and mediates hearing and vestibular function, whereas pcdh15b is expressed in retina and mediates retinal development and function.Citation24 Zebrafish are therefore not an ideal model for studying this condition. Furthermore, fish and amphibian retinae also have limitations in their use for studying retinal degeneration because—in contrast to mammals—photoreceptors can regenerate in many adult fish and amphibians.Citation25,Citation26 As is the case in most avenues of research, a compromise must be sought when choosing which model organism to use for retinal development and degeneration studies.
Photoreceptor Development and Inherited Retinal Conditions
Mutations in genes encoding proteins involved in photoreceptor development are associated with a broad group of inherited retinal dystrophies (), often as part of complex multi-organ syndromic conditions termed the ciliopathies.Citation27 Refer to Table S1 for further details. Photoreceptor biogenesis has been suggested to occur in six distinct stages,Citation28 based on the four phases of ciliogenesis in an epithelial cell,Citation29 followed by two stages of OS development. This whole process takes over two weeks to complete in the mouse retina.Citation30 In the mouse, it begins with the docking of paired centrioles (each consisting of nine microtubule triplets) at the apical surface of the undifferentiated progenitor cell early in photoreceptor development.Citation31 The distal end of the mother centriole is surrounded by the primary vesicle and a microtubule ciliary bud begins to grow from this centriole. At the same time, this centriole matures into the basal body by recruitment of appendages, such as ODF2,Citation32 and aligns perpendicular to the plasma membrane. Many proteins implicated in human retinal disease are localized to the basal body (), and may play a role in photoreceptor biogenesis. Examples include the TOPORS protein, loss of which is associated with failure of OS formation in zebrafish,Citation33 and retinitis pigmentosa (RP), a hereditary retinal degeneration in humans.Citation34 OFD1 is a distal centriole protein that regulates the length of centriolesCitation35 and ciliary axonemes.Citation36 Mutations in OFD1 cause oro-facial-digital syndrome, non-syndromic RP, and RP as a feature of Joubert syndrome (JBTS).Citation37-Citation39 ofd1 morphant zebrafish occasionally develop retinal coloboma.Citation40 RAB28 localizes to the basal body and is suggested to play a role in coordinating ciliogenesis and rhodopsin transport, with mutations in this gene causing cone-rod dystrophy.Citation41
Once the basal body has docked at the apical cell surface, binding of post-Golgi vesicles to the primary vesicle expands the membrane to form the ciliary vesicle, allowing the ciliary bud to extend to form the axoneme. This ciliary vesicle then fuses with the plasma membrane and the CC extends toward the RPE.Citation28,Citation42 The protein CC2D2A plays a role in extension of the CC membrane through RAB8-mediated vesicular trafficking. cc2d2a zebrafish morphants develop disorganized photoreceptor OS and accumulation of opsins and vesicles in IS.Citation43 Mutations in CC2D2A in humans lead to RP, often as part of multi-organ syndromes.Citation44-Citation46 Growth of the microtubule axoneme is dependent on chaperone proteins such as prefoldin-5 that, when mutated in mice, leads to photoreceptor degeneration.Citation47 Other chaperones including HSC70 (Hspa8) have also been found to be associated with photoreceptor axonemal proteins.Citation48 Glutamylation of axonemal tubulin is essential for normal photoreceptor development and function,Citation49 and the stabilization of axonemal microtubules by other post-translational modifications such as acetylation are also presumably required for these normal processes. FAM161A, a microtubule-associated protein, is thought to have a role in this stabilization process since patients with mutations in this gene develop RP.Citation50
Rod OS biogenesis begins as the distal end of the CC, followed by membrane vesicular fusion to form the discs of the rod OS. Disc assembly is an on-going process as the membrane discs of the OS are constantly sloughed off to prevent the accumulation of toxic by-products of phototransduction.Citation51 Normal disc assembly occurs at the base of the OS, near to the ciliary axoneme. OS plasma membrane invaginates to form nascent discs, and disc proteins are then localized to their correct compartment: the OS plasma membrane, disc rim or disc lamellar region. The correct formation of discs is dependent on membrane proteins, including rom-1 and peripherin that localize to disc rims,Citation52,Citation53 and the visual pigment rhodopsin.Citation54 RAB8 mediates transport of vesicles carrying rhodopsin to the base of the CC where they bind to the Qa-SNARE syntaxin 3, or to the IS where they bind the Qbc-SNARE SNAP-25 to fuse with the plasma membrane to deliver their cargo.Citation55 RP1 also plays a role in disc assembly. RP1 binds to singlet microtubules of the axoneme,Citation56 particularly at the point of disc membrane formation. In Rp1 mutant mice, disc membranes are abnormally organizedCitation57 and discs do not stack correctly. RP1 is phosphorylated by MAK, and this is thought to regulate extension of the ciliary axoneme to control CC and OS length. Loss of Mak in mice leads to increased length of photoreceptors and retinal degeneration.Citation58 Similar to RP1, RP1L1 binds to the photoreceptor axoneme, with consequences on OS morphology and photosensitivity, leading to progressive photoreceptor degeneration when this protein is lost in mutant mice.Citation59 Loss of this protein in humans is associated with occult macular dystrophy (OMD) and RP.Citation60,Citation61 Myosin 7a, that is mutated in both Leber congenital amaurosis (LCA), an early-onset retinal dystrophy,Citation62 and Usher syndrome type 1BCitation63 is also localized to the site of disc morphogenesis. Myosin 7a plays a role in transport of proteins from the IS to the OS for incorporation into discs, and opsin accumulates in the IS of Myo7a mutant mouse.Citation64 The orphan membrane-bound receptor TMEM67 (meckelin), the protein product of the TMEM67 gene, is also required for membrane disc assembly. Bpck mice, which have a spontaneous deletion of Tmem67, have structurally normal CC but dysmorphic, misaligned membrane discs, and mislocalized rhodopsin, arrestin, and transducin leading to rapid photoreceptor degeneration.Citation65 While the CC are structurally normal in bpck mice, rhodopsin transport is compromised and rhodopsin accumulates in the IS and ONL, likely reflecting a role of TMEM67 in mediating transport along the photoreceptor CC.Citation66 Mutations in human TMEM67 are associated with a suite of ciliopathies, many of which include retinal degeneration phenotypes.Citation67-Citation69
Many retinal proteins also need to be correctly localized to the CC membrane. The cilium membrane is the site of many G-protein coupled receptors (GPCRs), mediating many important roles in cellular signaling. Inositol polyphosphate-5-phosphatase E (INPP5E) is a phosphatase that hydrolyses phosphotidylinositols, membrane-bound intermediate molecules for PI3K signaling. INPP5E localizes to the IS but does not appear to be required for ciliogenesis. Instead, it maintains cilium stability, and loss of the protein results in anophthalmia in mutant miceCitation70 and microphthalmia in morphant zebrafish.Citation71 Human mutations are associated with several syndromic retinal dystrophies including mental retardation, obesity, retinal dystrophy and micropenis (MORM) syndrome, JBTS and CORS.Citation70,Citation72 Trafficking of INPP5E to the cilium depends on PDE6D, in concert with ARL13B and CEP164,Citation73 which when mutated in humans also lead to syndromic retinal dystrophies.Citation74,Citation75
The Tectonic proteins TCTN1 and TCTN2 play a role in targeting GPCRs to the cilium membrane, as well as other components of G-protein signaling such as the downstream effector adenylyl cyclase III (ACIII) and the putative channel protein polycystin-276. Tctn1 mutant mice develop microphthalmia and mutations in humans lead to JBTS.Citation76 Tctn2 mutant mice also develop micropthalmia, and mutations in humans are associated with JBTSCitation77 and Meckel-Gruber syndrome (MKS).Citation78
Transport in the Photoreceptor
All protein synthesis in the photoreceptor occurs in the IS, yet the OS is the region involved in photoreception, with the highest demand for protein. The mouse OS proteome contains almost 2000 proteins,Citation79 yet transcription and translation occur in neither the OS nor CC. An estimated 10% of protein is lost from the rod OS each day as membranes are shed into the RPE to prevent accumulation of toxic by-products of phototransduction.Citation30,Citation80 Thus, transport between the two segments along the CC is of critical importance to photoreceptor development and function.Citation51,Citation81
Some molecules, including even some proteins, move along the CC by passive diffusion. This is perhaps surprising given the presence of the septin “barrier” at the base of the cilium that is thought to regulate the movement of molecules along the cilium.Citation82 However, it has been shown that the CC does not significantly inhibit protein diffusion through the photoreceptor cell.Citation83 There remains a debate over whether transducin and arrestin, proteins involved in phototransduction, move between the OS and IS along the CC in response to light by either active or passive transport mechanisms.Citation84-Citation87 In at least some cases, proteins move by diffusion. For example, after light-induced translocation, transducin forms a stable complex with UNC119 and diffuses back into the OS.Citation88 Movement of transducin is also thought to be regulated by the Ca2+-binding centrin proteins that are localized to the basal body and axoneme of the CCCitation89 and are phosphorylated by CK2.Citation90 UNC119 also binds myristoylated ciliary proteins, such as nephrocystin-3 (NPHP3), to target them to the ciliary membrane and maintain the spatial organization of the cilium. UNC119 releases its protein cargo when it binds to the activated isoform of the small GTP-binding protein ARL3-GTP, but not when it binds ARL2. The Retinitis Pigmentosa 2 protein (RP2) localizes to the Golgi apparatus and the basal body of the photoreceptor, and coordinates vesicle trafficking and docking at the base of the cilium.Citation91 RP2 acts as the ARL3 GTPase activating protein (GAP)Citation92 and ARL2BP is an effector for both ARL2 and ARL3. Correct targeting of such proteins to ciliary membranes is functionally important for photoreceptors, and mutations in RP2, NPHP3 and ARL2BP can all lead to ciliopathies involving retinal degeneration in humans and model organisms.Citation93-Citation97
Intraflagellar Transport in the Photoreceptor Axoneme and Inherited Retinal Conditions
ARL3 is also involved in the main active transport process along the photoreceptor axoneme: the process of intraflagellar transport (IFT).Citation98,Citation99 IFT is required for the entire process of CC and OS development, and IFT proteins are involved at all stages of this development. A pool of IFT proteins is localized in the cytoplasm at the base of the axoneme during the formation of the ciliary vesicle and elongation of the axoneme early in CC and OS development, and IFT proteins are consistently associated with the developed axoneme.Citation28 IFT is also essential for normal photoreceptor function by mediating the transport of proteins of the phototransduction cascade to the OS.Citation87 Cargoes of IFT have been shown to include rhodopsin, chaperone proteins and the photoreceptor-specific membrane protein guanylyl cyclase 1 (GC1, Gucy2e).Citation48
IFT is a bi-directional transport process that moves cargo along the axoneme of the cilium from base to tip (anterograde IFT, from IS to OS in the case of the photoreceptor), and from tip to base (retrograde IFT, from OS to IS). It is likely that retrograde IFT plays a less important role than anterograde IFT in photoreceptor function, because proteins are shed from the distal tip of the OS rather than transported back to the IS for recycling. Nevertheless, retrograde IFT components are found to be associated with the CC, as the cell will always have a minimum requirement for retrograde IFT to return anterograde IFT proteins back to the IS. Kinesins catalyze anterograde IFT,Citation100,Citation101 whereas dyneins (specifically the axonemal dyneins and the DHC1b isoform of cytoplasmic dynein) power retrograde IFT.Citation102 The retrograde IFT motor component cytoplasmic dynein 2 is required for OS biogenesis and function, and is found localized to the photoreceptor axoneme.Citation103 Zebrafish lacking this cytoplasmic dynein develop short, disordered OS which accumulate vesicles.Citation104
Heterotrimeric kinesin-II, composed of subunits of KIF3A in addition to either KIF3B or KIF3C, has been shown to be particularly important for IFT and is considered the most important kinesin motor for powering anterograde IFT. KIF3A localizes to the axoneme of the CC and IS in fish,Citation105 Xenopus, monkey, and humansCitation106 and KIF3B localizes to the CC and IS in Xenopus retina.Citation106 Knockout of Kif3a or Kif3b in mouse results in mid-gestation embryonic lethality, reflecting the general importance of these motor subunits in anterograde IFT in all cilia.Citation107,Citation108 Photoreceptor defects are not seen in Kif3a or Kif3b heterozygote mice, suggesting that variants in these genes in humans do not contribute to degenerative human retinal conditions.Citation109 However, mice with a conditional knockout of Kif3a in rods and cones accumulate vesicles and opsin in the IS preceding photoreceptor death.Citation87 The accumulation of opsin is less severe in the rods yet cell death is more rapid in these cells, suggesting that kinesin-II plays different roles in opsin transport in rods and cones.Citation110 Kif3c knockout mice, however, are viableCitation111 and do not exhibit photoreceptor defects,Citation109 suggesting redundancy with either Kif3a or Kif3b. Consistent with this, kif3b morphant zebrafish embryos develop relatively short but otherwise functionally normal photoreceptor cilia, also suggesting some sort of functional redundancy. Overexpression of kif3c in these kif3b morphants confirmed partial functional redundancy of these subunits.Citation112 Homodimeric kinesin-II, composed of Kif17, is also thought to play a role in anterograde IFT, although its function varies in different cilia. Kif17 is thought to play a role in photoreceptors, as it is found in the axoneme of mouse photoreceptor cells and expression of a dominant negative form of kif17 in zebrafish affects the development of photoreceptor OS.Citation113 Similarly, kif17 morphant zebrafish develop short OS and aberrant discs.Citation114
In association with the motor proteins, two distinct IFT complexes are associated with the bidirectional transport (). Proteins involved in anterograde IFT are collectively known as IFT complex B proteins, and those involved in retrograde IFT are components of the IFT complex A. IFT complex B proteins IFT20, 52, 57 and 88 localize around the basal body and in discrete puncta along the axoneme of the CC of retinal photoreceptors.Citation115,Citation116 All of these IFT components are required for normal OS development and retinal function. Loss of ift52, 57, or 88 in zebrafish leads to an absence of OS and subsequent photoreceptor degeneration.Citation117 IFT57 is not essential for IFT, but is required for efficient IFT as it plays a specific role in dissociation of kinesin-II from IFT complex proteins.Citation118 IFT57 interacts with DYF-1/Fleer,Citation119 as does IFT74, which helps the IFT particle dock onto the anterograde kinesin motor.Citation120 Ift88 hypomorphic mutant mice develop rod cell photoreceptors with abnormal OS and mislocalized opsin, leading to progressive retinal degeneration.Citation115 Mutations in IFT80, another IFT complex B protein, cause the skeletal ciliopathy asphyxiating thoracic dystrophy (ATD, also known as Jeune syndrome) in humans, a condition that involves retinal degeneration.Citation121 Zebrafish ift80 morphants exhibit defects in photoreceptor OS formation and photoreceptor deathCitation122 but hypomorphic Ift80 mutations in mice (human mutations associated with ATD are hypomorphic) have normal retinas at P21, although retinal degeneration may occur later.Citation123
IFT20 is an IFTB component that has both distinct and complementary functions to other IFT proteins since it appears to shuttle between the basal body and the Golgi apparatus,Citation124 yet it is also essential for normal photoreceptor development and function. IFT20 localizes to the cytoplasm around the basal body of the CC of the retinal photoreceptorsCitation125 and Ift20 knockout mice do not traffic opsin appropriately and OS develop abnormally.Citation126 Similarly, the IFTB component Ttc26 is required for OS formation, and ttc26 zebrafish morphants have short or absent OS.Citation127 The exact role of IFTB component TRAF3IP1 (IFT54) and IFT172 in photoreceptor development is unclear, but Traf3ip1 mutant mice develop small eyes and Ift172 mutants do not develop eyes at all, suggesting the critical importance of these proteins in ocular development.Citation128,Citation129 It is likely that the other IFTB components are also required for photoreceptor development, but to date their roles in this process have not been studied, in some cases because loss of the protein is embryonically lethal in mice.Citation130
IFT complex A protein IFT140 localizes along the photoreceptor axoneme and is particularly abundant at the tip of the axoneme.Citation125 IFT140 is essential for normal photoreceptor development in humans. Mutations in human IFT140 are a cause of Mainzer-Saldino syndrome (MZSDS)Citation131 and ATD,Citation132 both of which are skeletal ciliopathies that are associated with retinal dystrophy phenotypes. TTC21B, another IFT complex A protein, also localizes to the photoreceptor axoneme, and loss of this protein in mutant mice leads to defective photoreceptor development.Citation133 Mutations in human TTC21B are also associated with ATD, involving photoreceptor degeneration.Citation134 WDR19, which links IFTA to the BBSome (see below), is proposed to be a regulatory sub-module of IFT.Citation135 WDR19 is also mutated in ATD and cranioectodermal dysplasia (CED, also known as Sensenbrenner syndrome), which occasionally involves photoreceptor dystrophy.Citation136 It remains unknown why a skeletal ciliopathy, namely ATD, can be caused by recessive mutations in both an IFTB protein (IFT80) and IFTA proteins, and why they predominately affect the endochondral ossification of the long bones.
Other proteins external to the core A and B complexes regulate IFT, including components of the BBSome that are also localized to the base of the photoreceptor axoneme. The BBSome, comprised of BBS1, 2, 4, 5, 7, 9 and TTC8 (BBS8), is thought to function as a coat complex to sort cilium proteins into membrane domains.Citation137 Another class of BBS proteins, the chaperonin-like proteins MKKS (BBS6), BBS10 and BBS12, are thought to regulate the assembly of the BBSomeCitation138 and LZTFL1 (BBS17) regulates trafficking of the BBSome into the cilium.Citation139 The activity of the BBSome is regulated by Arf-like GTPase ARL6 (BBS3)Citation137 which is found throughout the IS, OS and ONL.Citation140 Many BBS proteins have been shown to localize to the photoreceptors,Citation140-Citation143 and loss of most of the BBSome proteins leads to retinal degeneration in model organisms and humans as part of the multi-organ condition Bardet-Biedl syndrome (BBS).Citation141,Citation144-Citation159 For example, Bbs1 knock-in mutant mice develop disorganized OS, and exhibit degeneration of IS and OS.Citation160 Other work has exemplified the disease mechanism of disrupted trafficking that underlies the retinal degeneration in this ciliopathy. Bbbs4 null mice develop grossly normal photoreceptors at an early age but exhibit defective IFT. This leads to mislocalization of specific phototransduction proteins (rhodopsin, transducin and arrestin), but not the structural photoreceptor proteins peripherin or ROM1, which subsequently causes photoreceptor degeneration.Citation142 Bbs2 mutant mice similarly mislocalize rhodopsin, leading to retinal degeneration.Citation161 Late-onset photoreceptor degeneration is observed in Bbs6 mutant miceCitation162 and Bbs8 mutant mice also mislocalize rhodopsin to the IS, leading to retinal degeneration.Citation163 BBS7 is a core component of the BBSome and physically interacts with the BBS chaperonin complex, and Bbs7 mutant mice exhibit IS, OS and ONL degenerationCitation164 due to defects in membrane protein trafficking. However, in contrast, bbs9 morphant zebrafish have an even more severe phenotype and the retina does not develop OS or discrete layers.Citation165
Proteins Interacting with IFT Components in Photoreceptors
Many other proteins, while not core components of IFT particles, are associated with IFT complexes. These proteins include the Arf-like GTPase ARL13, which is thought to promote association between IFT complex A and complex B.Citation98 Arl13B localizes to the CC in miceCitation74 and Arl13bhnn mutant mice develop abnormal eyes, polydactyly and neural tube patterning defects.Citation166 Mutations in ARL13B are a cause of the severe neurodevelopmental condition Joubert syndrome (JBTS) that can include additional clinical features such as RP.Citation74 Lebercilin also interacts with complex A, complex B IFT proteins and retrograde IFT motor proteins, and is found to localize along the ciliary axoneme (). In photoreceptor cells it is localized at the base of the CC, where it is thought to regulate loading and unloading of IFT cargo. This hypothesis is supported by the finding that in mice lacking wild-type lebercilin, cone and rod opsins are mislocalized and transducin, the G protein mediating phototransduction, partially mislocalizes in response to light. This suggests that lebercilin plays a role in connecting the IFT core machinery to proteins involved in selecting and recruiting cargo.Citation167 Lebercilin is not considered a core component of the IFT machinery because siRNA knockdown of LCA5, the gene which encodes lebercilin in humans, does not affect ciliogenesis or IFT88 localization in ciliated retinal cell lines. These interactions are disrupted when the protein is truncated, leading to disrupted IFT. This results in Leber congenital amaurosis (LCA), an early-onset retinal dystrophy in humans.Citation167,Citation168 In at least some patients with mutations in this gene, photoreceptors in the central retina are structurally normalCitation169 but visual acuity is poor from an early age. This suggests that, while important for IFT along the photoreceptor axoneme, lebercilin is not essential for structural development of the photoreceptor.
Lebercilin interacts with ninein-like protein (NINL) isoform b and usherin (USH2A) isoform b at the basal body of the connecting cilium, where these proteins are thought to play a role in regulating the docking of IFT particles.Citation170 NINL isoform b is a centrosomal protein that functions in nucleation, anchoring and outgrowth of microtubules,Citation170 and usherin (encoded by USH2A) is essential for the long-term structural maintenance of photoreceptors. Mutations in USH2A cause Usher syndromeCitation171 and mice lacking Ush2a develop late-onset progressive photoreceptor degeneration.Citation172 The usherin protein localizes to the apical IS membrane that wraps around the CC, a specialized region also known as the apical inner segment collar or pericilium that is homologous to the periciliary ridge complex of amphibians. Myosin VIIa (USH1B) also localizes to this apical IS collar,Citation173 as does VLGR1 (USH2C), whereas the USH proteins harmonin (USH1C), SANS (USH1G), and whirlin (USH2D) act as structural proteins at the apical IS collar. The apical IS collar is a specialized membrane domain that functions in the docking and loading of IFT cargos, and USH proteins are therefore considered to play important roles at this stage of IFT.Citation174 Their loss or mutation results in deafness and photoreceptor degeneration in humans.Citation63,Citation175-Citation178
CEP290 is also believed to play a role in IFT along the CC because truncating mutations in CEP290 lead to mislocalization of rhodopsin and arrestin.Citation179 CEP290 is necessary for transport along the CC, but is not required for CC development as loss of CEP290 does not compromise normal CC structure.Citation179 CEP290 is localized to the basal body, pericentriolar matrix and axoneme of the CC. Mutations in CEP290 are associated with a suite of ciliopathies with retinal involvement, ranging from LCACitation180 to multi-organ JBTS.Citation181,Citation182 Mutations in this gene are the single most common cause of non-syndromic LCA, accounting for 21% of casesCitation180,Citation183. CEP290-mutated LCA patients retain normal overall retinal and photoreceptor architecture in the cone-rich central retina, but rod-rich pericentrical and peripheral regions are not conserved in this manner. CEP290 interacts with RAF-1 kinase inhibitory protein (RKIP), and accumulation of Rkip in photoreceptors of Cep290 mutant mice is thought to be one of the causes of photoreceptor degeneration in these mice.Citation184
CEP290 also interacts with RPGR, another protein that localizes to the CCCitation185 and is thought to interact with several protein complexes that mediate transport along the photoreceptor. CEP290 also interacts with the USH protein complex through whirlin,Citation186 and multiple components of the NPHP complex, possibly as two distinct complexes involving NPHP1, 2, 5 and NPHP4, 6, 8.Citation187 However, most of the insights about potential disease mechanisms have come from further study of the complexes containing RPGR. RPGR is mutated in 15% of RP cases in humansCitation188 and loss of this protein in mice leads to ectopic localization of cone opsins and the reduction in levels of rhodopsin in rods, leading to cone-rod degeneration.Citation189 RPGR contains a domain with homology to the RCC1 guanine nucleotide exchange factor (GEF) for Ran GTPase. This RCC1-like domain in RPGR acts as a GTP/GDP exchange factor for RAB8, a GTPase important for vesicular trafficking to the primary cilium.Citation190 Jouberin, the protein encoded by AHI1 and mutated in JBTS,Citation191 localizes to the CC and is thought to play a role in this RAB8-mediated polarized vesicular trafficking. Ahi1 mutant mice either do not develop OS or develop abnormal OS leading to subsequent photoreceptor degeneration, associated with defects in vesicular trafficking of transducin, ROM1 and opsin with a decrease in Rab8 expression.Citation192,Citation193 ahi1 morphant zebrafish develop abnormally shaped eyes with coloboma and defective retinal lamination.Citation194
CEP290 and RPGR also interact with RPGR-interacting protein 1 (RPGRIP1) in rod OS.Citation195 Loss of RPGRIP1 has no effect on central (cone-rich) retinal architectureCitation196 but it is required for rod OS structureCitation197 and mice lacking RPGRIP1 develop grossly oversized OS discs.Citation198 RPGRIP1 is localized to the distal part of the photoreceptor axoneme, and is required for the recruitment of RPGR, NPHP4 and SDCCAG8 to the CC: Rpgrip1nmf247 mutant mice lack these proteins at the CC.Citation199,Citation200 Mutations in RPGRIP1 in humans are associated with RP,Citation201 cone-rod dystrophy (CORD)Citation202 and LCA.Citation203 NPHP4 mutations lead to nephronophthisis (NPHP) and RP,Citation204 and SDCCAG8 is mutated in BBS and a retinal-renal ciliopathy.Citation205-Citation207 Mutations in the homolog of RPGRIP, RPGRIP1L, are also associated with a suite of disorders involving retinal dystrophy, including JBTS,Citation208 MKS and cerebello-oculo-renal syndrome (CORS).Citation209 A specific mutation in RPGRIP1L has been shown to be a modifier of retinal phenotype in these syndromic ciliopathies.Citation210 Mutation of alanine 229 to threonine compromises the interaction of RPGRIP1L with RPGR, with functional consequences on transport within the photoreceptors that leads to retinal degeneration. RPGRIP1L also interacts with MKS and NPHP proteins at the ciliary transition zone/basal body to initiate IFT-driven cilium extension early in ciliogenesis.Citation211 The Ftm mouse mutant (in which Rpgrip1l is mutated) develops microphthalmia, associated with a global reduction in cilia number and Shh signaling defects.Citation212
ALMS1, which is mutated in Alström syndrome (ALMS), a condition often presenting with CORDCitation213,Citation214 and LCA,Citation62 is thought to play a role in transport along the photoreceptor axoneme, as gene-trap Alms1 mutant mice accumulate vesicles in their IS and rhodopsin is mislocalized to the outer nuclear layer.Citation215 ALMS1 has been shown to play a role in endosome recycling in other cell types, which may reflect its role in photoreceptors.Citation216 B9D2, inversin (NPHP2) and IQCB1 (NPHP5) are also thought to play roles in the transport of specific proteins along the photoreceptor. IQCB1 (NPHP5) localizes to the CC, OS and basal bodies of human and mouse photoreceptors, it interacts with RPGR in the CC,Citation217 and mutations in NPHP5 are the most common cause of Senior-Løken syndrome (SLS), a ciliopathy that associates NPHP with RP or LCA.Citation217 Only about 10% of individuals with NPHP also have RP or LCA, constituting SLS, but 100% of NPHP patients with NPHP5 mutations have RP. This emphasizes the essential role of IQCB1 in the photoreceptor,Citation217 but, by contrast, RP is very rarely seen in NPHP2 patients, suggesting that inversin has a high level of functional redundancy in human photoreceptors.Citation218,Citation219 Loss of b9d2 or nphp2 (inversin) in zebrafish leads to mislocalized opsin but not mislocalized peripherin,Citation119 whereas B9D2 mutations in humans are a cause of the lethal ciliopathy MKS that can occasionally present with retinal involvement.Citation220 Nephrocystin (NPHP1) localizes to the CC, especially around the basal body, and may play a similar role to B9D2 and inversin.Citation6 Indeed, Nphp1 mutant mice exhibit defects in the both the sorting of proteins between the photoreceptor IS and OS, and compromised IFT.Citation221 Mutations in NPHP1 cause the ciliopathies NPHP,Citation222 JBTSCitation223 and SLS,Citation224 all of which include retinal degeneration in some patients. NPHP4 localizes to the base of the connecting cilium, proximal to SDCCAG8, RPGRIP1 and RPGRCitation200 and when mutated leads to CORD in dogs,Citation225 NPHP, SLS and CORS in humans.Citation204,Citation226,Citation227 Similar to Nphp1 mutant mice, mice with mutations in Nphp4 exhibit mislocalization of OS proteins (rhodopsin and ROM1) to the IS and inner nuclear layer. These mice do not develop normal OS despite developing normal CC, and their photoreceptors degenerate rapidly leading to early-onset loss of vision.
Interestingly, the synaptic ribbons of Nphp4 mutant mice also degenerate after developing normally, with associated disturbances in the localizations of synaptophysin (a major synaptic vesicle protein) and post-synaptic density protein 95 (PSD95, a presynaptic marker in the outer plexiform layer).Citation228 KIF3A and IFT88 are also localized to the ribbon synapse, and expression of dominant-negative KIF3B results in a complete loss of ribbon synapses,Citation113 suggesting that IFT has an essential functional role at the photoreceptor synapse.Citation115,Citation229 The presence of clarin-1 (USH3A), which is mutated in Usher syndrome, further implicates the role of cilia proteins at the ribbon synapse in inherited disease.Citation230
Future Perspectives and Challenges
Further extending the role of IFT and the cilium in the retina, Ataxia-telangiectasia and Rad3 (ATR) protein has recently been shown to be localized to the photoreceptor CC, and mice with mutations in this gene exhibit early-onset degeneration of rods and cones.Citation231 ATR is a DNA damage sensor, potentially extending the role of photoreceptors, and primary cilia in general, to involvement in the DNA damage response (DDR). Supporting this hypothesis, mutations in other DDR genes (CEP164 and ZNF423) have been found in ciliopathy patients with retinal degeneration. It has been suggested that defective CC could cause postnatal accumulation of UV light-induced DNA damage, subsequently leading to retinal degeneration.Citation75 However, the details of this novel ciliopathy disease mechanism remain unclear, and will undoubtedly form the basis of exciting advances in basic research.
These recent findings highlight our ever-expanding understanding of the importance of primary cilia in retinal development and function, and their role in human disease (). Refer to Table S1 for further details. Novel uncharacterised proteins are still being discovered to play a role in such processes, such as C8orf37, a ciliary protein which is mutated in CORD and RPCitation232 and C2orf71, a ciliary protein mutated in RP in humans and progressive retinal atrophy in several breeds of dog.Citation233,Citation234 While our understanding of primary cilia and specialized sensory cilia such as the photoreceptor has advanced greatly in the past two decades, there still remains much to discover. With the ever-increasing power and affordability of genetic sequencing technologies, there are now unprecedented opportunities for rapid gene discovery in this group of retinal conditions, providing further insights into disease mechanism. A better understanding of the genetic basis of eye disease provides opportunities for improved diagnostics and disease management, including gene therapy. Retinal disease is a convenient model for the study of gene therapy, since vectors carrying wild-type copies of mutated genes are easily delivered to the affected cells by subretinal injection, and this method has been used to successfully restore partial vision in patients with a variety of gene mutations. Such therapies are currently in development for the treatment of patients with LCA5 mutations (Ronald Roepman, personal communication) because in this ciliopathy, as in many retinal ciliopathies, retinal architecture is not compromised and blindness arises due to the lack of a single specific protein causing defects in protein trafficking. Preliminary gene therapy experiments in Bbs1 mice have given promising results, with improved rhodopsin trafficking, reduced photoreceptor degeneration and improved ERG.Citation235 Similarly, Bbs4 mutant mice have been successfully treated with gene therapy to restore rhodopsin localization, OS structure and prevention of photoreceptor degeneration.Citation236 While treating BBS has its difficulties (particularly in ensuring that the correct stoichiometry of the BBSome complex is restored), retinal gene therapy remains a promising therapeutic approach that needs further research to assess its clinical efficacy and utility.
However, gene therapy is costly and difficult to develop, and can only be used to treat individuals with specific mutations in specific genes. This is a considerable disadvantage for groups of conditions with broad genetic heterogeneity such as the ciliopathies. The wide phenotypic variability, including allelism in different clinical entities, and the genetic heterogeneity of these conditions remain major challenges in the field of research into photoreceptor development and disease, especially in the attempts to develop therapies for these conditions. Broader therapeutic interventions such as treatment with tauroursodeoycholic acid (TUDCA), an anti-apoptotic factor, may offer opportunities for all degenerative retinal ciliopathies in the medium term, and it is promising that this treatment has been used successfully to slow retinal degeneration in Bbs1 mice.Citation237
Summary
The photoreceptor OS is a highly modified primary cilium which has evolved to play a crucial role in vision. Development of the photoreceptor is a complex process that is an elaborate form of the ciliogenesis seen in other cell types. Most of the transport along the photoreceptor is performed by the cilium transport process of intraflagellar transport (IFT). IFT is essential for the movement of proteins involved in visual transduction in this cell type. For these reasons, proteins involved in primary cilium growth, structure, maintenance and function are of critical importance to retinal structure and function. The loss or mutation of these proteins results in a group of conditions known as the retinal ciliopathies that have defects in retinal development or retinal degeneration. Many of these proteins also have functions in the primary cilia of other cell types, so mutations are often associated with other complex multi-organ or developmental phenotypes, and retinal dystrophy forms just part of a syndromic ciliopathy. A range of model organisms and human genetic studies have advanced understanding of the role of the photoreceptor cilium in retinal health and disease, but a number of key challenges still remain in this field of research. Further insights into disease mechanisms in this group of conditions, as well as the identification of mutations in an ever-increasing number of genes, will be essential if gene therapy is to fulfil the early promise of many research studies and become an effective clinical treatment for the retinal ciliopathies.
| Abbreviations: | ||
| ACIII | = | adenylyl cyclase III |
| ARL2 | = | ADP-ribosylation factor-like 2 protein |
| ARL2BP | = | ADP-ribosylation factor-like 2 binding protein |
| ARL3 | = | ADP-ribosylation factor-like 3 protein |
| ATD | = | asphyxiating thoracic dystrophy |
| BBS | = | Bardet-Biedl syndrome |
| CC | = | connecting cilium |
| CED | = | cranioectodermal dysplasia (Sensenbrenner syndrome) |
| CORS | = | cerebelloocullorenal syndrome |
| GC1 | = | guanylyl cyclase 1 protein |
| IFT | = | intraflagellar transport |
| IS | = | inner segment (of a photoreceptor cell) |
| JBTS | = | Joubert syndrome |
| LCA | = | Leber congenital amaurosis |
| MKS | = | Meckel-Gruber syndrome |
| MKKS | = | McKusick-Kaufman syndrome |
| MORM | = | mental retardation, obesity, retinal dystrophy and micropenis |
| MZSDS | = | Mainzer-Saldino syndrome |
| NINL | = | ninein-like protein |
| NPHP | = | nephronophthisis |
| OMD | = | occult macular dystrophy |
| ONL | = | outer nuclear layer |
| OS | = | outer segment (of a photoreceptor cell) |
| PKD | = | polycystic kidney disease |
| ROM1 | = | retinal outer segment membrane protein 1 |
| RP2 | = | Retinitis Pigmentosa 2 protein |
| TMEM | = | transmembrane protein |
| SLS | = | Senior-Løken syndrome |
| SNARE | = | soluble N- ethylmaleimide sensitive factor receptor |
| USH | = | Usher syndrome |
Additional material
Download Zip (47.3 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to acknowledge funding from the European Community's Seventh Framework Programme FP7/2009 under grant agreement no: 241955, SYSCILIA http://syscilia.org/ and a Sir Jules Thorn Award for Biomedical Research (JTA/09).
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/organogenesis/article/26710
Related Research Data
References
- De Robertis E. Electron microscope observations on the submicroscopic organization of the retinal rods. J Biophys Biochem Cytol 1956; 2:319 - 30; http://dx.doi.org/10.1083/jcb.2.3.319; PMID: 13331964
- De Robertis E. Some observations on the ultrastructure and morphogenesis of photoreceptors. J Gen Physiol 1960; 43:1 - 13; http://dx.doi.org/10.1085/jgp.43.6.1; PMID: 13814989
- Tokuyasu K, Yamada E. The fine structure of the retina studied with the electron microscope. IV. Morphogenesis of outer segments of retinal rods. J Biophys Biochem Cytol 1959; 6:225 - 30; http://dx.doi.org/10.1083/jcb.6.2.225; PMID: 13838675
- Besharse JC, Forestner DM, Defoe DM. Membrane assembly in retinal photoreceptors. III. Distinct membrane domains of the connecting cilium of developing rods. J Neurosci 1985; 5:1035 - 48; PMID: 3156973
- Horst CJ, Johnson LV, Besharse JC. Transmembrane assemblage of the photoreceptor connecting cilium and motile cilium transition zone contain a common immunologic epitope. Cell Motil Cytoskeleton 1990; 17:329 - 44; http://dx.doi.org/10.1002/cm.970170408; PMID: 1706225
- Fliegauf M, Horvath J, von Schnakenburg C, Olbrich H, Müller D, Thumfart J, Schermer B, Pazour GJ, Neumann HPH, Zentgraf H, et al. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J Am Soc Nephrol 2006; 17:2424 - 33; http://dx.doi.org/10.1681/ASN.2005121351; PMID: 16885411
- Besharse JC, Horst CJ. The Photoreceptor Connecting Cilium A Model for the Transition Zone. In: Bloodgood RA, ed. Ciliary and Flagellar Membranes. New York: Springer US, 1990:389-447.
- Pugh EN Jr., Lamb TD. Phototransduction in Vertebrate Rods and Cones: Molecular Mechanisms of Amplification, Recovery and Light Adaptation. In: Stavenga DG, Grip WJd, E.N.Pugh J, eds. Handbook of Biological Physics. Amsterdam: Elsevier, 2000:183-254.
- LaVail MM. Rod outer segment disk shedding in rat retina: relationship to cyclic lighting. Science 1976; 194:1071 - 4; http://dx.doi.org/10.1126/science.982063; PMID: 982063
- Long KO, Fisher SK, Fariss RN, Anderson DH. Disc shedding and autophagy in the cone-dominant ground squirrel retina. Exp Eye Res 1986; 43:193 - 205; http://dx.doi.org/10.1016/S0014-4835(86)80087-2; PMID: 3758219
- Steinberg RH, Wood I. Clefts and microtubules of photoreceptor outer segments in the retina of the domestic cat. J Ultrastruct Res 1975; 51:307 - 403; http://dx.doi.org/10.1016/S0022-5320(75)80102-X; PMID: 1138108
- Roof D, Adamian M, Jacobs D, Hayes A. Cytoskeletal specializations at the rod photoreceptor distal tip. J Comp Neurol 1991; 305:289 - 303; http://dx.doi.org/10.1002/cne.903050210; PMID: 1902849
- Eckmiller MS. Renewal of the ciliary axoneme in cone outer segments of the retina of Xenopus laevis. Cell Tissue Res 1996; 285:165 - 9; http://dx.doi.org/10.1007/s004410050632; PMID: 8766870
- Yang J, Gao J, Adamian M, Wen X-H, Pawlyk B, Zhang L, Sanderson MJ, Zuo J, Makino CL, Li T. The ciliary rootlet maintains long-term stability of sensory cilia. Mol Cell Biol 2005; 25:4129 - 37; http://dx.doi.org/10.1128/MCB.25.10.4129-4137.2005; PMID: 15870283
- Yildiz O, Khanna H. Ciliary signaling cascades in photoreceptors. Vision Res 2012; 75:112 - 6; http://dx.doi.org/10.1016/j.visres.2012.08.007; PMID: 22921640
- Curcio CA, Sloan KR, Kalina RE, Hendrickson AE. Human photoreceptor topography. J Comp Neurol 1990; 292:497 - 523; http://dx.doi.org/10.1002/cne.902920402; PMID: 2324310
- Nathans J, Thomas D, Hogness DS. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science 1986; 232:193 - 202; http://dx.doi.org/10.1126/science.2937147; PMID: 2937147
- De Robertis E. Morphogenesis of the retinal rods; an electron microscope study. J Biophys Biochem Cytol 1956; 2:Suppl 209 - 18; http://dx.doi.org/10.1083/jcb.2.4.209; PMID: 13357544
- Cideciyan AV, Rachel RA, Aleman TS, Swider M, Schwartz SB, Sumaroka A, Roman AJ, Stone EM, Jacobson SG, Swaroop A. Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum Mol Genet 2011; 20:1411 - 23; http://dx.doi.org/10.1093/hmg/ddr022; PMID: 21245082
- Sahly I, Dufour E, Schietroma C, Michel V, Bahloul A, Perfettini I, Pepermans E, Estivalet A, Carette D, Aghaie A, et al. Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J Cell Biol 2012; 199:381 - 99; http://dx.doi.org/10.1083/jcb.201202012; PMID: 23045546
- Loosli F, Del Bene F, Quiring R, Rembold M, Martinez-Morales JR, Carl M, Grabher C, Iquel C, Krone A, Wittbrodt B, et al. Mutations affecting retina development in Medaka. Mech Dev 2004; 121:703 - 14; http://dx.doi.org/10.1016/j.mod.2004.03.004; PMID: 15210178
- Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, et al. Zebrafish hox clusters and vertebrate genome evolution. Science 1998; 282:1711 - 4; http://dx.doi.org/10.1126/science.282.5394.1711; PMID: 9831563
- Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CR, Lowry RB, Knaus R, Van Laer L, Bernier FP, et al. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet 2001; 10:1709 - 18; http://dx.doi.org/10.1093/hmg/10.16.1709; PMID: 11487575
- Seiler C, Finger-Baier KC, Rinner O, Makhankov YV, Schwarz H, Neuhauss SC, Nicolson T. Duplicated genes with split functions: independent roles of protocadherin15 orthologues in zebrafish hearing and vision. Development 2005; 132:615 - 23; http://dx.doi.org/10.1242/dev.01591; PMID: 15634702
- Vergara MN, Del Rio-Tsonis K. Retinal regeneration in the Xenopus laevis tadpole: a new model system. Mol Vis 2009; 15:1000 - 13; PMID: 19461929
- Hitchcock PF, Raymond PA. The teleost retina as a model for developmental and regeneration biology. Zebrafish 2004; 1:257 - 71; http://dx.doi.org/10.1089/zeb.2004.1.257; PMID: 18248236
- Adams NA, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet 2007; 28:113 - 25; http://dx.doi.org/10.1080/13816810701537424; PMID: 17896309
- Sedmak T, Wolfrum U. Intraflagellar transport proteins in ciliogenesis of photoreceptor cells. Biol Cell 2011; 103:449 - 66; http://dx.doi.org/10.1042/BC20110034; PMID: 21732910
- Pedersen LB, Veland IR, Schrøder JM, Christensen ST. Assembly of primary cilia. Dev Dyn 2008; 237:1993 - 2006; http://dx.doi.org/10.1002/dvdy.21521; PMID: 18393310
- LaVail MM. Kinetics of rod outer segment renewal in the developing mouse retina. J Cell Biol 1973; 58:650 - 61; http://dx.doi.org/10.1083/jcb.58.3.650; PMID: 4747920
- Knabe W, Kuhn HJ. Ciliogenesis in photoreceptor cells of the tree shrew retina. Anat Embryol (Berl) 1997; 196:123 - 31; http://dx.doi.org/10.1007/s004290050085; PMID: 9278157
- Schweizer S, Hoyer-Fender S. Mouse Odf2 localizes to centrosomes and basal bodies in adult tissues and to the photoreceptor primary cilium. Cell Tissue Res 2009; 338:295 - 301; http://dx.doi.org/10.1007/s00441-009-0861-3; PMID: 19756757
- Chakarova CF, Khanna H, Shah AZ, Patil SB, Sedmak T, Murga-Zamalloa CA, Papaioannou MG, Nagel-Wolfrum K, Lopez I, Munro P, et al. TOPORS, implicated in retinal degeneration, is a cilia-centrosomal protein. Hum Mol Genet 2011; 20:975 - 87; http://dx.doi.org/10.1093/hmg/ddq543; PMID: 21159800
- Chakarova CF, Papaioannou MG, Khanna H, Lopez I, Waseem N, Shah A, Theis T, Friedman J, Maubaret C, Bujakowska K, et al. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am J Hum Genet 2007; 81:1098 - 103; http://dx.doi.org/10.1086/521953; PMID: 17924349
- Singla V, Romaguera-Ros M, Garcia-Verdugo JM, Reiter JF. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell 2010; 18:410 - 24; http://dx.doi.org/10.1016/j.devcel.2009.12.022; PMID: 20230748
- D’Angelo A, De Angelis A, Avallone B, Piscopo I, Tammaro R, Studer M, Franco B. Ofd1 controls dorso-ventral patterning and axoneme elongation during embryonic brain development. PLoS One 2012; 7:e52937; http://dx.doi.org/10.1371/journal.pone.0052937; PMID: 23300826
- Coene KLM, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJF, Ngu LH, Budny B, van Wijk E, Gorden NT, et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet 2009; 85:465 - 81; http://dx.doi.org/10.1016/j.ajhg.2009.09.002; PMID: 19800048
- Webb TR, Parfitt DA, Gardner JC, Martinez A, Bevilacqua D, Davidson AE, Zito I, Thiselton DL, Ressa JHC, Apergi M, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet 2012; 21:3647 - 54; http://dx.doi.org/10.1093/hmg/dds194; PMID: 22619378
- Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet 2001; 68:569 - 76; http://dx.doi.org/10.1086/318802; PMID: 11179005
- Ferrante MI, Romio L, Castro S, Collins JE, Goulding DA, Stemple DL, Woolf AS, Wilson SW. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum Mol Genet 2009; 18:289 - 303; http://dx.doi.org/10.1093/hmg/ddn356; PMID: 18971206
- Roosing S, Rohrschneider K, Beryozkin A, Sharon D, Weisschuh N, Staller J, Kohl S, Zelinger L, Peters TA, Neveling K, et al, European Retinal Disease Consortium. Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal-recessive cone-rod dystrophy. Am J Hum Genet 2013; 93:110 - 7; http://dx.doi.org/10.1016/j.ajhg.2013.05.005; PMID: 23746546
- Greiner JV, Weidman TA, Bodley HD, Greiner CAM. Ciliogenesis in photoreceptor cells of the retina. Exp Eye Res 1981; 33:433 - 46; http://dx.doi.org/10.1016/S0014-4835(81)80094-2; PMID: 7297621
- Bachmann-Gagescu R, Phelps IG, Stearns G, Link BA, Brockerhoff SE, Moens CB, Doherty D. The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum Mol Genet 2011; 20:4041 - 55; http://dx.doi.org/10.1093/hmg/ddr332; PMID: 21816947
- Noor A, Windpassinger C, Patel M, Stachowiak B, Mikhailov A, Azam M, Irfan M, Siddiqui ZK, Naeem F, Paterson AD, et al. CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal-recessive mental retardation with retinitis pigmentosa. Am J Hum Genet 2008; 82:1011 - 8; http://dx.doi.org/10.1016/j.ajhg.2008.01.021; PMID: 18387594
- Tallila J, Jakkula E, Peltonen L, Salonen R, Kestilä M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet 2008; 82:1361 - 7; http://dx.doi.org/10.1016/j.ajhg.2008.05.004; PMID: 18513680
- Gorden NT, Arts HH, Parisi MA, Coene KLM, Letteboer SJF, van Beersum SEC, Mans DA, Hikida A, Eckert M, Knutzen D, et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet 2008; 83:559 - 71; http://dx.doi.org/10.1016/j.ajhg.2008.10.002; PMID: 18950740
- Lee Y, Smith RS, Jordan W, King BL, Won J, Valpuesta JM, Naggert JK, Nishina PM. Prefoldin 5 is required for normal sensory and neuronal development in a murine model. J Biol Chem 2011; 286:726 - 36; http://dx.doi.org/10.1074/jbc.M110.177352; PMID: 20956523
- Bhowmick R, Li M, Sun J, Baker SA, Insinna C, Besharse JC. Photoreceptor IFT complexes containing chaperones, guanylyl cyclase 1 and rhodopsin. Traffic 2009; 10:648 - 63; http://dx.doi.org/10.1111/j.1600-0854.2009.00896.x; PMID: 19302411
- Pathak N, Obara T, Mangos S, Liu Y, Drummond IA. The zebrafish fleer gene encodes an essential regulator of cilia tubulin polyglutamylation. Mol Biol Cell 2007; 18:4353 - 64; http://dx.doi.org/10.1091/mbc.E07-06-0537; PMID: 17761526
- Di Gioia SA, Letteboer SJF, Kostic C, Bandah-Rozenfeld D, Hetterschijt L, Sharon D, Arsenijevic Y, Roepman R, Rivolta C. FAM161A, associated with retinitis pigmentosa, is a component of the cilia-basal body complex and interacts with proteins involved in ciliopathies. Hum Mol Genet 2012; 21:5174 - 84; http://dx.doi.org/10.1093/hmg/dds368; PMID: 22940612
- Young RW. The renewal of photoreceptor cell outer segments. J Cell Biol 1967; 33:61 - 72; http://dx.doi.org/10.1083/jcb.33.1.61; PMID: 6033942
- Bascom RA, Manara S, Collins L, Molday RS, Kalnins VI, McInnes RR. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron 1992; 8:1171 - 84; http://dx.doi.org/10.1016/0896-6273(92)90137-3; PMID: 1610568
- Connell G, Bascom R, Molday L, Reid D, McInnes RR, Molday RS. Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc Natl Acad Sci U S A 1991; 88:723 - 6; http://dx.doi.org/10.1073/pnas.88.3.723; PMID: 1992463
- Lee ES, Burnside B, Flannery JG. Characterization of peripherin/rds and rom-1 transport in rod photoreceptors of transgenic and knockout animals. Invest Ophthalmol Vis Sci 2006; 47:2150 - 60; http://dx.doi.org/10.1167/iovs.05-0919; PMID: 16639027
- Mazelova J, Ransom N, Astuto-Gribble L, Wilson MC, Deretic D. Syntaxin 3 and SNAP-25 pairing, regulated by omega-3 docosahexaenoic acid, controls the delivery of rhodopsin for the biogenesis of cilia-derived sensory organelles, the rod outer segments. J Cell Sci 2009; 122:2003 - 13; http://dx.doi.org/10.1242/jcs.039982; PMID: 19454479
- Liu Q, Zuo J, Pierce EA. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J Neurosci 2004; 24:6427 - 36; http://dx.doi.org/10.1523/JNEUROSCI.1335-04.2004; PMID: 15269252
- Gao J, Cheon K, Nusinowitz S, Liu Q, Bei D, Atkins K, Azimi A, Daiger SP, Farber DB, Heckenlively JR, et al. Progressive photoreceptor degeneration, outer segment dysplasia, and rhodopsin mislocalization in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc Natl Acad Sci U S A 2002; 99:5698 - 703; http://dx.doi.org/10.1073/pnas.042122399; PMID: 11960024
- Omori Y, Chaya T, Katoh K, Kajimura N, Sato S, Muraoka K, Ueno S, Koyasu T, Kondo M, Furukawa T. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc Natl Acad Sci U S A 2010; 107:22671 - 6; http://dx.doi.org/10.1073/pnas.1009437108; PMID: 21148103
- Yamashita T, Liu J, Gao J, LeNoue S, Wang C, Kaminoh J, Bowne SJ, Sullivan LS, Daiger SP, Zhang K, et al. Essential and synergistic roles of RP1 and RP1L1 in rod photoreceptor axoneme and retinitis pigmentosa. J Neurosci 2009; 29:9748 - 60; http://dx.doi.org/10.1523/JNEUROSCI.5854-08.2009; PMID: 19657028
- Akahori M, Tsunoda K, Miyake Y, Fukuda Y, Ishiura H, Tsuji S, Usui T, Hatase T, Nakamura M, Ohde H, et al. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am J Hum Genet 2010; 87:424 - 9; http://dx.doi.org/10.1016/j.ajhg.2010.08.009; PMID: 20826268
- Davidson AE, Sergouniotis PI, Mackay DS, Wright GA, Waseem NH, Michaelides M, Holder GE, Robson AG, Moore AT, Plagnol V, et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat 2013; 34:506 - 14; http://dx.doi.org/10.1002/humu.22264; PMID: 23281133
- Wang X, Wang H, Cao M, Li Z, Chen X, Patenia C, Gore A, Abboud EB, Al-Rajhi AA, Lewis RA, et al. Whole-exome sequencing identifies ALMS1, IQCB1, CNGA3, and MYO7A mutations in patients with Leber congenital amaurosis. Hum Mutat 2011; 32:1450 - 9; http://dx.doi.org/10.1002/humu.21587; PMID: 21901789
- Weil D, Küssel P, Blanchard S, Lévy G, Levi-Acobas F, Drira M, Ayadi H, Petit C. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet 1997; 16:191 - 3; http://dx.doi.org/10.1038/ng0697-191; PMID: 9171833
- Liu X, Udovichenko IP, Brown SDM, Steel KP, Williams DS. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J Neurosci 1999; 19:6267 - 74; PMID: 10414956
- Collin GB, Won J, Hicks WL, Cook SA, Nishina PM, Naggert JK. Meckelin is necessary for photoreceptor intraciliary transport and outer segment morphogenesis. Invest Ophthalmol Vis Sci 2012; 53:7; http://dx.doi.org/10.1167/iovs.11-8766; PMID: 22125274
- Leightner AC, Hommerding CJ, Peng Y, Salisbury JL, Gainullin VG, Czarnecki PG, Sussman CR, Harris PC. The Meckel syndrome protein meckelin (TMEM67) is a key regulator of cilia function but is not required for tissue planar polarity. Hum Mol Genet 2013; 22:2024 - 40; http://dx.doi.org/10.1093/hmg/ddt054; PMID: 23393159
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet 2007; 80:186 - 94; http://dx.doi.org/10.1086/510499; PMID: 17160906
- Brancati F, Iannicelli M, Travaglini L, Mazzotta A, Bertini E, Boltshauser E, D’Arrigo S, Emma F, Fazzi E, Gallizzi R, et al, International JSRD Study Group. MKS3/TMEM67 mutations are a major cause of COACH Syndrome, a Joubert Syndrome related disorder with liver involvement. Hum Mutat 2009; 30:E432 - 42; http://dx.doi.org/10.1002/humu.20924; PMID: 19058225
- Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Wise EL, Wolf MTF, Utsch B, Becker C, et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J Med Genet 2009; 46:663 - 70; http://dx.doi.org/10.1136/jmg.2009.066613; PMID: 19508969
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compère P, Schiffmann SN, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 2009; 41:1027 - 31; http://dx.doi.org/10.1038/ng.427; PMID: 19668215
- Luo N, Lu J, Sun Y. Evidence of a role of inositol polyphosphate 5-phosphatase INPP5E in cilia formation in zebrafish. Vision Res 2012; 75:98 - 107; http://dx.doi.org/10.1016/j.visres.2012.09.011; PMID: 23022135
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel-Aleem A, Rosti RO, et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 2009; 41:1032 - 6; http://dx.doi.org/10.1038/ng.423; PMID: 19668216
- Humbert MC, Weihbrecht K, Searby CC, Li Y, Pope RM, Sheffield VC, Seo S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci U S A 2012; 109:19691 - 6; http://dx.doi.org/10.1073/pnas.1210916109; PMID: 23150559
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attié-Bitach T, Holden KR, Dobyns WB, et al, International Joubert Syndrome Related Disorders Study Group. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet 2008; 83:170 - 9; http://dx.doi.org/10.1016/j.ajhg.2008.06.023; PMID: 18674751
- Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012; 150:533 - 48; http://dx.doi.org/10.1016/j.cell.2012.06.028; PMID: 22863007
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 2011; 43:776 - 84; http://dx.doi.org/10.1038/ng.891; PMID: 21725307
- Sang LY, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen XH, Scales SJ, Kwong M, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011; 145:513 - 28; http://dx.doi.org/10.1016/j.cell.2011.04.019; PMID: 21565611
- Shaheen R, Faqeih E, Seidahmed MZ, Sunker A, Alali FE, AlQahtani K, Alkuraya FSA. A TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum Mutat 2011; 32:573 - 8; http://dx.doi.org/10.1002/humu.21507; PMID: 21462283
- Liu Q, Tan G, Levenkova N, Li T, Pugh EN Jr., Rux JJ, Speicher DW, Pierce EA. The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics 2007; 6:1299 - 317; http://dx.doi.org/10.1074/mcp.M700054-MCP200; PMID: 17494944
- Anderson RE, Maude MB, Kelleher PA, Maida TM, Basinger SF. Metabolism of phosphatidylcholine in the frog retina. Biochimica et Biophysica Acta (BBA) -. Lipids and Lipid Metabolism 1980; 620:212 - 26; http://dx.doi.org/10.1016/0005-2760(80)90203-9
- Wolfrum U, Schmitt A. Rhodopsin transport in the membrane of the connecting cilium of mammalian photoreceptor cells. Cell Motil Cytoskeleton 2000; 46:95 - 107; http://dx.doi.org/10.1002/1097-0169(200006)46:2<95::AID-CM2>3.0.CO;2-Q; PMID: 10891855
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 2010; 329:436 - 9; http://dx.doi.org/10.1126/science.1191054; PMID: 20558667
- Calvert PD, Schiesser WE, Pugh EN Jr.. Diffusion of a soluble protein, photoactivatable GFP, through a sensory cilium. J Gen Physiol 2010; 135:173 - 96; http://dx.doi.org/10.1085/jgp.200910322; PMID: 20176852
- Whelan JP, McGinnis JF. Light-dependent subcellular movement of photoreceptor proteins. J Neurosci Res 1988; 20:263 - 70; http://dx.doi.org/10.1002/jnr.490200216; PMID: 3172281
- Sokolov M, Lyubarsky AL, Strissel KJ, Savchenko AB, Govardovskii VI, Pugh EN Jr., Arshavsky VY. Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron 2002; 34:95 - 106; http://dx.doi.org/10.1016/S0896-6273(02)00636-0; PMID: 11931744
- Philp NJ, Chang W, Long K. Light-stimulated protein movement in rod photoreceptor cells of the rat retina. FEBS Lett 1987; 225:127 - 32; http://dx.doi.org/10.1016/0014-5793(87)81144-4; PMID: 2826235
- Marszalek JR, Liu XR, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LSB. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell 2000; 102:175 - 87; http://dx.doi.org/10.1016/S0092-8674(00)00023-4; PMID: 10943838
- Zhang H, Constantine R, Vorobiev S, Chen Y, Seetharaman J, Huang YJ, Xiao R, Montelione GT, Gerstner CD, Davis MW, et al. UNC119 is required for G protein trafficking in sensory neurons. Nat Neurosci 2011; 14:874 - 80; http://dx.doi.org/10.1038/nn.2835; PMID: 21642972
- Giessl A, Trojan P, Rausch S, Pulvermüller A, Wolfrum U. Centrins, gatekeepers for the light-dependent translocation of transducin through the photoreceptor cell connecting cilium. Vision Res 2006; 46:4502 - 9; http://dx.doi.org/10.1016/j.visres.2006.07.029; PMID: 17027897
- Trojan P, Rausch S, Giessl A, Klemm C, Krause E, Pulvermüller A, Wolfrum U. Light-dependent CK2-mediated phosphorylation of centrins regulates complex formation with visual G-protein. Biochim Biophys Acta 2008; 1783:1248 - 60; http://dx.doi.org/10.1016/j.bbamcr.2008.01.006; PMID: 18269917
- Evans RJ, Schwarz N, Nagel-Wolfrum K, Wolfrum U, Hardcastle AJ, Cheetham ME. The retinitis pigmentosa protein RP2 links pericentriolar vesicle transport between the Golgi and the primary cilium. Hum Mol Genet 2010; 19:1358 - 67; http://dx.doi.org/10.1093/hmg/ddq012; PMID: 20106869
- Veltel S, Gasper R, Eisenacher E, Wittinghofer A. The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nat Struct Mol Biol 2008; 15:373 - 80; http://dx.doi.org/10.1038/nsmb.1396; PMID: 18376416
- Shu X, Zeng Z, Gautier P, Lennon A, Gakovic M, Cheetham ME, Patton EE, Wright AF. Knockdown of the zebrafish ortholog of the retinitis pigmentosa 2 (RP2) gene results in retinal degeneration. Invest Ophthalmol Vis Sci 2011; 52:2960 - 6; http://dx.doi.org/10.1167/iovs.10-6800; PMID: 21282572
- Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AAB, Rosenberg T, et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet 1998; 19:327 - 32; http://dx.doi.org/10.1038/1214; PMID: 9697692
- Bergmann C, Fliegauf M, Brüchle NO, Frank V, Olbrich H, Kirschner J, Schermer B, Schmedding I, Kispert A, Kränzlin B, et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet 2008; 82:959 - 70; http://dx.doi.org/10.1016/j.ajhg.2008.02.017; PMID: 18371931
- Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, Sasmaz G, Trauer U, Reinhardt R, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet 2003; 34:455 - 9; http://dx.doi.org/10.1038/ng1216; PMID: 12872122
- Davidson AE, Schwarz N, Zelinger L, Stern-Schneider G, Shoemark A, Spitzbarth B, Gross M, Laxer U, Sosna J, Sergouniotis PI, et al. Mutations in ARL2BP, Encoding ADP-Ribosylation-Factor-Like 2 Binding Protein, Cause Autosomal-Recessive Retinitis Pigmentosa. Am J Hum Genet 2013; 93:321 - 9; http://dx.doi.org/10.1016/j.ajhg.2013.06.003; PMID: 23849777
- Li Y, Wei Q, Zhang Y, Ling K, Hu J. The small GTPases ARL-13 and ARL-3 coordinate intraflagellar transport and ciliogenesis. J Cell Biol 2010; 189:1039 - 51; http://dx.doi.org/10.1083/jcb.200912001; PMID: 20530210
- Insinna C, Besharse JC. Intraflagellar transport and the sensory outer segment of vertebrate photoreceptors. Dev Dyn 2008; 237:1982 - 92; http://dx.doi.org/10.1002/dvdy.21554; PMID: 18489002
- Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol 1998; 141:993 - 1008; http://dx.doi.org/10.1083/jcb.141.4.993; PMID: 9585417
- Snow JJ, Ou GS, Gunnarson AL, Walker MRS, Zhou HM, Brust-Mascher I, Scholey JM. Two anterograde intraflagellar transport motors cooperate to build sensory cilia on C. elegans neurons. Nat Cell Biol 2004; 6:1109 - 13; http://dx.doi.org/10.1038/ncb1186; PMID: 15489852
- Pazour GJ, Dickert BL, Witman GB. The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J Cell Biol 1999; 144:473 - 81; http://dx.doi.org/10.1083/jcb.144.3.473; PMID: 9971742
- Mikami A, Tynan SH, Hama T, Luby-Phelps K, Saito T, Crandall JE, Besharse JC, Vallee RB. Molecular structure of cytoplasmic dynein 2 and its distribution in neuronal and ciliated cells. J Cell Sci 2002; 115:4801 - 8; http://dx.doi.org/10.1242/jcs.00168; PMID: 12432068
- Krock BL, Mills-Henry I, Perkins BD. Retrograde intraflagellar transport by cytoplasmic dynein-2 is required for outer segment extension in vertebrate photoreceptors but not arrestin translocation. Invest Ophthalmol Vis Sci 2009; 50:5463 - 71; http://dx.doi.org/10.1167/iovs.09-3828; PMID: 19474410
- Beech PL, Pagh-Roehl K, Noda Y, Hirokawa N, Burnside B, Rosenbaum JL. Localization of kinesin superfamily proteins to the connecting cilium of fish photoreceptors. J Cell Sci 1996; 109:889 - 97; PMID: 8718680
- Whitehead JL, Wang SY, Bost-Usinger L, Hoang E, Frazer KA, Burnside B. Photoreceptor localization of the KIF3A and KIF3B subunits of the heterotrimeric microtubule motor kinesin II in vertebrate retina. Exp Eye Res 1999; 69:491 - 503; http://dx.doi.org/10.1006/exer.1999.0724; PMID: 10548469
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci U S A 1999; 96:5043 - 8; http://dx.doi.org/10.1073/pnas.96.9.5043; PMID: 10220415
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 1998; 95:829 - 37; http://dx.doi.org/10.1016/S0092-8674(00)81705-5; PMID: 9865700
- Jimeno D, Lillo C, Roberts EA, Goldstein LS, Williams DS. Kinesin-2 and photoreceptor cell death: requirement of motor subunits. Exp Eye Res 2006; 82:351 - 3; http://dx.doi.org/10.1016/j.exer.2005.10.026; PMID: 16337628
- Avasthi P, Watt CB, Williams DS, Le YZ, Li S, Chen CK, Marc RE, Frederick JM, Baehr W. Trafficking of membrane proteins to cone but not rod outer segments is dependent on heterotrimeric kinesin-II. J Neurosci 2009; 29:14287 - 98; http://dx.doi.org/10.1523/JNEUROSCI.3976-09.2009; PMID: 19906976
- Yang Z, Roberts EA, Goldstein LS. Functional analysis of mouse kinesin motor Kif3C. Mol Cell Biol 2001; 21:5306 - 11; http://dx.doi.org/10.1128/MCB.21.16.5306-5311.2001; PMID: 11463814
- Zhao C, Omori Y, Brodowska K, Kovach P, Malicki J. Kinesin-2 family in vertebrate ciliogenesis. Proc Natl Acad Sci U S A 2012; 109:2388 - 93; http://dx.doi.org/10.1073/pnas.1116035109; PMID: 22308397
- Insinna C, Humby M, Sedmak T, Wolfrum U, Besharse JC. Different roles for KIF17 and kinesin II in photoreceptor development and maintenance. Dev Dyn 2009; 238:2211 - 22; http://dx.doi.org/10.1002/dvdy.21956; PMID: 19384852
- Insinna C, Pathak N, Perkins B, Drummond I, Besharse JC. The homodimeric kinesin, Kif17, is essential for vertebrate photoreceptor sensory outer segment development. Dev Biol 2008; 316:160 - 70; http://dx.doi.org/10.1016/j.ydbio.2008.01.025; PMID: 18304522
- Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol 2002; 157:103 - 13; http://dx.doi.org/10.1083/jcb.200107108; PMID: 11916979
- Luby-Phelps K, Fogerty J, Baker SA, Pazour GJ, Besharse JC. Spatial distribution of intraflagellar transport proteins in vertebrate photoreceptors. Vision Res 2008; 48:413 - 23; http://dx.doi.org/10.1016/j.visres.2007.08.022; PMID: 17931679
- Tsujikawa M, Malicki J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron 2004; 42:703 - 16; http://dx.doi.org/10.1016/S0896-6273(04)00268-5; PMID: 15182712
- Krock BL, Perkins BD. The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle-kinesin-II dissociation in vertebrate photoreceptors. J Cell Sci 2008; 121:1907 - 15; http://dx.doi.org/10.1242/jcs.029397; PMID: 18492793
- Zhao C, Malicki J. Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J 2011; 30:2532 - 44; http://dx.doi.org/10.1038/emboj.2011.165; PMID: 21602787
- Ou GS, Blacque OE, Snow JJ, Leroux MR, Scholey JM. Functional coordination of intraflagellar transport motors. Nature 2005; 436:583 - 7; http://dx.doi.org/10.1038/nature03818; PMID: 16049494
- Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J, et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet 2007; 39:727 - 9; http://dx.doi.org/10.1038/ng2038; PMID: 17468754
- Hudak LM, Lunt S, Chang CH, Winkler E, Flammer H, Lindsey M, Perkins BD. The intraflagellar transport protein ift80 is essential for photoreceptor survival in a zebrafish model of jeune asphyxiating thoracic dystrophy. Invest Ophthalmol Vis Sci 2010; 51:3792 - 9; http://dx.doi.org/10.1167/iovs.09-4312; PMID: 20207966
- Rix S, Calmont A, Scambler PJ, Beales PL. An Ift80 mouse model of short rib polydactyly syndromes shows defects in hedgehog signalling without loss or malformation of cilia. Hum Mol Genet 2011; 20:1306 - 14; http://dx.doi.org/10.1093/hmg/ddr013; PMID: 21227999
- Follit JA, Tuft RA, Fogarty KE, Pazour GJ. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol Biol Cell 2006; 17:3781 - 92; http://dx.doi.org/10.1091/mbc.E06-02-0133; PMID: 16775004
- Sedmak T, Wolfrum U. Intraflagellar transport molecules in ciliary and nonciliary cells of the retina. J Cell Biol 2010; 189:171 - 86; http://dx.doi.org/10.1083/jcb.200911095; PMID: 20368623
- Keady BT, Le YZ, Pazour GJ. IFT20 is required for opsin trafficking and photoreceptor outer segment development. Mol Biol Cell 2011; 22:921 - 30; http://dx.doi.org/10.1091/mbc.E10-09-0792; PMID: 21307337
- Zhang Q, Liu Q, Austin C, Drummond I, Pierce EA. Knockdown of ttc26 disrupts ciliogenesis of the photoreceptor cells and the pronephros in zebrafish. Mol Biol Cell 2012; 23:3069 - 78; http://dx.doi.org/10.1091/mbc.E12-01-0019; PMID: 22718903
- Berbari NF, Kin NW, Sharma N, Michaud EJ, Kesterson RA, Yoder BK. Mutations in Traf3ip1 reveal defects in ciliogenesis, embryonic development, and altered cell size regulation. Dev Biol 2011; 360:66 - 76; PMID: 21945076
- Gorivodsky M, Mukhopadhyay M, Wilsch-Braeuninger M, Phillips M, Teufel A, Kim C, Malik N, Huttner W, Westphal H. Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev Biol 2009; 325:24 - 32; http://dx.doi.org/10.1016/j.ydbio.2008.09.019; PMID: 18930042
- Pasek RC, Berbari NF, Lewis WR, Kesterson RA, Yoder BK. Mammalian Clusterin associated protein 1 is an evolutionarily conserved protein required for ciliogenesis. Cilia 2012; 1:20; http://dx.doi.org/10.1186/2046-2530-1-20; PMID: 23351563
- Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, Collins F, Salih MA, Gerber S, Delphin N, Bigot K, et al. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet 2012; 90:864 - 70; http://dx.doi.org/10.1016/j.ajhg.2012.03.006; PMID: 22503633
- Schmidts M, Frank V, Eisenberger T, Al Turki S, Bizet AA, Antony D, Rix S, Decker C, Bachmann N, Bald M, et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney Disease. Hum Mutat 2013; 34:714 - 24; http://dx.doi.org/10.1002/humu.22294; PMID: 23418020
- Liu Q, Zhang Q, Pierce EA. Photoreceptor Sensory Cilia and Inherited Retinal Degeneration. In: Anderson RE, Hollyfield JG, LaVail MM, eds. Retinal Degenerative Diseases. New York: Springer US, 2010.
- Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, et al, NISC Comparative Sequencing Program. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet 2011; 43:189 - 96; http://dx.doi.org/10.1038/ng.756; PMID: 21258341
- Wei Q, Zhang Y, Li Y, Zhang Q, Ling K, Hu J. The BBSome controls IFT assembly and turnaround in cilia. Nat Cell Biol 2012; 14:950 - 7; http://dx.doi.org/10.1038/ncb2560; PMID: 22922713
- Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, Leh SM, Midtbø M, Filhol E, Bole-Feysot C, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet 2011; 89:634 - 43; http://dx.doi.org/10.1016/j.ajhg.2011.10.001; PMID: 22019273
- Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 2010; 141:1208 - 19; http://dx.doi.org/10.1016/j.cell.2010.05.015; PMID: 20603001
- Seo S, Baye LM, Schulz NP, Beck JS, Zhang QH, Slusarski DC, Sheffield VC. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc Natl Acad Sci U S A 2010; 107:1488 - 93; http://dx.doi.org/10.1073/pnas.0910268107; PMID: 20080638
- Seo S, Zhang Q, Bugge K, Breslow DK, Searby CC, Nachury MV, Sheffield VC. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet 2011; 7:e1002358; http://dx.doi.org/10.1371/journal.pgen.1002358; PMID: 22072986
- Pretorius PR, Baye LM, Nishimura DY, Searby CC, Bugge K, Yang B, Mullins RF, Stone EM, Sheffield VC, Slusarski DC. Identification and functional analysis of the vision-specific BBS3 (ARL6) long isoform. PLoS Genet 2010; 6:e1000884; http://dx.doi.org/10.1371/journal.pgen.1000884; PMID: 20333246
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 2003; 425:628 - 33; http://dx.doi.org/10.1038/nature02030; PMID: 14520415
- Abd-El-Barr MM, Sykoudis K, Andrabi S, Eichers ER, Pennesi ME, Tan PL, Wilson JH, Katsanis N, Lupski JR, Wu SM. Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet-Biedl syndrome. Vision Res 2007; 47:3394 - 407; http://dx.doi.org/10.1016/j.visres.2007.09.016; PMID: 18022666
- Kim JC, Ou YY, Badano JL, Esmail MA, Leitch CC, Fiedrich E, Beales PL, Archibald JM, Katsanis N, Rattner JB, et al. MKKS/BBS6, a divergent chaperonin-like protein linked to the obesity disorder Bardet-Biedl syndrome, is a novel centrosomal component required for cytokinesis. J Cell Sci 2005; 118:1007 - 20; http://dx.doi.org/10.1242/jcs.01676; PMID: 15731008
- Mykytyn K, Nishimura DY, Searby CC, Shastri M, Yen HJ, Beck JS, Braun T, Streb LM, Cornier AS, Cox GF, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet 2002; 31:435 - 8; PMID: 12118255
- Nishimura DY, Searby CC, Carmi R, Elbedour K, Van Maldergem L, Fulton AB, Lam BL, Powell BR, Swiderski RE, Bugge KE, et al. Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2). Hum Mol Genet 2001; 10:865 - 74; http://dx.doi.org/10.1093/hmg/10.8.865; PMID: 11285252
- Chiang AP, Nishimura D, Searby C, Elbedour K, Carmi R, Ferguson AL, Secrist J, Braun T, Casavant T, Stone EM, et al. Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3). Am J Hum Genet 2004; 75:475 - 84; http://dx.doi.org/10.1086/423903; PMID: 15258860
- Mykytyn K, Braun T, Carmi R, Haider NB, Searby CC, Shastri M, Beck G, Wright AF, Iannaccone A, Elbedour K, et al. Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet 2001; 28:188 - 91; http://dx.doi.org/10.1038/88925; PMID: 11381270
- Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May-Simera H, Li H, Blacque OE, Li L, Leitch CC, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 2004; 117:541 - 52; http://dx.doi.org/10.1016/S0092-8674(04)00450-7; PMID: 15137946
- Katsanis N, Beales PL, Woods MO, Lewis RA, Green JS, Parfrey PS, Ansley SJ, Davidson WS, Lupski JR. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat Genet 2000; 26:67 - 70; http://dx.doi.org/10.1038/79201; PMID: 10973251
- Slavotinek AM, Stone EM, Mykytyn K, Heckenlively JR, Green JS, Heon E, Musarella MA, Parfrey PS, Sheffield VC, Biesecker LG. Mutations in MKKS cause Bardet-Biedl syndrome. Nat Genet 2000; 26:15 - 6; http://dx.doi.org/10.1038/79116; PMID: 10973238
- Badano JL, Ansley SJ, Leitch CC, Lewis RA, Lupski JR, Katsanis N. Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am J Hum Genet 2003; 72:650 - 8; http://dx.doi.org/10.1086/368204; PMID: 12567324
- Nishimura DY, Swiderski RE, Searby CC, Berg EM, Ferguson AL, Hennekam R, Merin S, Weleber RG, Biesecker LG, Stone EM, et al. Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am J Hum Genet 2005; 77:1021 - 33; http://dx.doi.org/10.1086/498323; PMID: 16380913
- Stoetzel C, Laurier V, Davis EE, Muller J, Rix S, Badano JL, Leitch CC, Salem N, Chouery E, Corbani S, et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat Genet 2006; 38:521 - 4; http://dx.doi.org/10.1038/ng1771; PMID: 16582908
- Chiang AP, Beck JS, Yen H-J, Tayeh MK, Scheetz TE, Swiderski RE, Nishimura DY, Braun TA, Kim K-YA, Huang J, et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet–Biedl syndrome gene (BBS11). 2006; 103:6287 - 92
- Stoetzel C, Muller J, Laurier V, Davis EE, Zaghloul NA, Vicaire S, Jacquelin C, Plewniak F, Leitch CC, Sarda P, et al. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet 2007; 80:1 - 11; http://dx.doi.org/10.1086/510256; PMID: 17160889
- Muller J, Stoetzel C, Vincent MC, Leitch CC, Laurier V, Danse JM, Hellé S, Marion V, Bennouna-Greene V, Vicaire S, et al. Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum Genet 2010; 127:583 - 93; http://dx.doi.org/10.1007/s00439-010-0804-9; PMID: 20177705
- Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet 2008; 40:443 - 8; http://dx.doi.org/10.1038/ng.97; PMID: 18327255
- Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, Lewis RA, Johnson CA, Attie-Bittach T, Katsanis N, et al. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science 2010; 329:1337 - 40; http://dx.doi.org/10.1126/science.1191184; PMID: 20671153
- Marion V, Stutzmann F, Gérard M, De Melo C, Schaefer E, Claussmann A, Hellé S, Delague V, Souied E, Barrey C, et al. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet--Biedl syndrome with situs inversus and insertional polydactyly. J Med Genet 2012; 49:317 - 21; http://dx.doi.org/10.1136/jmedgenet-2012-100737; PMID: 22510444
- Davis RE, Swiderski RE, Rahmouni K, Nishimura DY, Mullins RF, Agassandian K, Philp AR, Searby CC, Andrews MP, Thompson S, et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci U S A 2007; 104:19422 - 7; http://dx.doi.org/10.1073/pnas.0708571104; PMID: 18032602
- Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A 2004; 101:16588 - 93; http://dx.doi.org/10.1073/pnas.0405496101; PMID: 15539463
- Fath MA, Mullins RF, Searby C, Nishimura DY, Wei J, Rahmouni K, Davis RE, Tayeh MK, Andrews M, Yang B, et al. Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum Mol Genet 2005; 14:1109 - 18; http://dx.doi.org/10.1093/hmg/ddi123; PMID: 15772095
- Tadenev AL, Kulaga HM, May-Simera HL, Kelley MW, Katsanis N, Reed RR. Loss of Bardet-Biedl syndrome protein-8 (BBS8) perturbs olfactory function, protein localization, and axon targeting. Proc Natl Acad Sci U S A 2011; 108:10320 - 5; http://dx.doi.org/10.1073/pnas.1016531108; PMID: 21646512
- Zhang Q, Nishimura D, Vogel T, Shao J, Swiderski R, Yin T, Searby C, Carter CS, Kim G, Bugge K, et al. BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. J Cell Sci 2013; 126:2372 - 80; http://dx.doi.org/10.1242/jcs.111740; PMID: 23572516
- Veleri S, Bishop K, Dalle Nogare DE, English MA, Foskett TJ, Chitnis A, Sood R, Liu P, Swaroop A. Knockdown of Bardet-Biedl syndrome gene BBS9/PTHB1 leads to cilia defects. PLoS One 2012; 7:e34389; http://dx.doi.org/10.1371/journal.pone.0034389; PMID: 22479622
- Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell 2007; 12:767 - 78; http://dx.doi.org/10.1016/j.devcel.2007.03.004; PMID: 17488627
- Boldt K, Mans DA, Won J, van Reeuwijk J, Vogt A, Kinkl N, Letteboer SJF, Hicks WL, Hurd RE, Naggert JK, et al. Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J Clin Invest 2011; 121:2169 - 80; http://dx.doi.org/10.1172/JCI45627; PMID: 21606596
- den Hollander AI, Koenekoop RK, Mohamed MD, Arts HH, Boldt K, Towns KV, Sedmak T, Beer M, Nagel-Wolfrum K, McKibbin M, et al. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat Genet 2007; 39:889 - 95; http://dx.doi.org/10.1038/ng2066; PMID: 17546029
- Jacobson SG, Aleman TS, Cideciyan AV, Sumaroka A, Schwartz SB, Windsor EAM, Swider M, Herrera W, Stone EM. Leber congenital amaurosis caused by Lebercilin (LCA5) mutation: retained photoreceptors adjacent to retinal disorganization. Mol Vis 2009; 15:1098 - 106; PMID: 19503738
- van Wijk E, Kersten FFJ, Kartono A, Mans DA, Brandwijk K, Letteboer SJF, Peters TA, Märker T, Yan XM, Cremers CW, et al. Usher syndrome and Leber congenital amaurosis are molecularly linked via a novel isoform of the centrosomal ninein-like protein. Hum Mol Genet 2009; 18:51 - 64; http://dx.doi.org/10.1093/hmg/ddn312; PMID: 18826961
- Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 1998; 280:1753 - 7; http://dx.doi.org/10.1126/science.280.5370.1753; PMID: 9624053
- Liu X, Bulgakov OV, Darrow KN, Pawlyk B, Adamian M, Liberman MC, Li T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci U S A 2007; 104:4413 - 8; http://dx.doi.org/10.1073/pnas.0610950104; PMID: 17360538
- Liu X, Vansant G, Udovichenko IP, Wolfrum U, Williams DS. Myosin VIIa, the product of the Usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil Cytoskeleton 1997; 37:240 - 52; http://dx.doi.org/10.1002/(SICI)1097-0169(1997)37:3<240::AID-CM6>3.0.CO;2-A; PMID: 9227854
- Maerker T, van Wijk E, Overlack N, Kersten FFJ, McGee J, Goldmann T, Sehn E, Roepman R, Walsh EJ, Kremer H, et al. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum Mol Genet 2008; 17:71 - 86; http://dx.doi.org/10.1093/hmg/ddm285; PMID: 17906286
- Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Lainé S, Delmaghani S, Adato A, Nadifi S, Zina ZB, et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet 2003; 12:463 - 71; http://dx.doi.org/10.1093/hmg/ddg051; PMID: 12588794
- Verpy E, Leibovici M, Zwaenepoel I, Liu X-Z, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, et al. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet 2000; 26:51 - 5; http://dx.doi.org/10.1038/79171; PMID: 10973247
- Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, Millán JM, Aller E, Mitter D, Bolz H. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet 2007; 121:203 - 11; http://dx.doi.org/10.1007/s00439-006-0304-0; PMID: 17171570
- Weston MD, Luijendijk MW, Humphrey KD, Möller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet 2004; 74:357 - 66; http://dx.doi.org/10.1086/381685; PMID: 14740321
- Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, Parapuram SK, Cheng H, Scott A, Hurd RE, et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet 2006; 15:1847 - 57; http://dx.doi.org/10.1093/hmg/ddl107; PMID: 16632484
- den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KEJ, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet 2006; 79:556 - 61; http://dx.doi.org/10.1086/507318; PMID: 16909394
- Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet 2006; 38:674 - 81; http://dx.doi.org/10.1038/ng1786; PMID: 16682973
- Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, et al, International Joubert Syndrome Related Disorders Study Group. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet 2006; 38:623 - 5; http://dx.doi.org/10.1038/ng1805; PMID: 16682970
- Cremers FPM, van den Hurk JAJM, den Hollander AI. Molecular genetics of Leber congenital amaurosis. Hum Mol Genet 2002; 11:1169 - 76; http://dx.doi.org/10.1093/hmg/11.10.1169; PMID: 12015276
- Murga-Zamalloa CA, Ghosh AK, Patil SB, Reed NA, Chan LS, Davuluri S, Peränen J, Hurd TW, Rachel RA, Khanna H. Accumulation of the Raf-1 kinase inhibitory protein (Rkip) is associated with Cep290-mediated photoreceptor degeneration in ciliopathies. J Biol Chem 2011; 286:28276 - 86; http://dx.doi.org/10.1074/jbc.M111.237560; PMID: 21685394
- Hong D-H, Pawlyk B, Sokolov M, Strissel KJ, Yang J, Tulloch B, Wright AF, Arshavsky VY, Li T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Vis Sci 2003; 44:2413 - 21; http://dx.doi.org/10.1167/iovs.02-1206; PMID: 12766038
- Wright RN, Hong D-H, Perkins BD. RpgrORF15 connects to the usher protein network through direct interactions with multiple whirlin isoforms. Invest Ophthalmol Vis Sci 2012; 53:1519 - 29; http://dx.doi.org/10.1167/iovs.11-8845; PMID: 22323458
- Murga-Zamalloa CA, Desai NJ, Hildebrandt F, Khanna H. Interaction of ciliary disease protein retinitis pigmentosa GTPase regulator with nephronophthisis-associated proteins in mammalian retinas. Mol Vis 2010; 16:1373 - 81; PMID: 20664800
- Meindl A, Dry K, Herrmann K, Manson E, Ciccodicola A, Edgar A, Carvalho MRS, Achatz H, Hellebrand H, Lennon A, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet 1996; 12::13; PMID: 8528243
- Hong D-H, Pawlyk BS, Shang J, Sandberg MA, Berson EL, Li T. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc Natl Acad Sci U S A 2000; 97:3649 - 54; http://dx.doi.org/10.1073/pnas.97.7.3649; PMID: 10725384
- Murga-Zamalloa CA, Atkins SJ, Peranen J, Swaroop A, Khanna H. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum Mol Genet 2010; 19:3591 - 8; http://dx.doi.org/10.1093/hmg/ddq275; PMID: 20631154
- Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet 2004; 36:1008 - 13; http://dx.doi.org/10.1038/ng1419; PMID: 15322546
- Westfall JE, Hoyt C, Liu Q, Hsiao Y-C, Pierce EA, Page-McCaw PS, Ferland RJ. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J Neurosci 2010; 30:8759 - 68; http://dx.doi.org/10.1523/JNEUROSCI.5229-09.2010; PMID: 20592197
- Louie CM, Caridi G, Lopes VS, Brancati F, Kispert A, Lancaster MA, Schlossman AM, Otto EA, Leitges M, Gröne HJ, et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat Genet 2010; 42:175 - 80; http://dx.doi.org/10.1038/ng.519; PMID: 20081859
- Simms RJ, Hynes AM, Eley L, Inglis D, Chaudhry B, Dawe HR, Sayer JA. Modelling a ciliopathy: Ahi1 knockdown in model systems reveals an essential role in brain, retinal, and renal development. Cell Mol Life Sci 2012; 69:993 - 1009; http://dx.doi.org/10.1007/s00018-011-0826-z; PMID: 21959375
- Roepman R, Bernoud-Hubac N, Schick DE, Maugeri A, Berger W, Ropers H-H, Cremers FPM, Ferreira PA. The retinitis pigmentosa GTPase regulator (RPGR) interacts with novel transport-like proteins in the outer segments of rod photoreceptors. Hum Mol Genet 2000; 9:2095 - 105; http://dx.doi.org/10.1093/hmg/9.14.2095; PMID: 10958648
- Jacobson SG, Cideciyan AV, Aleman TS, Sumaroka A, Schwartz SB, Roman AJ, Stone EM. Leber congenital amaurosis caused by an RPGRIP1 mutation shows treatment potential. Ophthalmology 2007; 114:895 - 8; http://dx.doi.org/10.1016/j.ophtha.2006.10.028; PMID: 17306875
- Won J, Gifford E, Smith RS, Yi H, Ferreira PA, Hicks WL, Li T, Naggert JK, Nishina PM. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum Mol Genet 2009; 18:4329 - 39; http://dx.doi.org/10.1093/hmg/ddp385; PMID: 19679561
- Zhao Y, Hong D-H, Pawlyk B, Yue G, Adamian M, Grynberg M, Godzik A, Li T. The retinitis pigmentosa GTPase regulator (RPGR)- interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci U S A 2003; 100:3965 - 70; http://dx.doi.org/10.1073/pnas.0637349100; PMID: 12651948
- Hong DH, Yue GH, Adamian M, Li TS. Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem 2001; 276:12091 - 9; http://dx.doi.org/10.1074/jbc.M009351200; PMID: 11104772
- Patil H, Tserentsoodol N, Saha A, Hao Y, Webb M, Ferreira PA. Selective loss of RPGRIP1-dependent ciliary targeting of NPHP4, RPGR and SDCCAG8 underlies the degeneration of photoreceptor neurons. Cell Death Dis 2012; 3:e355; http://dx.doi.org/10.1038/cddis.2012.96; PMID: 22825473
- Booij JC, Florijn RJ, ten Brink JB, Loves W, Meire F, van Schooneveld MJ, de Jong PT, Bergen AAB. Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J Med Genet 2005; 42:e67; http://dx.doi.org/10.1136/jmg.2005.035121; PMID: 16272259
- Hameed A, Abid A, Aziz A, Ismail M, Mehdi SQ, Khaliq S. Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J Med Genet 2003; 40:616 - 9; http://dx.doi.org/10.1136/jmg.40.8.616; PMID: 12920076
- Dryja TP, Adams SM, Grimsby JL, McGee TL, Hong D-H, Li T, Andréasson S, Berson EL. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet 2001; 68:1295 - 8; http://dx.doi.org/10.1086/320113; PMID: 11283794
- Otto E, Hoefele J, Ruf R, Mueller AM, Hiller KS, Wolf MTF, Schuermann MJ, Becker A, Birkenhäger R, Sudbrak R, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet 2002; 71:1161 - 7; http://dx.doi.org/10.1086/344395; PMID: 12205563
- Billingsley G, Vincent A, Deveault C, Héon E. Mutational analysis of SDCCAG8 in Bardet-Biedl syndrome patients with renal involvement and absent polydactyly. Ophthalmic Genet 2012; 33:150 - 4; http://dx.doi.org/10.3109/13816810.2012.689411; PMID: 22626039
- Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, Stoetzel C, Patil SB, Levy S, Ghosh AK, Murga-Zamalloa CA, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet 2010; 42:840 - 50; http://dx.doi.org/10.1038/ng.662; PMID: 20835237
- Schaefer E, Zaloszyc A, Lauer J, Durand M, Stutzmann F, Perdomo-Trujillo Y, Redin C, Bennouna Greene V, Toutain A, Perrin L, et al. Mutations in SDCCAG8/NPHP10 Cause Bardet-Biedl Syndrome and Are Associated with Penetrant Renal Disease and Absent Polydactyly. Mol Syndromol 2011; 1:273 - 81; http://dx.doi.org/10.1159/000331268; PMID: 22190896
- Arts HH, Doherty D, van Beersum SEC, Parisi MA, Letteboer SJF, Gorden NT, Peters TA, Märker T, Voesenek K, Kartono A, et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet 2007; 39:882 - 8; http://dx.doi.org/10.1038/ng2069; PMID: 17558407
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 2007; 39:875 - 81; http://dx.doi.org/10.1038/ng2039; PMID: 17558409
- Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet 2009; 41:739 - 45; http://dx.doi.org/10.1038/ng.366; PMID: 19430481
- Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol 2011; 192:1023 - 41; http://dx.doi.org/10.1083/jcb.201012116; PMID: 21422230
- Vierkotten J, Dildrop R, Peters T, Wang BL, Rüther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development 2007; 134:2569 - 77; http://dx.doi.org/10.1242/dev.003715; PMID: 17553904
- Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel CF, Sicolo N, Martin M, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet 2002; 31:74 - 8; PMID: 11941369
- Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, White C, Connolly V, Taylor JFN, Russell-Eggitt I, et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat Genet 2002; 31:79 - 83; PMID: 11941370
- Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, Murray SA, Zheng QY, Smith RS, Nishina PM, et al. Alms1-disrupted mice recapitulate human Alström syndrome. Hum Mol Genet 2005; 14:2323 - 33; http://dx.doi.org/10.1093/hmg/ddi235; PMID: 16000322
- Collin GB, Marshall JD, King BL, Milan G, Maffei P, Jagger DJ, Naggert JK. The Alström syndrome protein, ALMS1, interacts with α-actinin and components of the endosome recycling pathway. PLoS One 2012; 7:e37925; http://dx.doi.org/10.1371/journal.pone.0037925; PMID: 22693585
- Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O’Toole JF, Helou J, Attanasio M, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet 2005; 37:282 - 8; http://dx.doi.org/10.1038/ng1520; PMID: 15723066
- Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet 2003; 34:413 - 20; http://dx.doi.org/10.1038/ng1217; PMID: 12872123
- O'Toole JF, Otto EA, Frishberg Y, Hildebrandt F. Retinitis pigmentosa and renal failure in a patient with mutations in INVS. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association. European Renal Association 2006; 21:1989 - 91
- Dowdle WE, Robinson JF, Kneist A, Sirerol-Piquer MS, Frints SGM, Corbit KC, Zaghloul NA, van Lijnschoten G, Mulders L, Verver DE, et al. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am J Hum Genet 2011; 89:94 - 110; http://dx.doi.org/10.1016/j.ajhg.2011.06.003; PMID: 21763481
- Jiang S-T, Chiou Y-Y, Wang E, Chien Y-L, Ho H-H, Tsai F-J, Lin C-Y, Tsai S-P, Li H. Essential role of nephrocystin in photoreceptor intraflagellar transport in mouse. Hum Mol Genet 2009; 18:1566 - 77; http://dx.doi.org/10.1093/hmg/ddp068; PMID: 19208653
- Hildebrandt F, Otto E, Rensing C, Nothwang HG, Vollmer M, Adolphs J, Hanusch H, Brandis M. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet 1997; 17:149 - 53; http://dx.doi.org/10.1038/ng1097-149; PMID: 9326933
- Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DWW, McDonald R, Eddy A, Chance PF, Glass IA. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet 2004; 75:82 - 91; http://dx.doi.org/10.1086/421846; PMID: 15138899
- Caridi G, Murer L, Bellantuono R, Sorino P, Caringella DA, Gusmano R, Ghiggeri GM. Renal-retinal syndromes: association of retinal anomalies and recessive nephronophthisis in patients with homozygous deletion of the NPH1 locus. Am J Kidney Dis 1998; 32:1059 - 62; http://dx.doi.org/10.1016/S0272-6386(98)70083-6; PMID: 9856524
- Wiik AC, Wade C, Biagi T, Ropstad EO, Bjerkås E, Lindblad-Toh K, Lingaas F. A deletion in nephronophthisis 4 (NPHP4) is associated with recessive cone-rod dystrophy in standard wire-haired dachshund. Genome Res 2008; 18:1415 - 21; http://dx.doi.org/10.1101/gr.074302.107; PMID: 18687878
- Mollet G, Salomon R, Gribouval O, Silbermann F, Bacq D, Landthaler G, Milford D, Nayir A, Rizzoni G, Antignac C, et al. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat Genet 2002; 32:300 - 5; http://dx.doi.org/10.1038/ng996; PMID: 12244321
- Alazami A, Alshammari M, Baig M, Salih M, Hassan H, Alkuraya F. NPHP4 mutation is linked to cerebello-oculo-renal syndrome and male infertility. Clin Genet 2013; Forthcoming; http://dx.doi.org/10.1111/cge.12160; PMID: 23574405
- Won J, Marín de Evsikova C, Smith RS, Hicks WL, Edwards MM, Longo-Guess C, Li T, Naggert JK, Nishina PM. NPHP4 is necessary for normal photoreceptor ribbon synapse maintenance and outer segment formation, and for sperm development. Hum Mol Genet 2011; 20:482 - 96; http://dx.doi.org/10.1093/hmg/ddq494; PMID: 21078623
- Muresan V, Lyass A, Schnapp BJ. The kinesin motor KIF3A is a component of the presynaptic ribbon in vertebrate photoreceptors. J Neurosci 1999; 19:1027 - 37; PMID: 9920666
- Zallocchi M, Meehan DT, Delimont D, Askew C, Garige S, Gratton MA, Rothermund-Franklin CA, Cosgrove D. Localization and expression of clarin-1, the Clrn1 gene product, in auditory hair cells and photoreceptors. Hear Res 2009; 255:109 - 20; http://dx.doi.org/10.1016/j.heares.2009.06.006; PMID: 19539019
- Valdés-Sánchez L, De la Cerda B, Diaz-Corrales FJ, Massalini S, Chakarova CF, Wright AF, Bhattacharya SS. ATR localizes to the photoreceptor connecting cilium and deficiency leads to severe photoreceptor degeneration in mice. Hum Mol Genet 2013; 22:1507 - 15; http://dx.doi.org/10.1093/hmg/dds563; PMID: 23297361
- Estrada-Cuzcano A, Neveling K, Kohl S, Banin E, Rotenstreich Y, Sharon D, Falik-Zaccai TC, Hipp S, Roepman R, Wissinger B, et al, European Retinal Disease Consortium. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am J Hum Genet 2012; 90:102 - 9; http://dx.doi.org/10.1016/j.ajhg.2011.11.015; PMID: 22177090
- Nishimura DY, Baye LM, Perveen R, Searby CC, Avila-Fernandez A, Pereiro I, Ayuso C, Valverde D, Bishop PN, Manson FDC, et al. Discovery and functional analysis of a retinitis pigmentosa gene, C2ORF71. Am J Hum Genet 2010; 86:686 - 95; http://dx.doi.org/10.1016/j.ajhg.2010.03.005; PMID: 20398886
- Downs LM, Bell JS, Freeman J, Hartley C, Hayward LJ, Mellersh CS. Late-onset progressive retinal atrophy in the Gordon and Irish Setter breeds is associated with a frameshift mutation in C2orf71. Anim Genet 2013; 44:169 - 77; http://dx.doi.org/10.1111/j.1365-2052.2012.02379.x; PMID: 22686255
- Seo S, Mullins RF, Dumitrescu AV, Bhattarai S, Gratie D, Wang K, Stone EM, Sheffield VC, Drack AV. Subretinal gene therapy of mice with Bardet-Biedl syndrome type 1. Invest Ophthalmol Vis Sci 2013; 54:6118 - 32; http://dx.doi.org/10.1167/iovs.13-11673; PMID: 23900607
- Simons DL, Boye SL, Hauswirth WW, Wu SM. Gene therapy prevents photoreceptor death and preserves retinal function in a Bardet-Biedl syndrome mouse model. Proc Natl Acad Sci U S A 2011; 108:6276 - 81; http://dx.doi.org/10.1073/pnas.1019222108; PMID: 21444805
- Drack AV, Dumitrescu AV, Bhattarai S, Gratie D, Stone EM, Mullins R, Sheffield VC. TUDCA slows retinal degeneration in two different mouse models of retinitis pigmentosa and prevents obesity in Bardet-Biedl syndrome type 1 mice. Invest Ophthalmol Vis Sci 2012; 53:100 - 6; http://dx.doi.org/10.1167/iovs.11-8544; PMID: 22110077