
Abstract
The ciliopathies are a group of related inherited diseases characterized by malformations in organ development. The diseases affect multiple organ systems, with kidney, skeleton, and brain malformations frequently observed. Research over the last decade has revealed that these diseases are due to defects in primary cilia, essential sensory organelles found on most cells in the human body. Here we discuss the genetic and cell biological basis of one of the most severe ciliopathies, Meckel-Gruber syndrome, and explain how primary cilia contribute to the development of the affected organ systems.
Introduction
The organization of cells into tissues and organs is fundamental to animal biology. To form such a complex series of tissues, cells from the appropriate lineage must grow, divide, migrate, differentiate, and adhere in a sequence of highly ordered events during embryogenesis. Many of the signals that control these events are relatively well-understood; however research over the last decade has revealed an unexpected link between transduction of key developmental signaling pathways and a once almost forgotten organelle: the primary cilium. In this review we discuss the current understanding of how primary cilia mediate development of the kidney, skeleton, and central nervous system, and some of the consequences when cilium functions in these tissues go awry.
Cilia are highly evolutionarily conserved organelles which project from the surface of virtually every cell type in the vertebrate bodyCitation1 (the major exceptions being leukocytes). The cilium consists of the microtubule-based core structure known as the axoneme, which is surrounded by the ciliary membrane.Citation2 Cilia have classically been categorised as either primary or motile. Motile cilia are present in large numbers in the tracheal epithelium, ependymal cells of the brain, and the oviduct, where multi-ciliated cells use a coordinated beating motion to generate fluid movement. In contrast, primary cilia are sensory organelles that act as “antennae” to sample the extracellular environment, and are involved with transducing extracellular signals to the interior of the cell.Citation3 All cilia are extended from a basal body. In the case of primary cilia, this is derived from the older of the two centrioles that together form the centrosome and have important roles in cell division. The centrioles move to the cell membrane—apical or basal depending on the cell type—and dock into the cortical cytoskeleton,Citation4 after which they are termed basal bodies, and for both motile and non-motile cilia, a specialized transport system termed intraflagellar transport (IFT) is then used to build the cilium.Citation5
The primary cilium plays an important role both during development and in adult life. The distinction between motile and sensory cilia is blurred, however, as motile cilia can be sensory,Citation6 even though the precise signaling pathways that are transduced through motile and primary cilia may be distinct.Citation7
The Ciliopathies
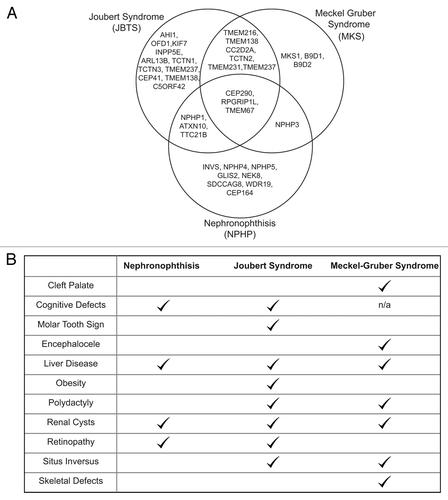
In accordance with the wide range of functions cilia perform, a number of inherited diseases, known as ciliopathies, have been increasingly linked with defects in genes that affect cilia assembly or function over recent years (reviewed in refs. Citation8 and Citation9). The protein products of these genes all localize to the cilium or basal body.Citation10 Ciliopathies share many clinical features, with renal, retinal, and hepatic involvement frequently observed alongside skeletal malformations and central nervous system developmental defects. The most severe are lethal in the early gestation or neonatal periods.Citation11 While many genes are associated with a single ciliopathy, mutation in others can give rise to a number of clinically different outcomes. For example, there is substantial overlap in the underlying genetic basis of the kidney disorder nephronophthisis (NPHP), the neurodevelopmental disorder Joubert syndrome (JBTS) and the lethal malformation disorder Meckel-Gruber syndrome (MKS) () even though the clinical presentation of the three diseases varies considerably () and genotype-phenotype correlations are frequently unclear.Citation12 It is likely that loss of function alleles might often lead to milder disease and nulls to a more severe phenotype; however, there is emerging evidence that mutation in a second ciliary gene may affect clinical outcome,Citation13 implicating modifier alleles or triallelic inheritence in the ciliopathies. One recent study on a large cohort of ciliopathy patients revealed extensive involvement of modifier alleles in conjunction with classical recessive inheritance.Citation14 Here, we focus on MKS as the most severe of these three syndromes. Diagnosis of MKS relies on the classical diagnostic triad of cystic renal disease, central nervous system malformation (almost invariably occipital encephalocele), and polydactyly, although hepatic fibrosis is also extremely common.Citation15 One of the most severe of the ciliopathies, MKS is lethal and death occurs in the perinatal period.
Figure 1. Genetic and Phenotypic overlap in Ciliopathies. (A) Venn diagram to show the high degree of allelism in the genetic basis of Joubert syndrome, nephronophthisis, and Meckel-Gruber syndrome. (B) Table of clinical phenotypes in Joubert Syndrome, nephronophthisis, and Meckel-Gruber syndrome.
Meckel Gruber Syndrome Proteins in Cilium Formation and Function
MKS shows extensive genetic heterogeneity. Mutations in MKS1, TMEM216, TMEM67, CEP290, RPGRIP1L, CC2D2A, NPHP3, TCTN2, B9D1 and B9D2, and TMEM231 thus far have been shown to be causative of MKS in humans,Citation16-Citation26 and likely pathogenic mutations in TMEM138, TMEM237, EVC2, C5orf42, and SEC8 have also been identified.Citation27,Citation28 The first genes underlying MKS were identified in 2006Citation21,Citation24 and, following the realization that MKS is linked to ciliary biology, initial efforts focused on localizing MKS proteins within cells and assessing the effect of deleting or depleting them on cilium formation and morphology. The picture is complicated and many MKS-linked proteins are found in multiple subcellular locations, including the basal body which templates the ciliary axonemeCitation29-Citation31 and centrioles/centrosomesCitation31,Citation32,Citation33,Citation34 in non-ciliated cells. Several MKS proteins are predicted to be transmembrane proteins, Citation26,Citation35,Citation36but the best studied of these, TMEM67, is also found at the basal body and the actin cytoskeleton as well as the apical and ciliary membrane.Citation35,Citation36 Similarly, TMEM216 is found at basal bodies, the ciliary membrane, and other microtubule-based cellular structures in addition to membranes.Citation26 Despite having different distribution patterns, TMEM67 interacts with both TMEM216 and MKS1 in mammalian cellsCitation26,Citation36and C. elegans.Citation29 There are conflicting data regarding their role in cilium assembly, which may reflect differences in experimental technique or model organism/tissue-specificity, with some studies showing defects in early stages of ciliogenesis and loss of ciliaCitation26,Citation36,Citation37,Citation38 and others observing increased cilium lengthCitation24,Citation39 or multi-centriolar, multi-ciliated cellsCitation40 on depletion of the protein, sometimes with alterations to cilium structure.Citation41 As well as their mutual interaction, MKS1 and TMEM67 also interact with nesprins and filamins, important scaffold proteins for rearrangement of the actin cytoskeletonCitation35,Citation42 and cells from Meckel-Gruber syndrome patients carrying mutations in either TMEM67 or TMEM216 display actin cytoskeletal defects as well as ciliogenesis defects.Citation26,Citation35 Less is known about many of the other MKS proteins. Several are required for ciliogenesis in model organismsCitation25,Citation30,Citation31,Citation33,Citation43,Citation44 and mutation or loss can also give rise to defects in wider cellular processes as microtubule organization,Citation33,Citation43 vesicle trafficking,Citation45 and signal transduction.Citation46,Citation47
The ciliary transition zone and ciliary compartmentalization
Lately the focus has shifted away from basal body and axonemal functions of MKS proteins to the transition zone, where multiple ciliopathy proteins form complexes involved in ciliary compartmentalisation. The commonality of ciliopathy proteins in this context provides an exciting linkage between the various syndromes.
It is not enough simply to build a cilium. For cilium-generated signaling to occur correctly, the cilium must be home to a distinct subset of proteins which must be able to transfer in and out of the cilium in response to different signals, and maintaining this compartmentalization represents a unique challenge. The membrane overlying the ciliary axoneme is contiguous with the plasma membrane, yet has a distinct composition,Citation48 suggesting that there must be a mechanism which prevents free diffusion between the two compartments. Several different mechanisms restrict entry and exit, and it is unclear how these co-operate or whether there are organism-, tissue- or cell-specific differences. Indeed, one study found no evidence of a diffusion barrier inhibiting entry to the cilium at all, but did find that certain proteins were selectively retained.Citation49 Several recent studies have identified overlapping protein complexes in this region which, when abrogated, result in a loss of correct ciliary compartmentalisation.Citation47,Citation50-Citation52 The majority of the proteins found in these complexes are ciliopathy proteins implicated in NPHP, JBTS, and MKS, which has led to the attractive hypothesis that these ciliopathies are linked via these complexes. The current model suggests there are two main complexes; a large MKS/JBTS complex consisting of approximately 16 proteins, and a second smaller complex consisting of NPHP1, 4 and 5.Citation2 All the MKS proteins except NPHP3 are found in the MKS/JBTS complex; MKS7/NPHP3 localizes to the inversin compartment just distal to the TZ.Citation53 In C. elegans, at least, the two complexes are linked via RPGRIP1L.Citation52
Vesicle transport of proteins to the cilium is limited by size restrictions imposed by the transition fibers, which extend from the distal end of the basal body and dock to the plasma membrane. The periciliary region may also represent a barrier to protein entry maintained via the apical actin cystoskeletonCitation49 and a ring of Septins 2 and 7 at the ciliary base,Citation54 which also mediates correct localization of the TZ protein complexes.Citation50 Another barrier exists at the base of the cilium; possibly a nuclear pore-like mechanism, consisting of many of the same pore proteins and utilizing an importin-mediated transport system responding to a Ran-GTP gradient from within the cilium.Citation55-Citation57
Several important questions remain to be addressed, perhaps most importantly how these complexes act as a barrier: are they a molecular sieve, or do they regulate some other mechanism contributing to ciliary entry and exit? The current model is complicated by the various organisms involved in the work; for example, in C. elegans at least two proteins must be abrogated—one from each complex—before a phenotype is seen except in the case of RPGRIP1L,Citation52 whereas in mammalian models (and apparently in human disease) mutation of a single gene is sufficient. It is also important to remember that while these proteins may function as a complex at the TZ after cilium assembly, many ciliopathy proteins have multiple localizations and roles outside of this TZ complex, including extra-ciliary functions as observed for IFT proteins in non-ciliated immune cells.Citation58 In the next few years, we will hopefully come to understand exactly how these complexes form, their proper function and regulation, and the contribution of each component.
Rodent models of MKS
We are only just beginning to understand the molecular and cellular functions of MKS proteins, and most of the roles we have described have only been uncovered in the last 2–3 y. Many mouse models of ciliopathies have been studied over the last few yearsCitation59 and these have helped yield new insights into the phenotypic defects observed following mutation in distinct ciliary proteins. Mouse and rat models covering many of the MKS genes have been produced.Citation17,Citation19,Citation24,Citation33,Citation37-Citation39,Citation47,Citation50,Citation60-Citation66 Many of the phenotypes observed can be explained by dysregulation of cilium-dependent signaling in these animals, presumably due to problems in the regulation of ciliary permeability at the transition zone. MKS mutants exhibit phenotypes that apparently reflect alterations in major developmental signaling pathways including Hedgehog (Hh) and Wnt signaling (see below) and several studies have found alterations in Hh signaling in these animals.Citation19,Citation37,Citation38,Citation50,Citation60,Citation62,Citation66,Citation67 The role of cilia in Hh signaling is now generally accepted; however their role in mediating canonical Wnt and non-canonical Wnt/planar cell polarity signaling remains controversial.Citation68 Nevertheless, MKS1, TMEM67, CEP290, and RPGRIP1L mutant mice show perturbation of either the canonical or non-canonical/planar cell polarity Wnt pathways,Citation38,Citation60,Citation63-Citation65 suggesting a link between some MKS proteins and Wnt signaling that merits further study. Several other important developmental signaling pathways have also been linked with cilia, including fibroblast growth factor (FGF),Citation69 platelet-derived growth factor (PDGF)Citation70 and Notch/Delta.Citation71 These remain less well understood and the effect of mutation in MKS genes on transduction of these and other pathways has not been examined. Further research will be required to disentangle the complex phenotypes and rationalise some of the conflicting data in the literature to gain a comprehensive understanding of how each mutation leads to the clinical presentation of MKS. In the meantime, a large body of evidence provides insights into why ciliopathy patients frequently present with developmental anomalies of the kidney, skeleton, and brain.
Cilia in Development of the Kidney, Skeleton, and Central Nervous System
Cystic kidney disease
Cystic kidney disease is characterized by abnormal proliferation and differentiation of the epithelial cells lining the tubules of the kidney, resulting in formation of fluid-filled cysts that prevent proper organ function and can eventually lead to renal failure. Kidney cysts are relatively common in adults over the age of 50Citation72 and can be symptomatic of various diseases including diabetes and high blood pressure. However, cystic kidney disease is the most common genetic cause of end-stage renal diseaseCitation73 and nephronophthisis is the main genetic cause of renal failure in patients under 30.Citation74 It is apparent that the kidney is highly susceptible to cyst formation. Numerous genes are linked to cystogenesis, the majority localizing to cilia and/or basal bodies, but the effector pathways involved are still not well understood. Renal cysts are a hallmark of the ciliopathies, leading to the suggestion that all genes causative of cystic kidney disease have a cilium-related function,Citation75 but cystic kidneys do occur in well-characterized diseases with no obvious ciliary role, such as Zellweger syndrome (a disorder of peroxisome biogenesis).
One of the first links between cilia and renal cysts was the discovery that polycystin-1 (PC1/Pkd1) and 2 (PC2/Pkd2), causative of Autosomal Dominant Polycystic Kidney Disease (PKD), localize to the primary cilium. The polycystins are not essential for ciliogenesisCitation76 but form a mechanosensory ion channel at the cilium which responds to fluid flow through the tubule of the nephron by inducing a calcium influx possibly involved in cell proliferation and differentiation.Citation77 Mutations in a third gene, fibrocystin (Pkhd1), cause the autosomal recessive form of the disease, ARPKD. Fibocystin localizes to primary cilia and basal bodies and interacts directly with polycystin-2.Citation78 However, loss of mechanosensation alone is insufficient for cyst formation,Citation79 and disease progression differs according to age of onset, suggesting an additional level of complexity. Disruption of IFT88 or Kif3a (both required for ciliogenesis) during development leads to acute cystic kidney disease shortly after birth, while abrogation of gene function in adult mice results in slow disease progression despite a comparable loss of cilia.Citation80,Citation81 Acute injury exacerbates this slow development of cysts in the adult.Citation80 It should also be noted that in at least two cases, cilia appear to be present in non-cystic kidney tissue but are lost after initiation of cystogenesis.Citation82,Citation83 Both polycystins also play roles in cell-cell adhesion and several signaling pathways, some of which are implicated in cystogenesis (for review see refs. Citation84 and Citation85).
The mTOR pathway, which integrates growth factor input with cell metabolism and proliferation, has risen to prominence in recent years; increased mTOR activation is observed in both ADPKD and ARPKD patients. There may be direct links to mTOR downstream of the polycystins,Citation86-Citation88 though other evidence shows calcium-independent links between the primary cilium and mTOR.Citation89 Initiation of cystogenesis occurs independently of mTOR in a Pkd1 mouse model.Citation90 Despite this, rapamycin treatment reduces cyst formation and prevents loss of kidney function in a variety of animal models, including at least one nephronophthisis model (reviewed in ref. Citation91), suggesting that the various mechanisms implicated in the different renal phenotypes might eventually converge upon mTOR,Citation92 possibly due to activation of renal repair mechanisms.Citation93 Unfortunately, clinical trials in ADPKD patients have thus far shown little efficacy.Citation94,Citation95 Two studies using cyclin-dependent kinase (CDK) inhibitors have shown promise in mouse models of ADPKD.Citation96,Citation97 These inhibitors prevent the excess cell proliferation which contributes to cyst enlargement and decrease the number of apoptotic cells – a feature also implicated in cyst formation.Citation98
The role of Wnt signaling in the kidney: Proliferation vs. polarity
The renal disease in the ciliopathies may vary in cyst location, age of onset and gross morphology—for example, the kidneys of patients with ADPKD, MKS or infantile nephronophthisis are grossly enlarged while the kidneys of patients with juvenile or adolescent nephronophthisis are of normal or reduced size. This age-dependent variation in phenotype provides further evidence that mechanisms other than the mechanosensory contribute to cystogenesis, and that correctly regulated cilium-dependent signaling and compartmentalisation are key. The Wnt signaling pathway has emerged as a key player in the development of cystic kidney disease. This complex family of secreted glycoproteins is implicated in cell growth, polarity, and cell fate determination.Citation99
Wnt signaling is usually broken down into three distinct pathways: canonical/ β-catenin dependent, and two β-catenin-independent pathways - planar cell polarity (PCP, also called Wnt/JNK) and Wnt/calcium. However, while it is convenient to categorize in this way, these pathways are not independent and recent evidence suggests that cellular responses are more complex, resulting in a move toward an integrative approach.Citation100,Citation101 The role of the cilium in Wnt signaling is controversial (discussed in ref. Citation68) but mutation of numerous ciliopathy genes results in perturbations in Wnt signaling,Citation102,Citation103 including MKS genes,Citation38,Citation60,Citation64,Citation65 though the exact nature of the changes varies. Inversin/NPHP2, for example, acts as a molecular switch from canonical to non-canonical signaling,Citation103 while AHI1/Jouberin is a positive regulator of canonical Wnt signaling and transports the effector β-catenin to the nucleus.Citation104
Canonical Wnt signaling induces the stabilization of β-catenin, which then translocates to the nucleus where it activates Wnt-specific target genes. Canonical Wnt signaling is linked to cell proliferation and is often hyperactivated in cancer.Citation105 Hyperactivation of canonical Wnt signaling also seems to play a central role in kidney cyst development; persistent β-catenin activation during development yields renal cysts similar to those seen in ADPKD.Citation106,Citation107 However, downregulation of canonical Wnt signaling also results in cyst formation.Citation108 There is evidence that polycystin-1 may both stabilize β-cateninCitation109 and inhibit its activity,Citation110 suggesting complex effects on canonical Wnt signaling in the pathogenesis of ADPKD. Together, these data imply that canonical Wnt signaling must be very carefully controlled through the development of the kidney, and that misregulation in either direction may lead to cyst development.
Non-canonical Wnt signaling has at least two branches; the planar cell polarity (PCP) pathway and Wnt/calcium. Wnt/Calcium results in a Wnt-induced rise in intracellular calcium levels, which activates several calcium-sensitive enzymes including Protein Kinase C (PKC), calcineurin, and calcium–calmodulin-dependent kinase II (CamKII) which in turn activate NFAT, NFκB, and CREB transcription factors.Citation111,Citation112 This pathway is also linked to a decrease in cyclic guanosine monophosphate (cGMP) levels via PDE6, a cGMP-specific phosphodiesterase. Wnt/calcium may be required for proper development of the kidney,Citation113 but as yet its roles in cystogenesis are not clear, and the majority of work has focused on the PCP pathway. The PCP pathway signals via the Rho family GTPases (including Rho, Rac, Cdc42) to control cell polarity and cytoskeletal arrangement. Loss of core PCP proteins results in cystogenesis,Citation114 suggesting this pathway is fundamental to kidney homeostasis. PCP is involved in oriented cell division (OCD), which controls the orientation of mitosis within the plane of an epithelial cell layer. In kidney tubules, division along the axis of the tubule results in tubule elongation, while division perpendicular to that axis results in tubule dilation, an early step in cyst formation. Mice lacking Kif3a lose OCD, implicating the cilium as a key organelle in PCP signaling in the kidney.Citation80 OCD is commonly lost in cystic kidneys, but the degree to which it is a cause or an effect of cystogenesis is controversial. Some models such as the pck rat (a model of ARPKD) and a Pkd1 mouse model show loss of OCD prior to cystogenesis.Citation115,Citation116 However, another study of Pkd1 and Pkd2 mutant mice show that tubule dilation, the first step in cystogenesis, precedes the loss of OCD, and the Pkhd1del4/del4 mouse model (ARPKD) loses OCD without ever forming cysts.Citation117 Therefore proliferation rather than polarity may be the main culprit in cystogenesis. In contrast, loss of OCD initiates cystogenesis prior to changes in proliferation in the inversin/NPHP2 mouse model of nephronophthisis.Citation118
It is clear that Wnt signaling plays an important role in the development of cystic kidneys, but the exact contribution of canonical and non-canonical Wnt signaling pathways in the initiation of cystogenesis remains complex, and either up- or down-regulation of the canonical Wnt signaling pathway leads to cyst development.Citation104,Citation106 It is not yet clear how Wnt signaling links to either mechanosensation or to mTOR activation, but it is apparent that a delicate balance of signaling is required for the correct formation and homeostasis of the kidney, and any perturbation of this balance results in cystogenesis.
Cilium-dependent Hedgehog signaling and skeletal patterning
Polydactyly has been a characteristic symptom of aberrant Hedgehog signaling for many years, but it was only in 2003 that it was finally discovered that cilia play a fundamental role in the Hh signaling pathway in vertebrates.Citation119 As such, polydactyly is perhaps the best-understood of the ciliopathy symptoms. There are three Hh ligands in mammals—Indian, Desert, and Sonic—which signal via similar pathways. Sonic Hedgehog signaling patterns the vertebrate limb in a concentration-dependent manner; deletion of Sonic Hedgehog (Shh) results in only a single digit, while overexpression results in formation of additional digits.Citation120 Shh is normally only expressed in the posterior portion of the limb bud, which specifies digits 3, 4, and 5; digit 2 does not express Shh but receives signal from the adjoining region.Citation120 As such, the single digit observed when Shh is lost represents digit 1 (in humans, the thumb). If the Shh signal is extended throughout more of the limb bud, additional digits are formed. Hh signaling is likewise essential for proper bone development and skeletal patterning, and therefore is also responsible for the various other skeletal defects common throughout the ciliopathies such as craniofacial malformations and short limbs.
Perhaps the main reason the role of cilia in Hh signaling came as such a surprise was that the majority of studies had been performed in Drosophila, where Hh signaling is cilia-independent. Likewise, the popular model organism C. elegans has a divergent Hh pathway compared with vertebrates, with many “key” pathway members absent.Citation121 It was not until studies of skeletal malformations in mice linked polydactyly with a variety of IFT genes that the importance of cilia in Hh signaling became apparentCitation119 and the role of cilia in Hh signaling has since then been relatively well-characterized, though some questions remain.
Hh ligands interact with the Patched-1 (Ptch-1) receptor on the primary cilium membrane, which stimulates translocation of Ptch-1 out of the primary cilium, allowing entry of the signaling protein Smoothened (Smo).Citation122 Activated Smo prevents processing of Gli transcription factors into transcriptional repressors, and thus allows transcription of Hh target genes. As the cilium is required for this response, it could be assumed that in the absence of a cilium, Shh signals are blocked; however, Shh absence results in loss rather than gain of digits, suggesting that the situation is more complex. In the absence of signal, the Gli transcription factors are normally processed to repressor forms (GliRs), which maintain the “off” state of Hh target genes. However, Hh ligands prevent this processing, leading to production of activated forms of Gli (GliAs). IFT proteins are required downstream of Ptch-1 and Smo for processing of Gli into both activator and repressor forms.Citation123-Citation125 As such, ciliary malfunction can lead to phenotypes characteristic of both hyper- and hypo-activation of Hh signaling; indeed, ciliopathies commonly present with both polydactyly in the limb, combined with brain abnormalities due to a weakened Hedgehog response in the neural tube.
The key role of IFT proteins in Hh signaling may be due less to a direct modulatory effect and more due to their roles in trafficking; large portions of the Hh signaling pathway are linked with dynamic relocalization of various proteins. Activation of Gli transcription factors relies on their relocalization to the ciliary tip;Citation126-Citation129 the current model suggests that here, Smo is able to release suppression by their binding partner, Suppressor of Fused (SuFu).Citation130,Citation131 In the absence of signal, Sufu sequesters Gli proteins in the cytoplasmCitation128,Citation132; both Sufu and Gli are trafficked to the ciliary tip via a Protein Kinase A-dependent mechanism shortly after Hh stimulation.Citation128,Citation133 Notably, Sufu abrogration in cells devoid of cilia leads to full activation of Hh signalingCitation134,Citation135; this is also the case in cells lacking Smo,Citation135 indicating that the Smo-mediated suppression of Sufu is key to pathway regulation, and that this suppression is cilium-dependent.
Recent evidence linking a number of ciliopathy proteins with ciliary compartmentalisation via the transition zone, together with the reliance of the Hh pathway on trafficking of signaling intermediates into and out of the primary cilium, suggests that many ciliopathy symptoms attributable to aberrant Hh signaling are caused by changes in ciliary permeability. At least one study has shown that Hh intermediates are not properly localized in response to Shh activation when TZ complexes are disrupted.Citation47 However, we do not yet understand the nature of the interaction between IFT complexes and transition zone complexes, nor the interactions of individual ciliopathy proteins with IFT-related processes. It is also essential that we develop a better understanding of the recently-uncovered “non-canonical” Hedgehog signaling mechanisms, defined as those which do not require the Gli effector proteins and which therefore may or may not require ciliary involvement.Citation136
Cilia in development of the CNS
The role of cilia within the nervous system is less well defined than within the kidney and skeleton but, given that many ciliopathies present with CNS malformations, it is clear that these organelles are of critical importance for brain development.Citation137,Citation138 While most cell types in the brain have a primary cilium, including all neurons, motile cilia are restricted to the ependymal epithelia that line the brain ventricles and direct cerebrospinal fluid (CSF) flow through the ventricles. Non canonical Wnt signaling is required for correct organization and function of these cilia,Citation139,Citation140 and defects lead to hydrocephalus. However, hydrocephalus can also develop prior to formation of a ciliated ependymaCitation141 due to misregulation of primary cilium-dependent ion transport and neuropeptide signaling within the choroid plexus cells,Citation141,Citation142 which produce the CSF.Citation142 Taken together, these studies indicate that primary cilia on choroid plexus cells act as chemosensors to regulate CSF production and transport into the brain ventricles, where the motile cilia are used to regulate CSF flow.
Most recent research has focused on the role of cilia in neural development. The vertebrate brain starts out as a sheet of neuroepithelial cells that migrate and differentiate to form the plethora of cell types found in the adult.Citation143 A complex network of signaling cascades regulates this processCitation144 and following induction, a series of cell shape changes and intercalation movements transform the sheet into the neural tube, which ultimately forms the brain and spinal cord.Citation145 Closure of the neural tube begins in the middle and works outwards. Failure to close the tube at any point leads to neural tube defects including spina bifida, anencephaly (a lethal condition caused by anterior neural tube closure defects and consequent degeneration of the forebrain and skull), and encephalocele (characterized by protrusion of the brain, cerebrospinal fluid and surrounding membrane through the skull). The neural tube defect seen in MKS (and more rarely in JBTS) is almost invariably encephalocele.
Cell migration, polarization and differentiation within the forming neural tube are regulated by several signaling pathways linked to primary cilia. Neural tube development is specified by opposing dorso-ventral gradients of Wnt (and bone morphogenetic proteins) and Shh, which regulate each other’s expression in a negative feedback loop (). During development, Shh is produced by the notochord to specify the ventral neural tube and form neuronal stem cells,Citation146 a process which requires the primary cilium.Citation147 Links between cilia and Wnt signaling in the brain are less clear-cut,Citation68 but it is clear that the various branches of Wnt signaling are critically important for brain development. During neural tube development, Wnts are expressed dorsally and regulate neural crest development and cellular differentiation.Citation148 Primary cilia may inhibit the canonical Wnt pathway: cells where cilium assembly or function is compromised have upregulated canonical Wnt signalingCitation149,Citation150 and the ciliopathy protein Inversin is proposed to act as a switch between canonical and non-canonical Wnt signaling at the basal body.Citation103 As such, loss of the primary cilium is likely to upregulate canonical Wnt signaling and downregulate non-canonical signaling, with downstream consequences for brain development. The non-canonical PCP pathway is vital to neural tube development and has also been linked to ciliogenesis in the brain,Citation151-Citation153 providing another potential level of cross-talk between Wnt and Hh. Therefore it is unsurprising that many ciliopathies present with neural tube defects.Citation154
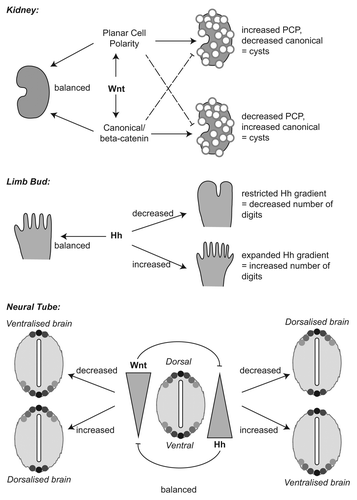
Figure 2. Hedgehog and Wnt Signaling in Kidney, Limb, and Neural Tube. A cartoon to show the effects of the Wnt signaling pathway on renal cystic disease, of the Hedgehog (Hh) pathway in the developing limb, and of both pathways in the neural tube. In the kidney, the balance between canonical (β-catenin dependent) and non-canonical Wnt signaling must be maintained; dysregulation of either branch, whether up- or downregulation, leads to development of cysts. In the limb bud, a gradient of Hh signaling is required to pattern the digits; where upregulation of Hh occurs, the gradient is expanded and additional digits are specified whereas when Hh is downregulated, the gradient becomes restricted and a reduced number of digits is specified. In the neural tube, a Wnt (and, to perhaps a greater extent, BMP) gradient is involved in dorsal cell fate determination; the gradient is counteracted by a ventral Hh gradient. Up- or downregulation of either pathway perturbs this balance and affects axis specification in the developing neural tube. Wnt signaling also has important effects on neuronal migration in the developing brain, leading to numerous other CNS effects.
Following neural tube closure, a population of multipotent neural crest cells de-adhere from the epithelium at the top of the neural tube and migrate until they encounter cues that regulate their differentiation into various cell types including neurons and glia of the peripheral nervous system, craniofacial cartilage and bone, smooth muscle, and pigment cells.Citation155 A subset of these cells form a primary cilium which, unusually, is formed on the basolateral rather than the apical cell surface, and the number of ciliated cells increases in frequency as neurogenesis proceeds, suggesting that the two may be linked.Citation156 The function of this cilium is unknown, but given its protrusion into the basement membrane underlying the neural crest cells it is tempting to speculate that it might be involved in sensing the signals that regulate changes from adhesive to migratory behavior that are key to early brain development. Wnt and Hh signaling are essential for neural crest cell migrationCitation157,Citation158 as well as neural tube patterning, and when this migration fails in ciliopathy patients, characteristic craniofacial defects are observed,Citation158,Citation159 suggesting that cilium dysfunction leads to wide-spread defects in developmental signaling required for neural crest cell migration and patterning.
Neurons, glial cells and oligodendrocytes of the CNS arise from several populations of neural stem cellsCitation160 and newborn neurons migrate long distances from their birthplace to reach their final destinations. This requires them to integrate and respond to a complex series of guidance cues that orchestrate the intricate migration patterns that are needed to form the brain.Citation161,Citation162 Primary cilia are required for neuron formation in the developing brainCitation67,Citation163-Citation165 but the role they play in neuronal migration is less clear, with one study showing that cilia develop only on post-migratory cortical neuronsCitation166 while other studies show that cilia are critical for migration and positioning of developing neurons.Citation167,Citation168 Thus, while it seems clear that primary cilia are needed at early stages of brain development for neural tube closure and neural crest cell migration, further work is required to clarify their role in neuronal migration. Nevertheless, several studies have shown that Hh signaling through primary cilia is needed for brain patterning, with mutations leading to loss or reduction of IFT components resulting in a variety of CNS defects.Citation165,Citation169,Citation170 Cilia-mediated Shh signaling is key to cerebellum development and regulates proliferation of neonatal granule cells, with loss of IFT function or Shh removal leading to cerebellar hypoplasia.Citation171,Citation172 The role of Wnt signaling is more difficult to resolve, as there is extensive crosstalk between the three branches of Wnt signaling during neuronal migration.Citation173 Disentangling the role of cilia in this process will require further investigation.
Finally, the role of primary cilia in the CNS is not restricted to embryogenesis and there is a growing body of evidence that indicates that they are also important in the postnatal and adult brain, where they mediate synapse formation in adult-born neuronsCitation174 and are involved in appetite regulation and obesity.Citation81,Citation175,Citation176 Moreover, defects in brain primary cilia have been linked to the development of neurodegenerative diseaseCitation177 and cancer.Citation178,Citation179 The picture is complicated and there is much to learn, but it is clear that, in common with primary cilia in the kidney and skeleton, primary cilia of the CNS may be regarded as antennae to receive signals from the extracellular environment and transmit them to the cell. Failure to build a cilium or to regulate cilium-dependent signaling can result in a multitude of developmental and adult brain defects.
Perspectives
It seems clear that the majority of symptoms found in ciliopathies are less to do with the loss of a cilium and more to do with defects in ciliary signaling—either developmental signaling or environment sensing. The linkage of many ciliopathy proteins together at the ciliary transition zone, and their role in ciliary compartmentalisation, is exciting. However, these are early days and the picture is likely to be much more complicated as the TZ is not the only mechanism contributing to ciliary signaling. Further, developmental signaling pathways such as Wnt and Hh are tremendously complicated, and there is extensive cross-talk, so it is important to consider the total signaling landscape rather than focusing on a single transduction cascade when assessing phenotypes. Moreover, it has only just become clear that the ciliary antenna can transmit signals as well as receive themCitation180; what it transmits and the contribution to ciliary function and organ development remains to be seen. However, it is important not to simply focus on developmental signaling pathways. These are largely restricted to metazoan organisms yet ciliopathy proteins play roles across eukaryotic evolution, implying more fundamental roles in ciliary biology. It will be essential to establish the function of ciliopathy proteins in a range of model organisms, and to integrate this information to provide a full picture.
Perhaps one of the key questions still remaining in the field is one of the hardest to answer—given the complexity of the interactions between the various ciliopathy genes and other ciliary and non-ciliary proteins, what are the relative contributions of perturbing a single protein vs. perturbation of an entire ciliary network? The intricacy of this ciliary network makes selection of appropriate controls extremely difficult—yet these are critical for distinguishing gene-specific roles from the role of the organelle itself. Furthermore, there is mounting evidence that “ciliary” proteins play extra-ciliary roles, and therefore dissecting out cilium-dependent vs. cilium-independent functions of these genes is essential for our understanding of ciliopathy. The picture may be far from complete, but many of the pieces of the jigsaw puzzle are now in place and the next few years will see some exciting advances in ciliopathy research that will advance our understanding of these devastating diseases.
| Abbreviations: | ||
| JBTS | = | Joubert syndrome |
| NPHP | = | Nephronophthisis |
| MKS | = | Meckel-Gruber syndrome |
| ARPKD | = | autosomal recessive polycystic kidney disease |
| ADPKD | = | autosomal dominant polycystic kidney disease |
| CNS | = | central nervous system |
| Hh | = | Hedgehog |
| Shh | = | Sonic Hedgehog |
| PCP | = | planar cell polarity |
| OCD | = | oriented cell division |
| IFT | = | Intraflagellar transport |
| TZ | = | transition zone |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Marie Harrisingh (University of Edinburgh) for helpful comments on the manuscript. Work in our laboratory is funded by the Medical Research Council.
Related Research Data
References
- Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol 2011; 12:222 - 34; http://dx.doi.org/10.1038/nrm3085; PMID: 21427764
- Garcia-Gonzalo FR, Reiter JF. Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol 2012; 197:697 - 709; http://dx.doi.org/10.1083/jcb.201111146; PMID: 22689651
- Davenport JR, Yoder BK. An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol 2005; 289:F1159 - 69; http://dx.doi.org/10.1152/ajprenal.00118.2005; PMID: 16275743
- Dawe HR, Farr H, Gull K. Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J Cell Sci 2007; 120:7 - 15; http://dx.doi.org/10.1242/jcs.03305; PMID: 17182899
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol 2002; 3:813 - 25; http://dx.doi.org/10.1038/nrm952; PMID: 12415299
- Bloodgood RA. Sensory reception is an attribute of both primary cilia and motile cilia. J Cell Sci 2010; 123:505 - 9; http://dx.doi.org/10.1242/jcs.066308; PMID: 20144998
- Roy S. Cilia and Hedgehog: when and how was their marriage solemnized?. Differentiation 2012; 83:S43 - 8; http://dx.doi.org/10.1016/j.diff.2011.11.010; PMID: 22154138
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 2006; 7:125 - 48; http://dx.doi.org/10.1146/annurev.genom.7.080505.115610; PMID: 16722803
- Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol 2011; 26:1039 - 56; http://dx.doi.org/10.1007/s00467-010-1731-7; PMID: 21210154
- Logan CV, Abdel-Hamed Z, Johnson CA. Molecular genetics and pathogenic mechanisms for the severe ciliopathies: insights into neurodevelopment and pathogenesis of neural tube defects. Mol Neurobiol 2011; 43:12 - 26; http://dx.doi.org/10.1007/s12035-010-8154-0; PMID: 21110233
- Adams M, Smith UM, Logan CV, Johnson CA. Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J Med Genet 2008; 45:257 - 67; http://dx.doi.org/10.1136/jmg.2007.054999; PMID: 18178628
- Coppieters F, Lefever S, Leroy BP, De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat 2010; 31:1097 - 108; http://dx.doi.org/10.1002/humu.21337; PMID: 20690115
- Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet 2009; 41:739 - 45; http://dx.doi.org/10.1038/ng.366; PMID: 19430481
- Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, et al, NISC Comparative Sequencing Program. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet 2011; 43:189 - 96; http://dx.doi.org/10.1038/ng.756; PMID: 21258341
- Alexiev BA, Lin X, Sun CC, Brenner DS. Meckel-Gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis. Arch Pathol Lab Med 2006; 130:1236 - 8; PMID: 16879033
- Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet 2007; 81:170 - 9; http://dx.doi.org/10.1086/519494; PMID: 17564974
- Bergmann C, Fliegauf M, Brüchle NO, Frank V, Olbrich H, Kirschner J, Schermer B, Schmedding I, Kispert A, Kränzlin B, et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet 2008; 82:959 - 70; http://dx.doi.org/10.1016/j.ajhg.2008.02.017; PMID: 18371931
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 2007; 39:875 - 81; http://dx.doi.org/10.1038/ng2039; PMID: 17558409
- Dowdle WE, Robinson JF, Kneist A, Sirerol-Piquer MS, Frints SG, Corbit KC, Zaghloul NA, van Lijnschoten G, Mulders L, Verver DE, et al. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am J Hum Genet 2011; 89:94 - 110; http://dx.doi.org/10.1016/j.ajhg.2011.06.003; PMID: 21763481
- Hopp K, Heyer CM, Hommerding CJ, Henke SA, Sundsbak JL, Patel S, Patel P, Consugar MB, Czarnecki PG, Gliem TJ, et al. B9D1 is revealed as a novel Meckel syndrome (MKS) gene by targeted exon-enriched next-generation sequencing and deletion analysis. Hum Mol Genet 2011; 20:2524 - 34; http://dx.doi.org/10.1093/hmg/ddr151; PMID: 21493627
- Kyttälä M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestilä M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet 2006; 38:155 - 7; http://dx.doi.org/10.1038/ng1714; PMID: 16415886
- Shaheen R, Ansari S, Mardawi EA, Alshammari MJ, Alkuraya FS. Mutations in TMEM231 cause Meckel-Gruber syndrome. J Med Genet 2013; 50:160 - 2; http://dx.doi.org/10.1136/jmedgenet-2012-101431; PMID: 23349226
- Shaheen R, Faqeih E, Seidahmed MZ, Sunker A, Alali FE, AlQahtani K, Alkuraya FSA. A TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum Mutat 2011; 32:573 - 8; http://dx.doi.org/10.1002/humu.21507; PMID: 21462283
- Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet 2006; 38:191 - 6; http://dx.doi.org/10.1038/ng1713; PMID: 16415887
- Tallila J, Jakkula E, Peltonen L, Salonen R, Kestilä M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet 2008; 82:1361 - 7; http://dx.doi.org/10.1016/j.ajhg.2008.05.004; PMID: 18513680
- Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, Iannicelli M, Travaglini L, Romani S, Illi B, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet 2010; 42:619 - 25; http://dx.doi.org/10.1038/ng.594; PMID: 20512146
- Shaheen R, Faqeih E, Alshammari MJ, Swaid A, Al-Gazali L, Mardawi E, Ansari S, Sogaty S, Seidahmed MZ, AlMotairi MI, et al. Genomic analysis of Meckel-Gruber syndrome in Arabs reveals marked genetic heterogeneity and novel candidate genes. Eur J Hum Genet 2013; 21:762 - 8; http://dx.doi.org/10.1038/ejhg.2012.254; PMID: 23169490
- Szymanska K, Berry I, Logan CV, Cousins SR, Lindsay H, Jafri H, Raashid Y, Malik-Sharif S, Castle B, Ahmed M, et al. Founder mutations and genotype-phenotype correlations in Meckel-Gruber syndrome and associated ciliopathies. Cilia 2012; 1:18; http://dx.doi.org/10.1186/2046-2530-1-18; PMID: 23351400
- Williams CL, Masyukova SV, Yoder BK. Normal ciliogenesis requires synergy between the cystic kidney disease genes MKS-3 and NPHP-4. J Am Soc Nephrol 2010; 21:782 - 93; http://dx.doi.org/10.1681/ASN.2009060597; PMID: 20150540
- Town T, Breunig JJ, Sarkisian MR, Spilianakis C, Ayoub AE, Liu X, Ferrandino AF, Gallagher AR, Li MO, Rakic P, et al. The stumpy gene is required for mammalian ciliogenesis. Proc Natl Acad Sci U S A 2008; 105:2853 - 8; http://dx.doi.org/10.1073/pnas.0712385105; PMID: 18287022
- Bialas NJ, Inglis PN, Li C, Robinson JF, Parker JD, Healey MP, Davis EE, Inglis CD, Toivonen T, Cottell DC, et al. Functional interactions between the ciliopathy-associated Meckel syndrome 1 (MKS1) protein and two novel MKS1-related (MKSR) proteins. J Cell Sci 2009; 122:611 - 24; http://dx.doi.org/10.1242/jcs.028621; PMID: 19208769
- Keller LC, Geimer S, Romijn E, Yates J 3rd, Zamora I, Marshall WF. Molecular architecture of the centriole proteome: the conserved WD40 domain protein POC1 is required for centriole duplication and length control. Mol Biol Cell 2009; 20:1150 - 66; http://dx.doi.org/10.1091/mbc.E08-06-0619; PMID: 19109428
- Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, Parapuram SK, Cheng H, Scott A, Hurd RE, et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet 2006; 15:1847 - 57; http://dx.doi.org/10.1093/hmg/ddl107; PMID: 16632484
- Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet 2006; 38:674 - 81; http://dx.doi.org/10.1038/ng1786; PMID: 16682973
- Dawe HR, Adams M, Wheway G, Szymanska K, Logan CV, Noegel AA, Gull K, Johnson CA. Nesprin-2 interacts with meckelin and mediates ciliogenesis via remodelling of the actin cytoskeleton. J Cell Sci 2009; 122:2716 - 26; http://dx.doi.org/10.1242/jcs.043794; PMID: 19596800
- Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N, Attie-Bitach T, et al. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet 2007; 16:173 - 86; http://dx.doi.org/10.1093/hmg/ddl459; PMID: 17185389
- Weatherbee SD, Niswander LA, Anderson KV. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet 2009; 18:4565 - 75; http://dx.doi.org/10.1093/hmg/ddp422; PMID: 19776033
- Wheway G, Abdelhamed Z, Natarajan S, Toomes C, Inglehearn C, Johnson CA. Aberrant Wnt signalling and cellular over-proliferation in a novel mouse model of Meckel-Gruber syndrome. Dev Biol 2013; 377:55 - 66; http://dx.doi.org/10.1016/j.ydbio.2013.02.015; PMID: 23454480
- Cook SA, Collin GB, Bronson RT, Naggert JK, Liu DP, Akeson EC, Davisson MT. A mouse model for Meckel syndrome type 3. J Am Soc Nephrol 2009; 20:753 - 64; http://dx.doi.org/10.1681/ASN.2008040412; PMID: 19211713
- Tammachote R, Hommerding CJ, Sinders RM, Miller CA, Czarnecki PG, Leightner AC, Salisbury JL, Ward CJ, Torres VE, Gattone VH 2nd, et al. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum Mol Genet 2009; 18:3311 - 23; http://dx.doi.org/10.1093/hmg/ddp272; PMID: 19515853
- Pluznick JL, Rodriguez-Gil DJ, Hull M, Mistry K, Gattone V, Johnson CA, Weatherbee S, Greer CA, Caplan MJ. Renal cystic disease proteins play critical roles in the organization of the olfactory epithelium. PLoS One 2011; 6:e19694; http://dx.doi.org/10.1371/journal.pone.0019694; PMID: 21614130
- Adams M, Simms RJ, Abdelhamed Z, Dawe HR, Szymanska K, Logan CV, Wheway G, Pitt E, Gull K, Knowles MA, et al. A meckelin-filamin A interaction mediates ciliogenesis. Hum Mol Genet 2012; 21:1272 - 86; http://dx.doi.org/10.1093/hmg/ddr557; PMID: 22121117
- Kim J, Krishnaswami SR, Gleeson JG. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet 2008; 17:3796 - 805; http://dx.doi.org/10.1093/hmg/ddn277; PMID: 18772192
- Ponsard C, Skowron-Zwarg M, Seltzer V, Perret E, Gallinger J, Fisch C, Dupuis-Williams P, Caruso N, Middendorp S, Tournier F. Identification of ICIS-1, a new protein involved in cilia stability. Front Biosci 2007; 12:1661 - 9; http://dx.doi.org/10.2741/2178; PMID: 17127412
- Bachmann-Gagescu R, Phelps IG, Stearns G, Link BA, Brockerhoff SE, Moens CB, Doherty D. The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum Mol Genet 2011; 20:4041 - 55; http://dx.doi.org/10.1093/hmg/ddr332; PMID: 21816947
- Noor A, Windpassinger C, Patel M, Stachowiak B, Mikhailov A, Azam M, Irfan M, Siddiqui ZK, Naeem F, Paterson AD, et al. CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal-recessive mental retardation with retinitis pigmentosa. Am J Hum Genet 2008; 82:1011 - 8; http://dx.doi.org/10.1016/j.ajhg.2008.01.021; PMID: 18387594
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 2011; 43:776 - 84; http://dx.doi.org/10.1038/ng.891; PMID: 21725307
- Bloodgood RA. The future of ciliary and flagellar membrane research. Mol Biol Cell 2012; 23:2407 - 11; http://dx.doi.org/10.1091/mbc.E12-01-0073; PMID: 22745341
- Francis SS, Sfakianos J, Lo B, Mellman I. A hierarchy of signals regulates entry of membrane proteins into the ciliary membrane domain in epithelial cells. J Cell Biol 2011; 193:219 - 33; http://dx.doi.org/10.1083/jcb.201009001; PMID: 21444686
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol 2012; 14:61 - 72; http://dx.doi.org/10.1038/ncb2410; PMID: 22179047
- Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011; 145:513 - 28; http://dx.doi.org/10.1016/j.cell.2011.04.019; PMID: 21565611
- Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol 2011; 192:1023 - 41; http://dx.doi.org/10.1083/jcb.201012116; PMID: 21422230
- Shiba D, Manning DK, Koga H, Beier DR, Yokoyama T. Inv acts as a molecular anchor for Nphp3 and Nek8 in the proximal segment of primary cilia. Cytoskeleton (Hoboken) 2010; 67:112 - 9; PMID: 20169535
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 2010; 329:436 - 9; http://dx.doi.org/10.1126/science.1191054; PMID: 20558667
- Dishinger JF, Kee HL, Jenkins PM, Fan S, Hurd TW, Hammond JW, Truong YN-T, Margolis B, Martens JR, Verhey KJ. Ciliary entry of the kinesin-2 motor KIF17 is regulated by importin-beta2 and RanGTP. Nat Cell Biol 2010; 12:703 - 10; http://dx.doi.org/10.1038/ncb2073; PMID: 20526328
- Hurd TW, Fan S, Margolis BL. Localization of retinitis pigmentosa 2 to cilia is regulated by Importin β2. J Cell Sci 2011; 124:718 - 26; http://dx.doi.org/10.1242/jcs.070839; PMID: 21285245
- Kee HL, Dishinger JF, Blasius TL, Liu CJ, Margolis B, Verhey KJ. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat Cell Biol 2012; 14:431 - 7; http://dx.doi.org/10.1038/ncb2450; PMID: 22388888
- Finetti F, Paccani SR, Riparbelli MG, Giacomello E, Perinetti G, Pazour GJ, Rosenbaum JL, Baldari CT. Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat Cell Biol 2009; 11:1332 - 9; http://dx.doi.org/10.1038/ncb1977; PMID: 19855387
- Norris DP, Grimes DT. Mouse models of ciliopathies: the state of the art. Dis Model Mech 2012; 5:299 - 312; http://dx.doi.org/10.1242/dmm.009340; PMID: 22566558
- Abdelhamed ZA, Wheway G, Szymanska K, Natarajan S, Toomes C, Inglehearn C, Johnson CA. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum Mol Genet 2013; 22:1358 - 72; http://dx.doi.org/10.1093/hmg/dds546; PMID: 23283079
- Collin GB, Won J, Hicks WL, Cook SA, Nishina PM, Naggert JK. Meckelin is necessary for photoreceptor intraciliary transport and outer segment morphogenesis. Invest Ophthalmol Vis Sci 2012; 53:967 - 74; http://dx.doi.org/10.1167/iovs.11-8766; PMID: 22247471
- Cui C, Chatterjee B, Francis D, Yu Q, SanAgustin JT, Francis R, Tansey T, Henry C, Wang B, Lemley B, et al. Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel-Gruber syndrome. Dis Model Mech 2011; 4:43 - 56; http://dx.doi.org/10.1242/dmm.006262; PMID: 21045211
- Lancaster MA, Schroth J, Gleeson JG. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol 2011; 13:700 - 7; http://dx.doi.org/10.1038/ncb2259; PMID: 21602792
- Leightner AC, Hommerding CJ, Peng Y, Salisbury JL, Gainullin VG, Czarnecki PG, Sussman CR, Harris PC. The Meckel syndrome protein meckelin (TMEM67) is a key regulator of cilia function but is not required for tissue planar polarity. Hum Mol Genet 2013; 22:2024 - 40; http://dx.doi.org/10.1093/hmg/ddt054; PMID: 23393159
- Mahuzier A, Gaudé HM, Grampa V, Anselme I, Silbermann F, Leroux-Berger M, Delacour D, Ezan J, Montcouquiol M, Saunier S, et al. Dishevelled stabilization by the ciliopathy protein Rpgrip1l is essential for planar cell polarity. J Cell Biol 2012; 198:927 - 40; http://dx.doi.org/10.1083/jcb.201111009; PMID: 22927466
- Vierkotten J, Dildrop R, Peters T, Wang B, Rüther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development 2007; 134:2569 - 77; http://dx.doi.org/10.1242/dev.003715; PMID: 17553904
- Breunig JJ, Sarkisian MR, Arellano JI, Morozov YM, Ayoub AE, Sojitra S, Wang B, Flavell RA, Rakic P, Town T. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci U S A 2008; 105:13127 - 32; http://dx.doi.org/10.1073/pnas.0804558105; PMID: 18728187
- Wallingford JB, Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev 2011; 25:201 - 13; http://dx.doi.org/10.1101/gad.2008011; PMID: 21289065
- Tanaka Y, Okada Y, Hirokawa N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 2005; 435:172 - 7; http://dx.doi.org/10.1038/nature03494; PMID: 15889083
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 2005; 15:1861 - 6; http://dx.doi.org/10.1016/j.cub.2005.09.012; PMID: 16243034
- Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E. A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 2011; 145:1129 - 41; http://dx.doi.org/10.1016/j.cell.2011.05.030; PMID: 21703454
- Ravine D, Gibson RN, Donlan J, Sheffield LJ. An ultrasound renal cyst prevalence survey: specificity data for inherited renal cystic diseases. Am J Kidney Dis 1993; 22:803 - 7; PMID: 8250026
- Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med 2009; 60:321 - 37; http://dx.doi.org/10.1146/annurev.med.60.101707.125712; PMID: 18947299
- Wolf MT, Hildebrandt F. Nephronophthisis. Pediatr Nephrol 2011; 26:181 - 94; http://dx.doi.org/10.1007/s00467-010-1585-z; PMID: 20652329
- Watnick T, Germino G. From cilia to cyst. Nat Genet 2003; 34:355 - 6; http://dx.doi.org/10.1038/ng0803-355; PMID: 12923538
- Winyard P, Jenkins D. Putative roles of cilia in polycystic kidney disease. Biochim Biophys Acta 2011; 1812:1256 - 62; http://dx.doi.org/10.1016/j.bbadis.2011.04.012; PMID: 21586324
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AEH, Lu W, Brown EM, Quinn SJ, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129 - 37; http://dx.doi.org/10.1038/ng1076; PMID: 12514735
- Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, Zhou J. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol 2007; 27:3241 - 52; http://dx.doi.org/10.1128/MCB.00072-07; PMID: 17283055
- Köttgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol 2008; 182:437 - 47; http://dx.doi.org/10.1083/jcb.200805124; PMID: 18695040
- Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet 2008; 17:1578 - 90; http://dx.doi.org/10.1093/hmg/ddn045; PMID: 18263895
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol 2007; 17:1586 - 94; http://dx.doi.org/10.1016/j.cub.2007.08.034; PMID: 17825558
- Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A 2003; 100:5286 - 91; http://dx.doi.org/10.1073/pnas.0836980100; PMID: 12672950
- Zullo A, Iaconis D, Barra A, Cantone A, Messaddeq N, Capasso G, Dollé P, Igarashi P, Franco B. Kidney-specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum Mol Genet 2010; 19:2792 - 803; http://dx.doi.org/10.1093/hmg/ddq180; PMID: 20444807
- Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int 2009; 76:149 - 68; http://dx.doi.org/10.1038/ki.2009.128; PMID: 19455193
- Chapin HC, Caplan MJ. The cell biology of polycystic kidney disease. J Cell Biol 2010; 191:701 - 10; http://dx.doi.org/10.1083/jcb.201006173; PMID: 21079243
- Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 2006; 103:5466 - 71; http://dx.doi.org/10.1073/pnas.0509694103; PMID: 16567633
- Dere R, Wilson PD, Sandford RN, Walker CL. Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS One 2010; 5:e9239; http://dx.doi.org/10.1371/journal.pone.0009239; PMID: 20169078
- Distefano G, Boca M, Rowe I, Wodarczyk C, Ma L, Piontek KB, Germino GG, Pandolfi PP, Boletta A. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol 2009; 29:2359 - 71; http://dx.doi.org/10.1128/MCB.01259-08; PMID: 19255143
- Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Gödel M, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol 2010; 12:1115 - 22; http://dx.doi.org/10.1038/ncb2117; PMID: 20972424
- Bonnet CS, Aldred M, von Ruhland C, Harris R, Sandford R, Cheadle JP. Defects in cell polarity underlie TSC and ADPKD-associated cystogenesis. Hum Mol Genet 2009; 18:2166 - 76; http://dx.doi.org/10.1093/hmg/ddp149; PMID: 19321600
- Torres VE, Boletta A, Chapman A, Gattone V, Pei Y, Qian Q, Wallace DP, Weimbs T, Wüthrich RP. Prospects for mTOR inhibitor use in patients with polycystic kidney disease and hamartomatous diseases. Clin J Am Soc Nephrol 2010; 5:1312 - 29; http://dx.doi.org/10.2215/CJN.01360210; PMID: 20498248
- Weimbs T. Polycystic kidney disease and renal injury repair: common pathways, fluid flow, and the function of polycystin-1. Am J Physiol Renal Physiol 2007; 293:F1423 - 32; http://dx.doi.org/10.1152/ajprenal.00275.2007; PMID: 17715262
- Weimbs T. Regulation of mTOR by polycystin-1: is polycystic kidney disease a case of futile repair?. Cell Cycle 2006; 5:2425 - 9; http://dx.doi.org/10.4161/cc.5.21.3408; PMID: 17102641
- Walz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Hörl WH, Obermüller N, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:830 - 40; http://dx.doi.org/10.1056/NEJMoa1003491; PMID: 20581392
- Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 2010; 363:820 - 9; http://dx.doi.org/10.1056/NEJMoa0907419; PMID: 20581391
- Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 2006; 444:949 - 52; http://dx.doi.org/10.1038/nature05348; PMID: 17122773
- Bukanov NO, Moreno SE, Natoli TA, Rogers KA, Smith LA, Ledbetter SR, Oumata N, Galons H, Meijer L, Ibraghimov-Beskrovnaya O. CDK inhibitors R-roscovitine and S-CR8 effectively block renal and hepatic cystogenesis in an orthologous model of ADPKD. Cell Cycle 2012; 11:4040 - 6; http://dx.doi.org/10.4161/cc.22375; PMID: 23032260
- Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int 2009; 76:149 - 68; http://dx.doi.org/10.1038/ki.2009.128; PMID: 19455193
- Kikuchi A, Yamamoto H, Sato A. Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol 2009; 19:119 - 29; http://dx.doi.org/10.1016/j.tcb.2009.01.003; PMID: 19208479
- Rao TP, Kühl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res 2010; 106:1798 - 806; http://dx.doi.org/10.1161/CIRCRESAHA.110.219840; PMID: 20576942
- van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development 2009; 136:3205 - 14; http://dx.doi.org/10.1242/dev.033910; PMID: 19736321
- Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, Jagger DJ, Leitch CC, Chapple JP, Munro PM, Fisher S, et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet 2005; 37:1135 - 40; http://dx.doi.org/10.1038/ng1644; PMID: 16170314
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Krönig C, Schermer B, Benzing T, Cabello OA, Jenny A, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 2005; 37:537 - 43; http://dx.doi.org/10.1038/ng1552; PMID: 15852005
- Lancaster MA, Louie CM, Silhavy JL, Sintasath L, Decambre M, Nigam SK, Willert K, Gleeson JG. Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat Med 2009; 15:1046 - 54; http://dx.doi.org/10.1038/nm.2010; PMID: 19718039
- Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 2013; 13:11 - 26; http://dx.doi.org/10.1038/nrc3419; PMID: 23258168
- Saadi-Kheddouci S, Berrebi D, Romagnolo B, Cluzeaud F, Peuchmaur M, Kahn A, Vandewalle A, Perret C. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene 2001; 20:5972 - 81; http://dx.doi.org/10.1038/sj.onc.1204825; PMID: 11593404
- Qian CN, Knol J, Igarashi P, Lin F, Zylstra U, Teh BT, Williams BO. Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium. J Biol Chem 2005; 280:3938 - 45; http://dx.doi.org/10.1074/jbc.M410697200; PMID: 15550389
- Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000; 407:535 - 8; http://dx.doi.org/10.1038/35035124; PMID: 11029008
- Kim E, Arnould T, Sellin LK, Benzing T, Fan MJ, Grüning W, Sokol SY, Drummond I, Walz G. The polycystic kidney disease 1 gene product modulates Wnt signaling. J Biol Chem 1999; 274:4947 - 53; http://dx.doi.org/10.1074/jbc.274.8.4947; PMID: 9988738
- Lal M, Song X, Pluznick JL, Di Giovanni V, Merrick DM, Rosenblum ND, Chauvet V, Gottardi CJ, Pei Y, Caplan MJ. Polycystin-1 C-terminal tail associates with β-catenin and inhibits canonical Wnt signaling. Hum Mol Genet 2008; 17:3105 - 17; http://dx.doi.org/10.1093/hmg/ddn208; PMID: 18632682
- Kestler HA, Kühl M. From individual Wnt pathways towards a Wnt signalling network. Philos Trans R Soc Lond B Biol Sci 2008; 363:1333 - 47; http://dx.doi.org/10.1098/rstb.2007.2251; PMID: 18192173
- De A. Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin (Shanghai) 2011; 43:745 - 56; http://dx.doi.org/10.1093/abbs/gmr079; PMID: 21903638
- Burn SF, Webb A, Berry RL, Davies JA, Ferrer-Vaquer A, Hadjantonakis AK, Hastie ND, Hohenstein P. Calcium/NFAT signalling promotes early nephrogenesis. Dev Biol 2011; 352:288 - 98; http://dx.doi.org/10.1016/j.ydbio.2011.01.033; PMID: 21295565
- Cao Y, Park A, Sun Z. Intraflagellar transport proteins are essential for cilia formation and for planar cell polarity. J Am Soc Nephrol 2010; 21:1326 - 33; http://dx.doi.org/10.1681/ASN.2009091001; PMID: 20576807
- Fischer E, Legue E, Doyen A, Nato F, Nicolas J-F, Torres V, Yaniv M, Pontoglio M. Defective planar cell polarity in polycystic kidney disease. Nat Genet 2006; 38:21 - 3; http://dx.doi.org/10.1038/ng1701; PMID: 16341222
- Luyten A, Su X, Gondela S, Chen Y, Rompani S, Takakura A, Zhou J. Aberrant regulation of planar cell polarity in polycystic kidney disease. J Am Soc Nephrol 2010; 21:1521 - 32; http://dx.doi.org/10.1681/ASN.2010010127; PMID: 20705705
- Nishio S, Tian X, Gallagher AR, Yu Z, Patel V, Igarashi P, Somlo S. Loss of oriented cell division does not initiate cyst formation. J Am Soc Nephrol 2010; 21:295 - 302; http://dx.doi.org/10.1681/ASN.2009060603; PMID: 19959710
- Sugiyama N, Tsukiyama T, Yamaguchi TP, Yokoyama T. The canonical Wnt signaling pathway is not involved in renal cyst development in the kidneys of inv mutant mice. Kidney Int 2011; 79:957 - 65; http://dx.doi.org/10.1038/ki.2010.534; PMID: 21248711
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003; 426:83 - 7; http://dx.doi.org/10.1038/nature02061; PMID: 14603322
- Ehlen HW, Buelens LA, Vortkamp A. Hedgehog signaling in skeletal development. Birth Defects Res C Embryo Today 2006; 78:267 - 79; http://dx.doi.org/10.1002/bdrc.20076; PMID: 17061262
- Bürglin TR, Kuwabara PE. Homologs of the Hh signalling network in C. elegans. WormBook 2006; 28:1 - 14; PMID: 18050469
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007; 317:372 - 6; http://dx.doi.org/10.1126/science.1139740; PMID: 17641202
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol 2005; 287:378 - 89; http://dx.doi.org/10.1016/j.ydbio.2005.08.050; PMID: 16229832
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A 2005; 102:11325 - 30; http://dx.doi.org/10.1073/pnas.0505328102; PMID: 16061793
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 2005; 132:3103 - 11; http://dx.doi.org/10.1242/dev.01894; PMID: 15930098
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 2005; 1:e53; http://dx.doi.org/10.1371/journal.pgen.0010053; PMID: 16254602
- Endoh-Yamagami S, Evangelista M, Wilson D, Wen X, Theunissen J-W, Phamluong K, Davis M, Scales SJ, Solloway MJ, de Sauvage FJ, et al. The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr Biol 2009; 19:1320 - 6; http://dx.doi.org/10.1016/j.cub.2009.06.046; PMID: 19592253
- Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol 2010; 191:415 - 28; http://dx.doi.org/10.1083/jcb.201004108; PMID: 20956384
- Kim J, Kato M, Beachy PA. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci U S A 2009; 106:21666 - 71; http://dx.doi.org/10.1073/pnas.0912180106; PMID: 19996169
- Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 2010; 24:670 - 82; http://dx.doi.org/10.1101/gad.1902910; PMID: 20360384
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 2010; 11:331 - 44; http://dx.doi.org/10.1038/nrg2774; PMID: 20395968
- Ding Q, Fukami Si, Meng X, Nishizaki Y, Zhang X, Sasaki H, Dlugosz A, Nakafuku M, Hui Cc. Mouse suppressor of fused is a negative regulator of sonic hedgehog signaling and alters the subcellular distribution of Gli1. Curr Biol 1999; 9:1119 - 22; http://dx.doi.org/10.1016/S0960-9822(99)80482-5; PMID: 10531011
- Zeng H, Jia J, Liu A. Coordinated translocation of mammalian Gli proteins and suppressor of fused to the primary cilium. PLoS One 2010; 5:e15900; http://dx.doi.org/10.1371/journal.pone.0015900; PMID: 21209912
- Chen M-H, Wilson CW, Li Y-J, Law KKL, Lu C-S, Gacayan R, Zhang X, Hui CC, Chuang P-T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev 2009; 23:1910 - 28; http://dx.doi.org/10.1101/gad.1794109; PMID: 19684112
- Jia J, Kolterud A, Zeng H, Hoover A, Teglund S, Toftgård R, Liu A. Suppressor of Fused inhibits mammalian Hedgehog signaling in the absence of cilia. Dev Biol 2009; 330:452 - 60; http://dx.doi.org/10.1016/j.ydbio.2009.04.009; PMID: 19371734
- Brennan D, Chen X, Cheng L, Mahoney M, Riobo NA. Noncanonical Hedgehog signaling. Vitam Horm 2012; 88:55 - 72; http://dx.doi.org/10.1016/B978-0-12-394622-5.00003-1; PMID: 22391299
- Louvi A, Grove EA. Cilia in the CNS: the quiet organelle claims center stage. Neuron 2011; 69:1046 - 60; http://dx.doi.org/10.1016/j.neuron.2011.03.002; PMID: 21435552
- Ruat M, Roudaut H, Ferent J, Traiffort E. Hedgehog trafficking, cilia and brain functions. Differentiation 2012; 83:S97 - 104; http://dx.doi.org/10.1016/j.diff.2011.11.011; PMID: 22169886
- Del Bigio MR. Ependymal cells: biology and pathology. Acta Neuropathol 2010; 119:55 - 73; http://dx.doi.org/10.1007/s00401-009-0624-y; PMID: 20024659
- Kishimoto N, Sawamoto K. Planar polarity of ependymal cilia. Differentiation 2012; 83:S86 - 90; http://dx.doi.org/10.1016/j.diff.2011.10.007; PMID: 22101065
- Banizs B, Pike MM, Millican CL, Ferguson WB, Komlosi P, Sheetz J, Bell PD, Schwiebert EM, Yoder BK. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 2005; 132:5329 - 39; http://dx.doi.org/10.1242/dev.02153; PMID: 16284123
- Narita K, Kawate T, Kakinuma N, Takeda S. Multiple primary cilia modulate the fluid transcytosis in choroid plexus epithelium. Traffic 2010; 11:287 - 301; http://dx.doi.org/10.1111/j.1600-0854.2009.01016.x; PMID: 19958467
- Breunig JJ, Haydar TF, Rakic P. Neural stem cells: historical perspective and future prospects. Neuron 2011; 70:614 - 25; http://dx.doi.org/10.1016/j.neuron.2011.05.005; PMID: 21609820
- Faigle R, Song H. Signaling mechanisms regulating adult neural stem cells and neurogenesis. Biochim Biophys Acta 2013; 1830:2435 - 48; http://dx.doi.org/10.1016/j.bbagen.2012.09.002; PMID: 22982587
- Keller R, Shook D, Skoglund P. The forces that shape embryos: physical aspects of convergent extension by cell intercalation. Phys Biol 2008; 5:015007; http://dx.doi.org/10.1088/1478-3975/5/1/015007; PMID: 18403829
- Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci 2008; 11:277 - 84; http://dx.doi.org/10.1038/nn2059; PMID: 18297065
- Murdoch JN, Copp AJ. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res A Clin Mol Teratol 2010; 88:633 - 52; http://dx.doi.org/10.1002/bdra.20686; PMID: 20544799
- Heydeck W, Zeng H, Liu A. Planar cell polarity effector gene Fuzzy regulates cilia formation and Hedgehog signal transduction in mouse. Dev Dyn 2009; 238:3035 - 42; http://dx.doi.org/10.1002/dvdy.22130; PMID: 19877275
- Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, Chuang PT, Reiter JF. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol 2008; 10:70 - 6; http://dx.doi.org/10.1038/ncb1670; PMID: 18084282
- Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet 2007; 39:1350 - 60; http://dx.doi.org/10.1038/ng.2007.12; PMID: 17906624
- Heydeck W, Liu A. PCP effector proteins inturned and fuzzy play nonredundant roles in the patterning but not convergent extension of mammalian neural tube. Dev Dyn 2011; 240:1938 - 48; http://dx.doi.org/10.1002/dvdy.22696; PMID: 21761479
- Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P. Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet 2008; 40:69 - 77; http://dx.doi.org/10.1038/ng.2007.54; PMID: 18066062
- Park TJ, Haigo SL, Wallingford JB. Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nat Genet 2006; 38:303 - 11; http://dx.doi.org/10.1038/ng1753; PMID: 16493421
- Vogel TW, Carter CS, Abode-Iyamah K, Zhang Q, Robinson S. The role of primary cilia in the pathophysiology of neural tube defects. Neurosurg Focus 2012; 33:E2; http://dx.doi.org/10.3171/2012.6.FOCUS12222; PMID: 23025443
- Huang X, Saint-Jeannet JP. Induction of the neural crest and the opportunities of life on the edge. Dev Biol 2004; 275:1 - 11; http://dx.doi.org/10.1016/j.ydbio.2004.07.033; PMID: 15464568
- Wilsch-Bräuninger M, Peters J, Paridaen JT, Huttner WB. Basolateral rather than apical primary cilia on neuroepithelial cells committed to delamination. Development 2012; 139:95 - 105; http://dx.doi.org/10.1242/dev.069294; PMID: 22096071
- De Calisto J, Araya C, Marchant L, Riaz CF, Mayor R. Essential role of non-canonical Wnt signalling in neural crest migration. Development 2005; 132:2587 - 97; http://dx.doi.org/10.1242/dev.01857; PMID: 15857909
- Tapadia MD, Cordero DR, Helms JA. It’s all in your head: new insights into craniofacial development and deformation. J Anat 2005; 207:461 - 77; http://dx.doi.org/10.1111/j.1469-7580.2005.00484.x; PMID: 16313388
- Tobin JL, Di Franco M, Eichers E, May-Simera H, Garcia M, Yan J, Quinlan R, Justice MJ, Hennekam RC, Briscoe J, et al. Inhibition of neural crest migration underlies craniofacial dysmorphology and Hirschsprung’s disease in Bardet-Biedl syndrome. Proc Natl Acad Sci U S A 2008; 105:6714 - 9; http://dx.doi.org/10.1073/pnas.0707057105; PMID: 18443298
- Franco SJ, Müller U. Shaping our minds: stem and progenitor cell diversity in the mammalian neocortex. Neuron 2013; 77:19 - 34; http://dx.doi.org/10.1016/j.neuron.2012.12.022; PMID: 23312513
- Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell 2007; 128:29 - 43; http://dx.doi.org/10.1016/j.cell.2006.12.021; PMID: 17218253
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci 2004; 27:392 - 9; http://dx.doi.org/10.1016/j.tins.2004.05.001; PMID: 15219738
- Amador-Arjona A, Elliott J, Miller A, Ginbey A, Pazour GJ, Enikolopov G, Roberts AJ, Terskikh AV. Primary cilia regulate proliferation of amplifying progenitors in adult hippocampus: implications for learning and memory. J Neurosci 2011; 31:9933 - 44; http://dx.doi.org/10.1523/JNEUROSCI.1062-11.2011; PMID: 21734285
- Besse L, Neti M, Anselme I, Gerhardt C, Rüther U, Laclef C, Schneider-Maunoury S. Primary cilia control telencephalic patterning and morphogenesis via Gli3 proteolytic processing. Development 2011; 138:2079 - 88; http://dx.doi.org/10.1242/dev.059808; PMID: 21490064
- Willaredt MA, Hasenpusch-Theil K, Gardner HA, Kitanovic I, Hirschfeld-Warneken VC, Gojak CP, Gorgas K, Bradford CL, Spatz J, Wölfl S, et al. A crucial role for primary cilia in cortical morphogenesis. J Neurosci 2008; 28:12887 - 900; http://dx.doi.org/10.1523/JNEUROSCI.2084-08.2008; PMID: 19036983
- Arellano JI, Guadiana SM, Breunig JJ, Rakic P, Sarkisian MR. Development and distribution of neuronal cilia in mouse neocortex. J Comp Neurol 2012; 520:848 - 73; http://dx.doi.org/10.1002/cne.22793; PMID: 22020803
- Baudoin JP, Viou L, Launay PS, Luccardini C, Espeso Gil S, Kiyasova V, Irinopoulou T, Alvarez C, Rio JP, Boudier T, et al. Tangentially migrating neurons assemble a primary cilium that promotes their reorientation to the cortical plate. Neuron 2012; 76:1108 - 22; http://dx.doi.org/10.1016/j.neuron.2012.10.027; PMID: 23259947
- Higginbotham H, Eom TY, Mariani LE, Bachleda A, Hirt J, Gukassyan V, Cusack CL, Lai C, Caspary T, Anton ES. Arl13b in primary cilia regulates the migration and placement of interneurons in the developing cerebral cortex. Dev Cell 2012; 23:925 - 38; http://dx.doi.org/10.1016/j.devcel.2012.09.019; PMID: 23153492
- Gorivodsky M, Mukhopadhyay M, Wilsch-Braeuninger M, Phillips M, Teufel A, Kim C, Malik N, Huttner W, Westphal H. Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev Biol 2009; 325:24 - 32; http://dx.doi.org/10.1016/j.ydbio.2008.09.019; PMID: 18930042
- Stottmann RW, Tran PV, Turbe-Doan A, Beier DR. Ttc21b is required to restrict sonic hedgehog activity in the developing mouse forebrain. Dev Biol 2009; 335:166 - 78; http://dx.doi.org/10.1016/j.ydbio.2009.08.023; PMID: 19732765
- Chizhikov VV, Davenport J, Zhang Q, Shih EK, Cabello OA, Fuchs JL, Yoder BK, Millen KJ. Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci 2007; 27:9780 - 9; http://dx.doi.org/10.1523/JNEUROSCI.5586-06.2007; PMID: 17804638
- Spassky N, Han YG, Aguilar A, Strehl L, Besse L, Laclef C, Ros MR, Garcia-Verdugo JM, Alvarez-Buylla A. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol 2008; 317:246 - 59; http://dx.doi.org/10.1016/j.ydbio.2008.02.026; PMID: 18353302
- Clark CE, Nourse CC, Cooper HM. The tangled web of non-canonical Wnt signalling in neural migration. Neurosignals 2012; 20:202 - 20; http://dx.doi.org/10.1159/000332153; PMID: 22456117
- Kumamoto N, Gu Y, Wang J, Janoschka S, Takemaru K, Levine J, Ge S. A role for primary cilia in glutamatergic synaptic integration of adult-born neurons. Nat Neurosci 2012; 15:399 - 405, S1; http://dx.doi.org/10.1038/nn.3042; PMID: 22306608
- Heydet D, Chen LX, Larter CZ, Inglis C, Silverman MA, Farrell GC, Leroux MR. A truncating mutation of Alms1 reduces the number of hypothalamic neuronal cilia in obese mice. Dev Neurobiol 2013; 73:1 - 13; http://dx.doi.org/10.1002/dneu.22031; PMID: 22581473
- Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet 2009; 18:1323 - 31; http://dx.doi.org/10.1093/hmg/ddp031; PMID: 19150989
- Keryer G, Pineda JR, Liot G, Kim J, Dietrich P, Benstaali C, Smith K, Cordelières FP, Spassky N, Ferrante RJ, et al. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J Clin Invest 2011; 121:4372 - 82; http://dx.doi.org/10.1172/JCI57552; PMID: 21985783
- Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 2009; 15:1062 - 5; http://dx.doi.org/10.1038/nm.2020; PMID: 19701203
- Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH Jr., Dlugosz AA, Reiter JF. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med 2009; 15:1055 - 61; http://dx.doi.org/10.1038/nm.2011; PMID: 19701205
- Wood CR, Huang K, Diener DR, Rosenbaum JL. The cilium secretes bioactive ectosomes. Curr Biol 2013; 23:906 - 11; http://dx.doi.org/10.1016/j.cub.2013.04.019; PMID: 23623554