
Abstract
The primary cilium is an antenna-like organelle that plays a vital role in organ generation and maintenance. It protrudes from the cell surface where it receives signals from the surrounding environment and relays them into the cell. These signals are then integrated to give the required outputs in terms of proliferation, differentiation, migration and polarization that ultimately lead to organ development and homeostasis. Defects in cilia function underlie a wide range of diverse but related human developmental or degenerative diseases. Collectively known as ciliopathies, these disorders present with varying severity and multiple organ involvement. The appreciation of the medical importance of the primary cilium has stimulated a huge effort into studies of the underlying cellular mechanisms. These in turn have revealed that ciliopathies result not only from defective assembly or organization of the primary cilium, but also from impaired ciliary signaling. This special edition of Organogenesis contains a set of review articles that highlight the role of the primary cilium in organ development and homeostasis, much of which has been learnt from studies of the associated human diseases. Here, we provide an introductory overview of our current understanding of the structure and function of the cilium, with a focus on the signaling pathways that are coordinated by primary cilia to ensure proper organ generation and maintenance.
Introduction
Although present as a single copy on most cells, primary cilia very much resemble the motile cilia that are found on respiratory epithelia or the sperm flagellum. However, whilst the functions of motile cilia are well appreciated in terms of generating propulsion or creating flow, the primary cilium was for many years considered a rather obscure structure of no great importance. The last decade or so has seen a complete reversal of this opinion and it is now known that primary cilia detect mechanical, light and chemical signals that contribute to the development and function of most, if not all, organs of the human bodyCitation1,Citation2 The major driver for this dramatic volte-face has been the realization that multiple, often severe, monogenic inherited diseases arise from defects in genes that encode ciliary proteins. Moreover, many of these diseases, collectively termed ciliopathies, are syndromic and exhibit multiple organ involvement. They can also display as either early-onset developmental dysplastic disorders or late-onset degenerative diseases.Citation3-Citation5 Below, we describe in brief what we currently know about the cell biology of primary cilia structure and function, and why their deregulation has such a major impact on organogenesis.
Cilia Structure and Organization
Cilia extend from and are continuous with the plasma membrane. At their core, they contain a microtubule-based axoneme composed of a ring of nine microtubule doublets.Citation6 These extend directly off a basal body that sits just beneath the cell surface (). The basal body is derived from the mature, or mother, centriole, which contributes to formation of the centrosome in a dividing cell.Citation7,Citation8 However, when proliferating cells enter quiescence, the mother centriole becomes associated with a Golgi-derived vesicle, migrates to the cell surface and attaches to the plasma membrane via appendages present at the distal end of the centriole.Citation9 At this point it becomes referred to as a basal body. Basal bodies, like centrioles, are composed of nine triplets of microtubules. These directly template the ring of nine axonemal microtubule doublets that elongate to form the cilium pushing out the plasma membrane as they grow. Why and how microtubule triplets convert to doublets between the basal body and the ciliary axoneme isn’t clear. However, at this junction, a specialized structure called the transition zone is assembled that includes Y-shaped fibers, which radiate out from the microtubules to the plasma membrane. This transition zone creates a barrier, known as the ciliary gate, that somehow regulates passage of both soluble and membrane-bound proteins in and out of the cilium.Citation10-Citation13
Figure 1. A schematic overview of the primary cilium illustrating the signaling pathways that are controlled through this organelle, and the clinical phenotypes, organ involvement and syndromic ciliopathies associated with defects in primary cilium signaling. MKS, Meckel-Gruber Syndrome; JBTS, Joubert Syndrome: BBS, Bardet Biedl Syndrome; NPHP, nephronophthisis; OFD, Oral-Facial-Digital Syndrome; SLS, Senior-Løken Syndrome; AS, Alstrom Syndrome; SRPS, Short Rib-Polydactyly Syndrome; EVS, Ellis-van Creveld Syndrome.
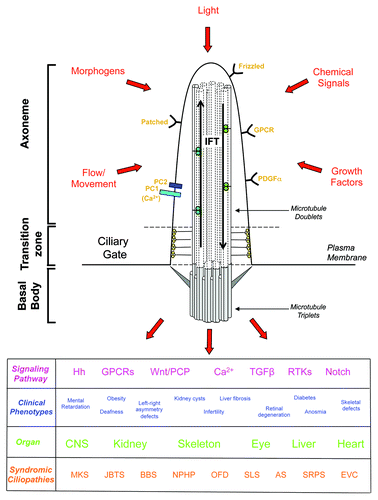
Due to the structural equivalence of the centriole and the basal body, many proteins implicated in centrosome organization in dividing cells also contribute to ciliogenesis in quiescent cells. Similarly, some proteins that regulate centriole biogenesis also contribute to axonemal microtubule extension. Hence, there has been an exciting convergence of the centrosome and cilia research fields in recent years, which has greatly contributed to our overall molecular understanding of these elegant organelles.Citation7,Citation8 Furthermore, if quiescent cells re-enter the cell cycle, then resorption of the primary cilium occurs and the basal body is converted back into a centriole that contributes to centrosome organization. As centrosomes generate the poles of the mitotic spindle, there is potential then for basal bodies, and possibly even cilia, to influence the orientation of cell division. This is important as oriented cell division (OCD) plays a key role in tissue patterning and organ development, perhaps best exemplified in the elongation of collecting duct tubules in the kidney.Citation14 Defects in OCD are one potential cause of kidney cyst development, where tubules expand laterally rather than longitudinally. However, the mechanistic connections between primary cilia and OCD remain frustratingly elusive.
Cilia lack the machinery required for protein synthesis. Both soluble and membrane-bound ciliary proteins therefore need to be trafficked to the basal body and taken up into the cilium. How this happens in a selective manner such that a highly specialized ciliary proteome is generated is the subject of the review by Malicki and Avidor-Reiss in this issue.Citation15 Furthermore, the delivery of these proteins to the appropriate site within the cilium, as well as the generation and maintenance of the microtubule axoneme, requires a specialized trafficking system, known as intraflagellar transport or IFT.Citation11,Citation16-Citation18 Complexes of IFT proteins carry cargoes from the base to the tip of the cilium in an anterograde direction that is dependent on plus-end directed kinesin-2 motors, and from the tip to the base in a retrograde direction via minus-end directed cytoplasmic dynein-2 motors. Proteins whose primary role seems to be in IFT fall into two groups that make up distinct complexes, the so-called IFT ‘A’- and ‘B’-particles. IFT-A components connect cargoes to dynein for retrograde transport, while IFT-B components connect cargoes to kinesin for anterograde transport. However, these two sets of trafficking machinery associate with each other so that once they have carried cargo in one direction, they are themselves returned to the opposite end.
Motile cilia differ from primary cilia in that motile cilia have an additional central pair of microtubules within their axoneme.Citation6-Citation8,Citation11,Citation19 This ‘9+2’ arrangement (as opposed to the ‘9+0’ of primary cilia), together with the presence of specialized ciliary motors and accessory proteins, provide motile cilia with the capacity to actively bend and thereby undergo coordinated beating patterns that create flow over the cell surface. Motile cilia are often longer than primary cilia and present in multiple copies, such as those found on the epithelial surface of the respiratory tract or oviduct, or the ventricular ependyma in the brain. As stated above, the primary purpose here is to create a luminal flow within the tissue for the purpose of, say, removing surface debris or setting up a chemical gradient. Indeed, there is a specialized type of motile cilium found in a particular site within developing vertebrate embryos, the embryonic node, whose job is to set up the morphogenetic gradients required to determine left-right body asymmetry. Nodal cilia, like primary cilia, have a ‘9+0’ microtubule axoneme. However, although they lack the central pair, they do have ciliary motors associated with the microtubule doublets that allow them to generate the rotary motion that is required for their function in body patterning.
Ciliary Signaling, Organ Development, and Human Disease
It has been long understood that defects in motile cilia have pathological consequences. Loss of motility of the respiratory multiciliated epithelia, for example, gives rise to primary ciliary dyskinesis (PCD) or immotile ciliary syndromes, such as Kartegener’s Syndrome, that lead to persistent respiratory infections.Citation19 The presence of motile cilia in the oviduct and brain, as well as the central role that the flagellum plays in sperm motility, also provide an explanation for why ciliary defects cause infertility and certain neurological diseases, such as hydrocephalus. Moreover, it is possible to understand from a mechanical perspective why loss of nodal cilia function leads to laterality defects in terms of body patterning, such as heterotaxy, situs ambiguous, and situs inversus. These left-right asymmetry defects can have a particularly important impact on heart development, as discussed in the accompanying article by Christensen and colleagues.Citation20 However, as these authors point out, some congenital heart defects may arise during later development as a result of defective signaling from primary cilia in cardiac tissues. Indeed, what is now becoming clear is the diversity and pleiotropy of organ involvement that is seen in ciliopathies whose major pathological consequences seem to arise from loss of function of primary cilia.
Primary cilia are typically 5–10 μm in length and extend into the surrounding extracellular environment. They are therefore ideally positioned to detect changes in chemical factors, morphogens or growth factors present in this medium. For example, the concentration of G-protein coupled receptors (GPCRs) on olfactory cilia allow detection of odorants that provide the basis for smell.Citation21 Primary cilia can also sense fluid movement across the cell surface. Thus, urine flow within kidney tubules causes bending of primary cilia that leads to Ca2+ influx through the polycystin-1 (PC-1)/polycystin-2 (PC-2) calcium channel. Mutations in the genes encoding these proteins, Pkd1 and Pkd2, respectively, are responsible for autosomal dominant polycystic kidney disease (ADPKD). Furthermore, retinal photoreceptors have specially adapted primary cilia that are packed with membranes loaded with phototransducing pigments, such as rhodopsin, and the role of primary cilia in photoreceptor development and function is discussed in detail by Johnson and colleagues in this issue.Citation22 Hence, the sensory function of primary cilia is now well established in terms of their ability to detect chemicals, mechanical signals and light.Citation23-Citation26 What remains less clear and is a matter of intense research is how they transduce these signals into coordinated cellular responses that ultimately determine tissue and organ generation.
A number of cilia-based signaling pathways have now been identified that respond to morphogens and growth factors and clearly play critical roles in controlling cell fate. Of particular importance in ciliary signaling are the Wnt and Hedgehog pathways. Wnt signaling occurs in two principal flavours: the canonical Wnt pathway whose activation regulates gene expression through stabilization of the β-catenin transcription factor, and the non-canonical Wnt pathway, otherwise known as planar cell polarity or PCP, whose activation rather leads to β-catenin degradation. The switch between canonical and non-canonical Wnt signaling seems to be very much controlled through binding of different Wnt ligands to receptors on the primary cilium.Citation27-Citation31 However, as well as dictating gene expression patterns, PCP signaling has a direct effect on cytoskeletal organization, which contributes to the polarized ‘convergent extension’ movements that occur within an epithelial cell layer during embryonic development. The specific roles proposed for Wnt and PCP signaling in kidney development is explained in more detail in the accompanying article by Goggolidou.Citation32 Hedgehog signaling also involves receptor binding at the primary cilium, leading to regulation of gene expression through proteolytic switching between activator and suppressor forms of the Gli transcription factors.Citation33-Citation36 Both Hedgehog and Wnt signaling are crucial to tissue patterning during embryogenesis explaining why their loss gives rise to dysplastic development defects. However, these pathways also have key roles in tissue maintenance and homeostasis, and their deregulation is implicated not only in degenerative diseases but also in cancer progression. Furthermore, the primary cilium plays a pivotal role in a number of major growth factor-regulated signaling pathways, including the PDGF, FGF, and Notch pathways that control proliferation, with receptors for these ligands concentrated on ciliary membranes.Citation37-Citation39
Some defects in primary cilium function seem to predominantly interfere with the function of a single organ. Among the best studied of these are the dominant forms of polycystic kidney disease, e.g., ADPKD, and retinopathies, such as retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA). However, of particular interest and scientific importance are the recessive ciliopathy syndromes, which affect a range of organs with differing severity. These include Alstrom Syndrome (AS), Bardet Biedl Syndrome (BBS), Joubert Syndrome (JBTS), Meckel-Gruber Syndrome (MKS), Jeune Syndrome (also known as asphyxiating thoracic dysplasia, ATD), Senior-Løken Syndrome (SLS), Nephronophthisis (NPHP), Oral-Facial-Digital Syndrome (OFD), Ellis-van Creveld Syndrome (EVS), and Short Rib-Polydactyly Syndrome (SRPS). These diseases affect the kidneys, liver, eyes, ears, brain, bones, and reproductive system and therefore can involve a devastating combination of symptoms ranging from cystic kidneys, liver fibrosis, blindness, deafness, anosmia, polydactyly, cranio-facial defects, and situs inversus to infertility, mental retardation, obesity, and diabetes. Historically, patients were originally diagnosed as having a particular syndrome based on the extent of different organ involvement. However, it is now apparent that patients assigned to these syndromes really represent different points across a single overarching disease spectrum. The underlying message is that primary ciliary function, which is dependent on correct assembly, architecture and signaling, is key to the generation and maintenance of most organs of the body.
Phenotypic Diversity in Ciliary Diseases
Most, albeit not all, ciliopathies can be classified as monogenic recessive disorders. In other words, they are loss of function conditions for which both alleles must be mutated to manifest in disease. Even in the case of ADPKD, which is the most common monogenic disease and inherited in a typical dominant Mendelian manner, it is thought that patients must pick up a mutation in the second allele (so-called ‘two hit’ hypothesis) to develop the disease. However, one of the most intriguing features of the ciliopathies is the diversity of clinical symptoms and organ involvement that can present from mutation of the same gene. At its simplest level, a patient with a missense mutation that leads to mild loss of function may have a late onset degenerative disease due to problems in tissue repair and maintenance. In contrast, a patient with a null mutation in the same gene may have a much more severe developmental dysplastic disorder that leads to embryonic or perinatal lethality.
In reality, though, the situation is much more complex for a number of reasons. First, being generally recessive diseases, two mutations are required and hence it is a combinatorial effect of the two alleles that is manifest, some of which may be point mutations, some may be truncating and some may be null. Second, most of these diseases are multi-allelic, with the gene products assembling into functional complexes that regulate primary cilia function. Hence, many different combinations of mutations are possible leading to the range of symptoms, some of which were originally classified as different diseases, but which are now understood to represent an overlapping spectrum of related syndromes. Third, polymorphic differences in genes encoding ciliary proteins may act as phenotypic modifiers in the presence of other disease-causing mutations. Moreover, such polymorphisms, which alone do not cause disease, may in combination lead to a symptomatic disorder, in what is now understood as ‘oligogenic disease’.
The pleiotropy in severity and organ involvement is striking in the recessive NPHP-like ciliopathies that although being rare are the most common cause of end-stage renal disease in children and young adults.Citation40,Citation41 These diseases range from a relatively mild NPHP, which may first present during late childhood and solely involve the kidneys, through to the more severe SLS, which also involves retinopathies, and JBTS, that has kidney, retinal, and CNS involvement, to the most severe MKS, where perinatal lethality results from severe developmental defects in the brain. So far, at least 15 NPHP genes have been identified which encode components of the primary cilia. From a molecular point of view, the proteins encoded by these genes assemble into a set of complexes that have distinct roles in cilia assembly, trafficking, and signaling. For example as several localize specifically to the transition zone or the region of the cilium close to the transition zone, then it has been proposed that they may contribute to the gatekeeper function of this region regulating protein entry and exit from the cilium.Citation42 Mutations in different genes of this group could therefore have similar consequences if they prevent formation of a particular complex, or they have could have somewhat different consequences if they differentially affect the function of that complex. Equally, mutations that significantly perturb signaling pathways required for development are going to lead to early-onset dysplastic disease, whereas mutations that primarily affect pathways required for cell and tissue homeostasis are more likely to lead to degenerative disease. Furthermore, hypomorphic point mutations are likely to have more subtle effects than null mutations. This molecular understanding is now beginning to explain the overlapping but wide spectrum of symptoms that can result within these ciliopathy syndromes. The role of primary cilia in kidney, skeletal, and CNS development and the diverse impact of mutations in MKS genes on these organs is discussed at length in the article by Dawe and colleagues in this special issue.Citation43
Animal Models of Ciliopathies
Much of our mechanistic understanding of primary cilia organization and function has come from studies in cultured cells, and the development of 2D and 3D culture systems has allowed exploration of cilia function in more complex processes, such as cell polarization and OCD. There can be no arguing though that genetic approaches in nematode worms (C. elegans), green algae (Chlamydomonas), zebrafish, toads (Xenopus laevis), and mice have proven to be of enormous value both in terms of understanding the biology and modeling human disease. The first clue that ciliary dysfunction may underlie a human inherited disease came just over a decade ago from finding that the C. elegans homolog of a human cystic kidney disease protein localized to ciliated neurons.Citation44 Soon after, it was found that the IFT88 component required for flagellar assembly in Chlamydomonas was homologous to the polaris protein, encoded by the Tg737 gene, whose mutation was the cause of the oak ridge polycystic kidney (orpk) disease mouse model.Citation45 Each system has particular advantages that has leant itself toward study of ciliary function or organ development. For example, while lower eukaryotes, such as C. elegans or Chlamydomonas, provide simple genetic systems to study cilia structure and organization, Xenopus and zebrafish act as vertebrate models for both motile and immotile cilia function, as well as ciliary signaling and organ development. Xenopus larvae are particularly useful for studying basal body distribution and mechanisms involved in generating polarized beating cilia, while zebrafish are fast becoming a system of choice for monitoring organogenesis due to the beautiful imaging that can be applied to the transparent developing embryos.Citation46,Citation47 The advantages of C. elegans to explore primary cilium organization, and its compartmentalization into specific sub-domains, is reviewed in detail by Blacque and Sanders in this issue.Citation48
Not all animals and plants form primary cilia. Most obviously, yeast do not have cilia and therefore cannot be used as model systems. In this regard, it is worth noting that yeast also do not have bona fide centrioles or basal bodies, rather using a layered structure known as the spindle pole body as their primary microtubule organizing center. On the whole, organisms that form cilia have a basal body or centriole-like structure as their microtubule-organizing center, whereas those that do not use morphological distinct structures for organization of their microtubule cytoskeleton. Higher plants are another example of organisms that lack cilia. It was on this basis, that Dutcher and colleagues undertook an elegant comparative genomics study a few years ago aimed at identifying novel disease-related cilia genes.Citation49 Their approach was to look for genes shared between distantly related organisms that had cilia, namely humans and Chlamydomonas, but were missing from the higher plant Arabidopsis, that lacks a cilium. Strikingly, using this approach they identified a novel gene, BBS5, which upon screening of patients was found to be mutated in the recessive BBS ciliopathy.
Mouse models of human disease have long been a vital resource to the scientific community and cloning of the genes involved has often preceded identification of mutations in the homologous human genes in patients. This is certainly the case for inherited ciliopathies, where causative genes have been identified in mouse models for most of the syndromic recessive ciliopathies described above, as well as non-syndromic cilia-related disorders, such as polycystic kidney diseases and degenerative retinopathies.Citation50 Study of these relevant models, both in terms of whole animals and derived cells, has allowed a much deeper understanding of the cell biology that underlies the different progressive stages and manifestations of the disease, as well as providing a key resource for the testing of novel therapeutic approaches.
Future Perspectives
The importance of the primary cilium as a sensory organelle is now clear with outputs from ciliary signaling influencing cell fate in many diverse ways. It is therefore to be expected that primary cilia will have a major impact not just on the monogenic inherited disorders classified as ciliopathies, but on the many complex human diseases that affect the large proportion of society, including heart disease, cancer and diabetes. Indeed, it is becoming apparent that the primary cilium may have an important role to play in specific aspects of cancer progression, such as in epithelial-mesenchymal transitions or metastasis.Citation51-Citation53
Particularly exciting have been recent studies shedding light on a crucial link between the disease pathogenesis of ciliopathies and the DNA damage response (DDR). Specifically, it has been found that proteins whose mutation is known to cause ciliopathies have roles in G2/M checkpoint pathways and the replication stress response.Citation54-Citation58 These include Mre11, Cep164, ZNF423, ATR, and Nek8/NPHP9. Many of these proteins show localization to both primary cilia and nuclei, with specific association in some cases with DNA damage foci. While the underlying mechanisms require further study, the hypothesis here would be that excessive levels of DNA damage during organ development lead to dysplasia, while failure to repair damage can lead to the degenerative aspects of organ disease. Particularly intriguing is the fact that kidney epithelia is subject to significant DNA damage due to its constant exposure to toxic agents in urine, potentially explaining the high level of renal involvement in these diseases. However, it should be emphasized that the role of the primary cilium itself in the DDR and whether ciliary signaling pathways are distinct from or somehow integrated with DNA damage signaling are important issues yet to be resolved. Indeed, these studies raise the possibility that some pathological consequences of these mutations may relate specifically to abrogation of DDR pathways and have little to do with cilia. Furthermore, we should be careful not to get carried away and ascribe all membrane-derived signaling events to this organelle, as many signaling components found within cilia are also distributed elsewhere in the cell. For example, there is quite some debate at the moment about how important the primary cilium is in Wnt signaling with reports that neither canonical nor non-canonical Wnt signaling is perturbed in mouse embryos lacking cilia.Citation31 Hence, it will be crucially important moving forward to determine the real and precise importance of primary cilia in these pathways and diseases.
Nevertheless, one can expect our increased mechanistic knowledge of primary cilia biology to have a major impact on diagnosis, prognosis, and treatment of cilia-based diseases. The better understanding of clinical symptoms associated with ciliopathy syndromes, the increasing number of genes known to be linked to these diseases, and the rapid, high-throughput exome sequencing technologies now available to screen for mutations means that the underlying molecular defect in affected patients should be much more quickly identified. As for finding new and effective therapies, this will remain a major challenge. Preventing the dysplastic development that underlies the most severe diseases will certainly be extremely difficult and rely very much on predictive diagnosis and very early intervention. However, treatment of degenerative conditions should be possible in the near future, although the small number of cases for most recessive ciliopathies makes clinical trials difficult to organize. Gene therapy may well be the long-term goal for the loss-of-function genetic diseases.Citation59 But in the short term the more realistic aim is to identify effective small molecule therapeutics that may block defective ciliary signaling or, perhaps, aberrant cell proliferation. For example, roscovitine and rapamycin, inhibitors of the Cdk and mTor kinases respectively, are examples of targeted agents that have shown potential benefit in animal ciliopathy models.Citation60,Citation61 Anti-apoptotic agents may also prove useful in degenerative diseases, as it is the induction of apoptosis upon accumulation of cellular defects and DNA damage that generally initiates the degenerative process.
Although the recessive ciliopathy syndromes are generally rare, other cilia-based diseases, such as ADPKD, are very common and a major health burden to society. Yet ADPKD shares many clinical aspects with recessive ciliopathy syndromes, such as interstitial fibrosis and inflammation within the kidney, meaning that treatments for one may be applicable to the others. Hence, finding new and better treatments to these diseases could be of enormous benefit if they led, for example, to replacement of the current and very costly kidney replacement treatments of dialysis and transplantation. It is clearly early days, but it is a reasonable aspiration that a detailed molecular understanding of how the primary cilium contributes to organ generation and maintenance will eventually allow development of personalized therapies for individual patients with defined mutations.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors’ laboratories are support by grants to A.M.F. from Kidney Research UK and the Association for International Cancer Research (AICR), and to R.B. from Cancer Research UK and the MRC.
Related Research Data
References
- Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell 2009; 137:32 - 45; http://dx.doi.org/10.1016/j.cell.2009.03.023; PMID: 19345185
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 2006; 7:125 - 48; http://dx.doi.org/10.1146/annurev.genom.7.080505.115610; PMID: 16722803
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364:1533 - 43; http://dx.doi.org/10.1056/NEJMra1010172; PMID: 21506742
- Oh EC, Katsanis N. Cilia in vertebrate development and disease. Development 2012; 139:443 - 8; http://dx.doi.org/10.1242/dev.050054; PMID: 22223675
- Marshall WF. The cell biological basis of ciliary disease. J Cell Biol 2008; 180:17 - 21; http://dx.doi.org/10.1083/jcb.200710085; PMID: 18180369
- Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol 2007; 69:377 - 400; http://dx.doi.org/10.1146/annurev.physiol.69.040705.141236; PMID: 17009929
- Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA. Centrosomes and cilia in human disease. Trends Genet 2011; 27:307 - 15; http://dx.doi.org/10.1016/j.tig.2011.05.004; PMID: 21680046
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell 2009; 139:663 - 78; http://dx.doi.org/10.1016/j.cell.2009.10.036; PMID: 19914163
- Kobayashi T, Dynlacht BD. Regulating the transition from centriole to basal body. J Cell Biol 2011; 193:435 - 44; http://dx.doi.org/10.1083/jcb.201101005; PMID: 21536747
- Czarnecki PG, Shah JV. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol 2012; 22:201 - 10; http://dx.doi.org/10.1016/j.tcb.2012.02.001; PMID: 22401885
- Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol 2011; 12:222 - 34; http://dx.doi.org/10.1038/nrm3085; PMID: 21427764
- Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep 2012; 13:608 - 18; http://dx.doi.org/10.1038/embor.2012.73; PMID: 22653444
- Shiba D, Yokoyama T. The ciliary transitional zone and nephrocystins. Differentiation 2012; 83:S91 - 6; http://dx.doi.org/10.1016/j.diff.2011.11.006; PMID: 22169048
- Carroll TJ, Yu J. The kidney and planar cell polarity. Curr Top Dev Biol 2012; 101:185 - 212; http://dx.doi.org/10.1016/B978-0-12-394592-1.00011-9; PMID: 23140630
- Malicki J, Avidor-Reiss T. From the cytoplasm into the cilium: bon voyage. Organogenesis 2014; Forthcoming
- Scholey JM. Intraflagellar transport motors in cilia: moving along the cell’s antenna. J Cell Biol 2008; 180:23 - 9; http://dx.doi.org/10.1083/jcb.200709133; PMID: 18180368
- Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol 2008; 85:23 - 61; http://dx.doi.org/10.1016/S0070-2153(08)00802-8; PMID: 19147001
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol 2002; 3:813 - 25; http://dx.doi.org/10.1038/nrm952; PMID: 12415299
- Lee L. Mechanisms of mammalian ciliary motility: Insights from primary ciliary dyskinesia genetics. Gene 2011; 473:57 - 66; http://dx.doi.org/10.1016/j.gene.2010.11.006; PMID: 21111794
- Koeford K, Veland IR, Pedersen LB, Larsen LA, Christensen ST. Cilia and coordination of signaling networks during heart development. Organogenesis 2013; Forthcoming
- Jenkins PM, McEwen DP, Martens JR. Olfactory cilia: linking sensory cilia function and human disease. Chem Senses 2009; 34:451 - 64; http://dx.doi.org/10.1093/chemse/bjp020; PMID: 19406873
- Wheway G, Parry DA, Johnson CA. The role of primary cilia in the development and disease of the retina. Organogenesis 2013; 10; In press PMID: 24162842
- Berbari NF, O’Connor AK, Haycraft CJ, Yoder BK. The primary cilium as a complex signaling center. Curr Biol 2009; 19:R526 - 35; http://dx.doi.org/10.1016/j.cub.2009.05.025; PMID: 19602418
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 2010; 11:331 - 44; http://dx.doi.org/10.1038/nrg2774; PMID: 20395968
- Lancaster MA, Gleeson JG. The primary cilium as a cellular signaling center: lessons from disease. Curr Opin Genet Dev 2009; 19:220 - 9; http://dx.doi.org/10.1016/j.gde.2009.04.008; PMID: 19477114
- Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science 2006; 313:629 - 33; http://dx.doi.org/10.1126/science.1124534; PMID: 16888132
- Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet 2007; 39:1350 - 60; http://dx.doi.org/10.1038/ng.2007.12; PMID: 17906624
- Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P. Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet 2008; 40:69 - 77; http://dx.doi.org/10.1038/ng.2007.54; PMID: 18066062
- Oishi I, Kawakami Y, Raya A, Callol-Massot C, Izpisúa Belmonte JC. Regulation of primary cilia formation and left-right patterning in zebrafish by a noncanonical Wnt signaling mediator, duboraya. Nat Genet 2006; 38:1316 - 22; http://dx.doi.org/10.1038/ng1892; PMID: 17013396
- Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB. Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat Genet 2008; 40:871 - 9; http://dx.doi.org/10.1038/ng.104; PMID: 18552847
- Wallingford JB, Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev 2011; 25:201 - 13; http://dx.doi.org/10.1101/gad.2008011; PMID: 21289065
- Goggolidou P. Wnt and planar cell polarity signaling in cystic renal disease. Organogenesis 2013; 10; In press PMID: 24162855
- Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 2013; 14:416 - 29; http://dx.doi.org/10.1038/nrm3598; PMID: 23719536
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature 2005; 437:1018 - 21; http://dx.doi.org/10.1038/nature04117; PMID: 16136078
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003; 426:83 - 7; http://dx.doi.org/10.1038/nature02061; PMID: 14603322
- Wong SY, Reiter JF. The primary cilium at the crossroads of mammalian hedgehog signaling. Curr Top Dev Biol 2008; 85:225 - 60; http://dx.doi.org/10.1016/S0070-2153(08)00809-0; PMID: 19147008
- Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E. A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 2011; 145:1129 - 41; http://dx.doi.org/10.1016/j.cell.2011.05.030; PMID: 21703454
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 2005; 15:1861 - 6; http://dx.doi.org/10.1016/j.cub.2005.09.012; PMID: 16243034
- Tanaka Y, Okada Y, Hirokawa N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 2005; 435:172 - 7; http://dx.doi.org/10.1038/nature03494; PMID: 15889083
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol 2009; 20:23 - 35; http://dx.doi.org/10.1681/ASN.2008050456; PMID: 19118152
- Hurd TW, Hildebrandt F. Mechanisms of nephronophthisis and related ciliopathies. Nephron Exp Nephrol 2011; 118:e9 - 14; http://dx.doi.org/10.1159/000320888; PMID: 21071979
- Omran H. NPHP proteins: gatekeepers of the ciliary compartment. J Cell Biol 2010; 190:715 - 7; http://dx.doi.org/10.1083/jcb.201008080; PMID: 20819931
- Barker AR, Thomas R, Dawe HR. Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton and central nervous system development. Organogenesis 2013; 10; In press PMID: 24322779
- Barr MM, Sternberg PW. A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature 1999; 401:386 - 9; http://dx.doi.org/10.1038/43913; PMID: 10517638
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol 2000; 151:709 - 18; http://dx.doi.org/10.1083/jcb.151.3.709; PMID: 11062270
- Drummond I, Austin-Tse C. Zebrafish cilia. Methods Enzymol 2013; 525:219 - 44; http://dx.doi.org/10.1016/B978-0-12-397944-5.00011-0; PMID: 23522472
- Werner ME, Mitchell BJ. Using Xenopus skin to study cilia development and function. Methods Enzymol 2013; 525:191 - 217; http://dx.doi.org/10.1016/B978-0-12-397944-5.00010-9; PMID: 23522471
- Blacque OE, Sanders AAWM. Compartments within a compartment: what C. elegans can tell us about ciliary subdomain composition, biogenesis, funtion and disease. Organogenesis 2014; Forthcoming http://dx.doi.org/10.4161/org.28830
- Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May-Simera H, Li H, Blacque OE, Li L, Leitch CC, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 2004; 117:541 - 52; http://dx.doi.org/10.1016/S0092-8674(04)00450-7; PMID: 15137946
- Norris DP, Grimes DT. Mouse models of ciliopathies: the state of the art. Dis Model Mech 2012; 5:299 - 312; http://dx.doi.org/10.1242/dmm.009340; PMID: 22566558
- Ten Dijke P, Egorova AD, Goumans MJ, Poelmann RE, Hierck BP. TGF-β signaling in endothelial-to-mesenchymal transition: the role of shear stress and primary cilia. Sci Signal 2012; 5:pt2; http://dx.doi.org/10.1126/scisignal.2002722; PMID: 22355187
- Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 2009; 15:1062 - 5; http://dx.doi.org/10.1038/nm.2020; PMID: 19701203
- Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH Jr., Dlugosz AA, Reiter JF. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med 2009; 15:1055 - 61; http://dx.doi.org/10.1038/nm.2011; PMID: 19701205
- Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012; 150:533 - 48; http://dx.doi.org/10.1016/j.cell.2012.06.028; PMID: 22863007
- Choi HJ, Lin JR, Vannier JB, Slaats GG, Kile AC, Paulsen RD, Manning DK, Beier DR, Giles RH, Boulton SJ, et al. NEK8 links the ATR-regulated replication stress response and S phase CDK activity to renal ciliopathies. Mol Cell 2013; 51:423 - 39; http://dx.doi.org/10.1016/j.molcel.2013.08.006; PMID: 23973373
- Valdés-Sánchez L, De la Cerda B, Diaz-Corrales FJ, Massalini S, Chakarova CF, Wright AF, Bhattacharya SS. ATR localizes to the photoreceptor connecting cilium and deficiency leads to severe photoreceptor degeneration in mice. Hum Mol Genet 2013; 22:1507 - 15; http://dx.doi.org/10.1093/hmg/dds563; PMID: 23297361
- Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, Chaki M, Diaz K, Lach FP, Bennett GR, Gee HY, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet 2012; 44:910 - 5; http://dx.doi.org/10.1038/ng.2347; PMID: 22772369
- Sivasubramaniam S, Sun X, Pan YR, Wang S, Lee EY. Cep164 is a mediator protein required for the maintenance of genomic stability through modulation of MDC1, RPA, and CHK1. Genes Dev 2008; 22:587 - 600; http://dx.doi.org/10.1101/gad.1627708; PMID: 18283122
- McIntyre JC, Davis EE, Joiner A, Williams CL, Tsai IC, Jenkins PM, McEwen DP, Zhang L, Escobado J, Thomas S, et al, NISC Comparative Sequencing Program. Gene therapy rescues cilia defects and restores olfactory function in a mammalian ciliopathy model. Nat Med 2012; 18:1423 - 8; http://dx.doi.org/10.1038/nm.2860; PMID: 22941275
- Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 2006; 444:949 - 52; http://dx.doi.org/10.1038/nature05348; PMID: 17122773
- Torres VE, Boletta A, Chapman A, Gattone V, Pei Y, Qian Q, Wallace DP, Weimbs T, Wüthrich RP. Prospects for mTOR inhibitor use in patients with polycystic kidney disease and hamartomatous diseases. Clin J Am Soc Nephrol 2010; 5:1312 - 29; http://dx.doi.org/10.2215/CJN.01360210; PMID: 20498248