
Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare congenital disease that causes bone formation within the muscles, tendons, ligaments and connective tissues. There is no cure for this disorder and only treatment of the symptoms is available. The purpose of this study was to review the literature and describe the clinical, cellular and molecular aspects of FOP. The material used for the study was obtained by reviewing scientific articles published in various literature-indexed databases. In view of its rarity and of the lack of insightful information and the unpredictability of its course, FOP is a challenging disorder for professionals who are confronted by it. However, this rare disease raises a great deal of interest because understanding the mechanism of mature bone formation can encourage research lines related to bone regeneration and the prevention of heterotopic ossification.
Introduction
The study of bone tissue injury and repair has established a series of events that lead to the replacement of newly produced bone and subsequent remodeling. Bone defects often result from a great loss of bone mass due to traumas, pathological fractures, abnormal skeletal development, and tumor resection. Current therapeutic procedures employed for the treatment of critical bone defects have several limitations.Citation1-3 These bone lesions can be classified as fractures with or without loss of bone mass. The repair process varies significantly in each of these cases, with striking consequences for the patient.
In the regeneration process of a fracture without loss of bone mass, bone neoformation, stabilized and consolidated by bone callus formation, is followed by tissue remodeling, in addition to revascularization and replacement of necrotic areas. External factors can markedly affect the regeneration process, but tissues act according to biological rules which control cell proliferation and differentiation, as well as extracellular matrix production, processes that usually occur independently of external interferences but are influenced by them.Citation4-6
However, fractures without loss of bone mass generally require the use of grafts or implants, since usually the size of the defect is beyond the natural capacity of tissue regeneration. The implants are used as support for bone regeneration, interacting with the interface of “receptor” fragments and stimulating the process of tissue regeneration. These devices, developed and designed to be implanted, are known as biomaterials.Citation7-9
Interdisciplinary approaches have been used in order to offer a therapeutic alternative to patients with loss of bone mass. One of them is the field of tissue engineering. Tissue engineering, a term created at the 1987 meeting of the National Science Foundation (NSF), refers to a combination of Cellular and Tissue Biology methods with Engineering and Surgery in order to elucidate the structure-function relationships of normal and damaged tissues, aiming at their repair or replacement. These methods can be combined with biomaterials used as scaffolds, which are designed to fill, replace or repair damaged tissues.Citation7-12 This approach has yielded very promising results, although a lot of study is still necessary to understand the mechanisms of tissue regeneration in a situation of injury with loss of bone mass.
On the other hand, there are some pathological situations in which heterotopic bone formation occurs. The development of heterotopic bone from soft tissue is a common feature of at least three distinct genetic disorders that affect humans: fibrodysplasia ossificans progressiva (FOP), Albright's hereditary osteodystrophy, and progressive osseous heteroplasia. These three disorders differ in terms of osteogenic induction, histopathology, anatomic distribution, and disease progression.Citation13,14
In the particular case of FOP, a process identical to post-fracture bone regeneration occurs. The organism can spend months without creating new bone, as if the disease were dormant; nevertheless, without any previous warning, new bones start to emerge in unexpected places.Citation15 Many researchers have attempted to elucidate the cellular and molecular mechanisms that correlate traumatized soft tissues with ectopic bone formation.Citation16 In the present study we tried to establish a parallel between mechanisms of bone repair and the known physiopathology data of FOP. We believe that these two lines of research are complementary and might be able to open new perspectives, since conditions such as FOP show a biological potential for bone formation in an abundant and comprehensive manner. Moreover, information obtained in the field of regenerative medicine, which uses the biological response of host tissue to implants, may provide new data that can be used in disorders such as FOP.
Epidemiology of FOP
The first cases of FOP were described by Patin in 1692 and by Freke in 1739.Citation17,18 In 1918, Rosenstirn conducted an extensive review of the medical literature, describing 115 cases of FOP.Citation14,17 The disease was first named myositis ossificans progressiva,Citation19,20 meaning a muscular inflammation that gradually turned into bones. However, this process affects not only muscles, but also soft parts such as articular capsules and ligaments.Citation21 Thus, the name was changed to fibrodysplasia ossificans progressiva by Victor McKusick in 1970.Citation14,22 In 2001, the International Association for Fibroplasia Ossificans Progressiva reported less than 200 people affected by the disease.Citation23 In 2008, Kaplan et al.Citation24 reported about 700 known cases worldwide.
FOP is one of the rarest diseases affecting humans,Citation25 with an estimated incidence of about 0.61 case per one million inhabitants.Citation26 Although most reports concern Caucasian patients, the disease affects all ethnic groups, with a predilection for males at a proportion of 4:1.Citation19,27 Since in most cases there is no family history, some studies have suggested that the disease is due to a sporadic mutation.Citation19 Some cases of FOP have been reported in the Brazilian medical literature. To date, the Brazilian Association for FOP (FOP Brasil, https://pt-br.facebook.com/pages/FOP-BRASIL/252108424811319) has registered 49 cases of the disease.Citation28 Information about FOP Brazil can be obtained by accessing the FOP page on the internet. Internationally, IFOPA (http://www.ifopa.org/) is a website that provides extensive information about the epidemiology of the disease, as well as international support, serving a large proportion of identified FOP patients around the world.
Clinical Characteristics of FOP
FOP is a rare disorder of dominant autosomal origin which manifests in connective tissue.Citation14,17,18 The disease is characterized by congenital malformation of the largest toes and heterotopic ossification leading to progressive immobility.Citation14,16
In most patients, peculiar characteristics are high rates of congenital anomalies associated more frequently with the thumb and hallux, which are present at birth in 75% to 90% of cases.Citation21 When malformations of the great toes, such as hallux valgus, and pre-osseous tumor-like swellings are present, the diagnosis is FOP.Citation29 Other alterations include macrodactyly, interphalangeal ankylosis and clinodactyly, narrow lumbar canal, decreased humerus epicondylar angle, and pseudoexostosis.Citation13,30 Sites of ectopic bone formation are tendons, fascia, aponeurosis, and skeletal muscles.Citation17,20,31 Diaphragm, extraocular muscles, or smooth and cardiac muscles are not affected by this disorder.Citation20,32
The affected individual usually presents progressive ossification of connective tissue, which causes a significant limitation of osteoarticular mobility,Citation25,26 more commonly of the hip, knee, shoulder and elbow,Citation16 with the patient being locked in a single position and even being unable to sit. This aspect characterizes the most advanced stage of the disease, called “stone man,” reported in only about 600 patients. At this stage, the patient rapidly progresses to death as a consequence of respiratory problems. Also, characteristic problems are reported in the face of patients.Citation31,32
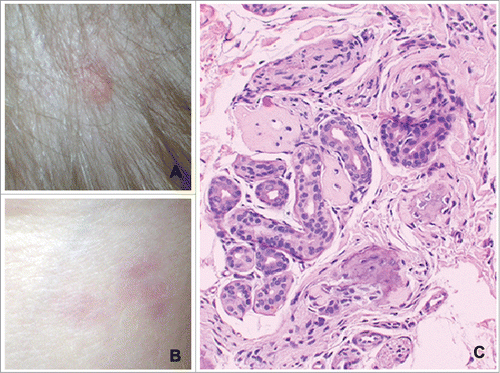
Exacerbation of FOP can occur spontaneously or can be precipitated by some types of trauma, including intramuscular injections, vaccines, local anesthesia, muscle biopsy, and peripheral intravenous puncture.Citation33 In cases of trauma, there seems to be a probable explanation about the factor that triggers the disease, considering the massive local release of cytokines.Citation34 In FOP, a biopsy of calcification nodules must be avoided, since it can result in recurrent ossification at this site, at times worse than the original lesion. Another clinical expression of FOP is swelling of the limbs, defined by an increase in limb circumference or increased tissue turgescence at one or more sites.Citation35 Although trauma is considered to be one of the acute factors leading to the disease, some episodes have been observed in which trauma was not a trigger.Citation17,36 The soft tissue masses coalesce, fibrose and calcify, forming bony bridges within a few weeks,Citation27 as shown in where muscle tissue is being calcified, undergoing ossification. Histologically, ectopically formed bone tissue is similar to normal bone tissue.Citation16,22
Figure 1. Images of a patient with FOP. (A) Ossification nodule at the scalp level; (B) photograph of the back, with multiple erythematous papules; (C) biopsy of the skin on the back: aspects of ossification in the dermis around sweat glands. Reprinted with permission from reference 52.
The progression of disability is irregular, with severe limitation in backbone and shoulder movement occurring after about 10 y of disease, with one or both hips being involved after 20 y, and with most patients being confined to a chair or to bed with joint restriction after 30 y. The progression of disability does not appear to be influenced by any form of medical treatment. Patient care should focus on the prevention of aggravating factors, including muscle traumas, biopsy of nodules, special ectopic bone surgery, intramuscular injections, and dental treatment.Citation37
Ideally, the diagnosis should be made during the neonatal period, so that any procedure that can trigger ossification could be avoided.Citation23,28,33 Noninvasive tests are indicated for an early diagnosis and monitoring of disease progression.Citation36 Bone scintigraphy reveals areas of ectopic calcification before their radiographic detection is possible, and can be used to determine the extent of the disease. Computed tomography and magnetic resonance methods seem to be promising for the detection of new foci not apparent on radiographs.
In FOP there are no specific changes in laboratory tests. Acute phase tests, as well as the metabolism of calcium, phosphorus and parathormone, are normal. Biochemical analyses of bone mineral metabolism are usually normal, although serum alkaline phosphatase activity can be increased. Blood composition and renal and parathyroid hormone levels are all within normal limits.Citation30,38 The urinary levels of basic fibroblast growth factor may be elevated during outbreaks of the disease, coinciding with the pre-osseous angiogenic phase.Citation38
Different diagnostic tests are limited since the phenotype, clinical history and radiographic findings practically define the framework for FOP.Citation14 The first radiographic finding is the presence of bone expansions or masses in soft tissue. Mineralization occurs after 3 to 4 wk, with the final appearance of bone columns that replace soft tissues.Citation19 As described above, the diagnosis of FOP is made when malformations of the great toes, such as hallux valgus, and pre-osseous tumor-like swellings are present.Citation29
So far, there is no definitive treatment for FOP and management should be conservative, avoiding any condition that can potentially cause ectopic ossification.Citation36 Of great importance for the quality of life of the patients and for slowing the progression of the disease are major efforts to avoid trauma and any invasive procedures, including blood drawing, as soon as FOP is diagnosed, since they can lead more rapidly to disability and death.
The rarity of FOP in humans prevents the scientific assessment of therapeutic efficacy or even the study of the natural history of the disease. A natural animal model would allow clarification of its pathophysiology, permitting the evaluation of therapeutic interventions.Citation39 However, Rothschild et al.Citation18 observed that the mouse- deer (genus Tragulus), found in Southeast Asia, has an osseous sheath covering the lower back and thigh region corresponding to the clinical definition of FOP. Heterotrophic bone deposition was found to be present in adult males of this species, both in wild animals and in animals kept in zoos, and was considered to be the first known example of spontaneous FOP in a non-human mammal, offering the opportunity to examine experimentally many of the important attributes of this pathology.
The histopathology of the disease varies according to its duration and changes are only observed in affected anatomic areas. These focal and evolutionary behaviors explain the normality of the biopsies obtained from patients at an early stage and probably from an unaffected area.Citation19,38
Normal Bone Formation
Bone formation can classically occur by two processes, i.e., endochondral and intramembranous ossification. In the normal endochondral ossification process, bone is formed from a piece of hyaline cartilage, which undergoes changes, degeneration and progressive replacement by bone tissue. This process begins by perichondrium intramembranous ossification and leads to changes in the cartilage which can be distinguished morphologically as distinct regions: (1) zone of normal cartilage, where hyaline cartilage without any visible change is observed; (2) seriate cartilage area, where the chondrocytes divide and form parallel columns of flattened cells pulled in the longitudinal direction; (3) hypertrophic cartilage zone, which has large chondrocytes and deposition of lipid and glycogen. The cartilaginous matrix is reduced to regions between hypertrophic cells. (4) Zone of calcified cartilage, where points of cartilage mineralization and death of chondrocytes occur. (5) Ossification zone, where blood capillaries originating from the periosteum invade the cavities left by dead chondrocytes. Osteoprogenitor cells differentiate into osteoblasts that form a continuous layer on the remains of calcified cartilage matrix. On these remains, osteoblasts deposit bone matrix which calcifies and traps cells that turn into osteocytes.Citation4-6 The process of intramembranous ossification starts when a small group of adjacent mesenchymal stem cells (MSCs) begin to replicate and form a small, dense cluster of cells. Once this nodule has been formed, the MSCs within it stop replicating. At this point, morphological changes to osteoprogenitor cells begin to occur in the MSCs, their shape becomes more columnar and the number of Golgi complexes and amount of rough endoplasmic reticulum increase. All cells within the nodule develop into osteoblasts, which create an extracellular matrix, named osteoid. Some of the osteoblasts become incorporated within the osteoid to become osteocytes. Then, the matrix becomes mineralized, resulting in a nodule consisting of mineralized osteoid that contains osteocytes and is lined with active osteoblasts.Citation4-6
The entire process of osteogenesis and differentiation of bone cells is highly regulated. We are beginning to know what molecules are involved in this process. Osteoblast differentiation is regulated by the hedgehog gene family, including the Ihh (Indian hedgehog) and Shh (Sonic Hedgehog) genes, besides the Cbfa1 factor and bone morphogenetic proteins (BMPs). Furthermore, precursor cells present molecules in the cell membrane that can act as markers of differentiation. The expression of Cbfa1 appears to be the first sign of osteogenic differentiation. Collagen I and osteopontin are also expressed quite precociously.40 Alkaline phosphatase, bone sialoprotein and osteocalcin are commonly related to the final stages of osteoblast differentiation.Citation4,Citation40-42
The very first inquiries about processes that determine bone neoformation at sites lacking bone tissue were based on classic findings. In 1931, Huggins defined the phenomenon of osteoinduction by demonstrating in dogs that self-graft of transitional epithelium of the urinary bladder in abdominal wall muscle was able to promote ectopic bone formation. In 1931 and 1937, Levander recognized the same phenomenon by injecting an alcoholic bone extract into muscle tissue, obtaining bone neoformation.Citation43 In 1965, ectopic bone formation was shown to occur when demineralized bone matrix was implanted into rabbits, rats, mice and guinea pigs.Citation44 Based on these studies, the search for “osteoinductive material” inside bone matrix has become an increasingly expanding research field, and subsequent studies have shown that low molecular weight proteins could be extracted from demineralized bone matrix, which were then named bone morphogenetic proteins (BMPs).Citation43,45 The inducer and induced cells were host derived and the inducer cells appear to be descendants of histiocytesCitation46 and perivascular connective tissue cells.Citation16,25,46
TGF-β/BMPs have widely recognized roles in bone formation during mammalian development. A BMP morphogen gradient is established in the embryo, and BMP drives the differentiation of ectodermal cells and mediates dorsal pattern formation to establish the dorsal-ventral axis.Citation47 Here we will focus on the BMP pathway because of its tight connection with FOP. BMP-2, 4, 5, 6, and 7 have osteogenic capacity. The addition of BMP-2 increases osteocalcin.Citation48 BMP-7 induces the expression of osteoblast differentiation markers such as alkaline phosphatase, Runx2/Cbfa1 mRNA, a bone-specific transcription factor, and osteocalcin.Citation49
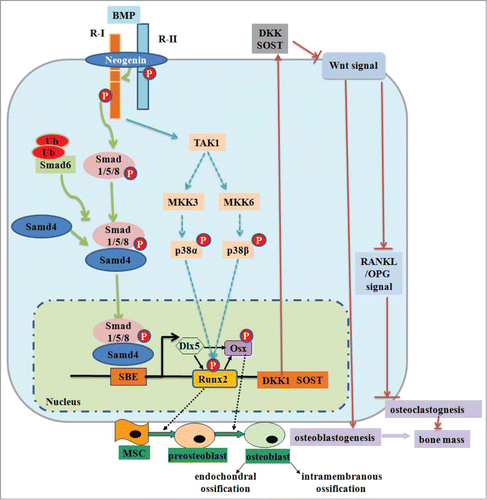
BMP stimulation occurs through Smads, which are intracellular proteins that transduce extracellular signals and form TGF-β/BMP ligands to the nucleus from where they activate downstream gene transcription. Smad-dependent- signaling is initiated by binding of BMP to receptor type II (R-II) and receptor type I (R-I) and subsequent signal transduction to their Smads. Activated Smads form a complex with Smad4 and then translocate into the nucleus where they interact with other transcription factors to trigger target gene expression. This signaling pathway has an important point of control. The transmembrane protein neogenin regulates BMP receptor association and Smad1/5/8 signaling.Citation47 Once this signaling pathway is activated, Smads regulate the expression of transcription factors and transcriptional coactivators that are important in osteoblast differentiation, such as Dlx5, Runx2 and Osx. This system is modulated by Smad6 that binds to the type I BMP receptor and prevents the activation of Smad1/5/8.Citation47 The non-Smad-dependent TGF-β-activated kinase 1 (TAK1) signaling pathway also regulates bone formation. In this pathway, BMP type IA receptor (BMPR-IA) signaling upregulates sclerostin protein (a product of the Sost gene) expression primarily through Smad-dependent signaling, while it upregulates DKK1 through Smad-dependent and non-Smad-dependent signaling. The sclerostin and DKK1 proteins can act as downstream molecules of BMP signaling to inhibit Wnt signaling and, therefore, act as negative regulators of bone formation.Citation47 This pathway of bone differentiation is illustrated in .
Figure 2. BMP signaling and negative regulation in bone formation. Reprinted with permission from reference 47.
Bone Neoformation in FOP
Cellular and molecular biology studies of FOP are being directed at the factors that cause the differentiation of pluripotent or oligopotent cells into osteoblasts. More recent studies have focused on the signaling pathway of BMPs and on the identification of progenitor cells responsible for ectopic bone formation.
Most FOP cases are a consequence of new gene mutations.Citation26 This disease is considered to be due to a gene mutated during embryonic development, causing bone congenital malformations, and that can be reactivated during the postnatal period, causing progressive heterotopic ossification.Citation29,50 In order to identify the chromosomal locus for the FOP gene, extensive genetic linkage analysis has been conducted using a subset of five families with the most striking and unambiguous features of FOP.
This approach identified the FOP gene on chromosome 2, more precisely in the 2q23–24 region. At this locus there is a gene that encodes activin A receptor I (ACVR1), also known as activin-like kinase 2 (ALK2), a type of BMP I receptor. DNA assays with the ACVR1 gene have determined that the same mutation (c.617G > A, p.R206H) occurs in the glycine/serine activation domain and appears in all affected individuals examined.Citation50,51
In another case study of an 18-mo-old patient diagnosed with FOP, the results suggested that another mutation in the GNAS1 gene located on chromosome 20, consisting of a base deletion in exon 7, can trigger disease development. The exact function of this gene on chromosome 20 is still unknown, although the gene seems to encode the α subunit of G protein, which is related to the negative control of osteogenesis.Citation52
A slow and progressive local fibroblastic proliferation occurs in FOP. A lymphocytic infiltrate consisting of macrophages and fibroblasts is usually observed in early lesions, later evolving to connective tissue areas with central ossification in which osteoblasts, osteocytes and osteoclasts differentiate.Citation53,54 Fibroblast proliferation is commonly observed in several areas of histological muscle sections, leading to muscle fiber degeneration.
A predominant muscle and subcutaneous connective tissue mononuclear infiltrate is present, with extensive fibroblast proliferation replacing damaged muscle fibers, leading to areas of newly formed bone tissue. Fibrous tissue, bone tissue or cartilage can be detected in the center of this infiltrate. Endochondral ossification in the affected areas has also been reported.Citation22,54
In a case study, BlaszczykCitation53 diagnosed a patient with osteosarcoma. Osteosarcoma is a primary malignant mesenchymal tumor whose cells produce osseous matrix.Citation55 Histological analysis revealed fibroblast proliferation combined with an extensive inflammatory infiltrate in subcutaneous tissue, tendons, ligaments, fascia and muscles, leading to heterotopic bone formation, the peculiar characteristic of FOP. In a study of a male cat with a clinical condition similar to FOP, histological analysis of ectopic bone showed mature bone tissue with preserved trabeculae and a hypercellular bone marrow.Citation56
Mesenchymal cells, which differentiate into bone-forming cells in response to stimulation by BMPs, are often denominated inducible osteoprogenitor cells. These cells are abundantly present in skeletal muscle and connective tissue, where bone formation can be induced more easily.Citation43
BMPs are members of a protein superfamily designated transforming growth factor (TGF), which has at least 43 members participating in many biological functions, including cell growth and differentiation in embryonic pattern formation.Citation54,57 According to Ripamonti and Reddi,Citation58 in addition to their functions in post-fetal osteogenesis, BMPs might play multiple roles in embryonic and organogenic development, including craniofacial and dental tissue skeletogenesis. In in vitro assays using rat osteoprogenitor cells, Hughes et al.Citation59 demonstrated that proteins BMP-6, BMP-4, and BMP-2 could stimulate osteoblastic differentiation.
Patients with FOP produce large amounts of BMP-4 and small amounts of BMP-4 antagonists. BMP-4 is a protein synthesized by skeletal muscles, and might also be produced at sites of soft tissue injury.Citation51,Citation60-62 Under normal conditions, such as negative feedback, BMP-4 stimulates the production of some BMP antagonists, resulting in the inhibition of their activities and interrupting the bone differentiation pattern. In patients with FOP, this process does not occur due to the lack of inhibitory proteins and to excess BMP-4.Citation60-62 The BMP-4 gene is located in the chromosome region 14q22-q23, where mutations in this gene or in its promoter region have been reported.Citation40,57,58 Thus, a mutation in the BMP-4 gene leads to deregulation of the signaling pathways of these BMPs (strong inducers of endochondral bone formation), consequently favoring bone neoformation.
BMPs are extracellular signaling proteins that regulate the transcription of specific genes by binding to complexes of type I and type II serine/threonine kinase receptors on the plasma membrane. The binding to activated receptors mediates signaling through signal transduction factors specific for BMPs (Smad1, Smad5, and Smad8) and through the MAPK signaling pathway.Citation63 As mentioned previously, BMP-4 is overexpressed in FOP. The cells also do not positively regulate the expression of BMP antagonists in response to stimulation by this protein.
Other studies have reported a change in BMP signaling in FOP, such as the prevention of internalization and high levels of the cell surface receptor BMPR-IA (a BMP type I receptor), in addition to deregulation of heparan sulfate proteoglycans that modulate the BMP signaling pathway in cells with FOP. Increased levels of heparan sulfate proteoglycans receptor and BMP activity can lead to the abnormal formation of bone. The BMP receptor signaling pathway is mediated by at least two known intracellular pathways: Smad 1/5/8 and p38MAPK,Citation63 which appear to coexist in different cellular systems that lead to FOP.
ACVR1/ALK2 is one of four type I receptors that mediate BMP signaling.Citation29,54,64 In response to receptor-ligand binding, these receptors phosphorylate signal transduction molecules that regulate the cytoplasmic expression or repression of target genes. ACVR1 is expressed in many tissues, including skeletal muscle and cartilage, consistent with the sites of heterotopic ossification in FOP.Citation41 This gene appears to be also related to chondrogenesis and heterotopic ossification in chicken embryos.Citation64
ACVR1/ALK2 is a type I receptor for BMP. The structural modeling of the protein predicts that the recurrent mutation occurs in the GS domain (glycine/serine), consistent with hyperactivity of the BMP signaling pathway as the underlying cause of ectopic chondrogenesis and osteogenesis occurring in FOP. According to Kaplan et al.,Citation38 this mutation is consistent with a wealth of previous findings of an overactive BMP signaling pathway in cells from patients with FOP and provides a rational basis for understanding both the postnatal heterotopic ossification and the skeletal malformations found in this disease. The resulting ACVR1/ALK2 mutant protein appears to alter the sensitivity of the receptor/ligand and BMP in turn modifies the differentiation of connective tissue progenitor cells. Moreover, stromal cells in the early stages of FOP lesions appear to be involved and are locally recruited into vascular cells, which maintain the potential to differentiate along a pathway of endochondral ossification in an environment permissive for BMP.Citation29
By using rat models of BMP signaling, it was possible to identify progenitor cells that contribute to different stages of heterotopic endochondral ossification. Progenitor cells consistent with markers such as endothelial precursors (which express the Tie2 marker) were present at all stages of endochondral ossification, while progenitors of skeletal and smooth muscle cells showed minimal or no contribution in any phase.Citation25 On the other hand, cells with Tie2 also seem to be related to the formation of fibroproliferative lesions.Citation65 These results suggest that a scenario of chronic stimulation of BMP activity, associated with muscle damage and inflammation, can trigger the formation of heterotopic bone and that vascular cells are essential for the construction of ectopic bone.
Since tissue trauma in patients with FOP induces bone metaplasia, tissue biopsies are very rare and are obtained just prior to the establishment of a diagnosis of FOP. The heterotopic bone formation in FOP occurs via an endochondral lymphocytic infiltration, muscle degradation, fibroproliferative and angiogenic stages, and then cartilage and bone formation.Citation29
The histological stages of FOP lesions have been well described. Initial lesions are characterized by intense perivascular lymphocytic infiltration of B and T cells. Subsequent migration of mononuclear inflammatory cells precedes widespread muscle necrosis. After a brief inflammatory phase, an intense fibroproliferative reaction associated with angiogenesis and neovascularization is observed. As the injury matures, fibroproliferative tissue undergoes avascular condensation into cartilage, followed by a phase of revascularization and osteogenesis in a characteristic process of endochondral ossification. The resulting heterotopic ossification consists of normal mature lamellar bone with histological marrow elements. Although in some aspects heterotopic bone formation in FOP is similar to bone formation during embryonic development of the skeleton or bone regeneration in fractures during postnatal life, important differences are the lack of inflammation in embryonic skeletal induction and the relative absence of lymphocytic inflammatory cells in fractures.Citation38,54
Current Approaches to Bone Tissue Engineering
In some respects, a repetition of the ossification process occurs during bone repair. Despite histological similarities, the comparison of the two processes is not yet fully understood. The exact mechanism of osteoinduction by biomaterials in tissue engineering or guided tissue regeneration remains largely unknown. It is also unknown whether the mechanisms of osteoinduction by BMPs and biomaterials are the same. Outstanding differences in osteoinduction by BMPs and biomaterials have been shown in a recent review.Citation66 These differences are: (1) bone induced by biomaterials is almost always intramembranous while bone induced by BMP is mainly formed via endochondral cells; (2) in small animals such as rodents, bone is very rarely induced by biomaterials, but easily by BMPs; (3) while bone is never observed at the periphery of biomaterials and is always formed inside their pores, BMP-induced bone formation is regularly seen outside the carrier and the soft tissue away from the surface of the carrier.
BMPs are widely used in tissue engineering. Recombinant forms of human BMP type 2 (rhBMP-2) were added to bioresorbable PLGA scaffolds which served as substrates for the growth of osteoblasts. A significant production of bone matrix was observed.Citation67,68 When rhBMP-2 was adsorbed to collagen I matrices and implanted in bone lesions, bone neoformation and the integration between the implant and injured bone were observed.Citation69 Subsequently, several groups have shown that BMPs have the ability to induce new bone formation through endochondral cells when implanted at ectopic sites in experimental animals.Citation66 Although the results are quite encouraging, clinically this type of therapy is limited by the size of the fracture produced,Citation67 unlike what happens in FOP where bone formation is quite extensive.
Therefore, progenitor cells, together with stimulating factors such as BMP, create an environment favorable to bone formation, which might be a target for the development of therapeutic interventions for the treatment of heterotrophic ossification.Citation54 A better understanding of this molecular pathophysiology is important for tissue engineering procedures which, in addition to a scaffold, also contain MSCs, progenitor cells or even osteoblasts in an implant situation, which in turn generates an inflammatory response. With a better knowledge of the physiopathology of FOP, these parallels open space for the optimization of bone fracture repair.
TGF-β and BMPs utilize parallel and related signaling pathways. However, the interaction between these pathways in bone remains unclear. TGF-β inhibition has been previously reported to promote osteogenic differentiation in vitro in implant materials,Citation70 suggesting that it may have a capacity to augment orthopedic repair.Citation71 The effects of BMP-2, TGF-β1, and TGF-β receptor (ALK-4/5/7) inhibitor on osteogenic differentiation have been recently reported in a murine pre-osteoblast cell line. It was found that BMP-2 and TGF-β receptor inhibitor increased the expression of osteogenic markers in vitro, while TGF-β1 decreased their expression. On the other hand, neither in vivo system was able to improve bone formation or repair with TGF-β receptor inhibitor treatment. Thus, ALK-4/5/7 inhibitors can promote osteogenic differentiation in vitro, but this may not readily translate to in vivo orthopedic applications. These contradictory results indicate that we still have much to learn about bone differentiation before we can develop an effective therapeutic approach.Citation72
How FOP Can Improve Bone Tissue Engineering
The rarity of FOP, coupled with difficulties in obtaining enlightening studies and the unpredictability of its course, make it a challenging disease for researchers who are faced with it, raising many doubts about which therapeutic measure to adopt in order to provide benefits without additional damage. As we have seen throughout this article, FOP is a disorder that leads to quite impressive skeleton formation from soft tissues. This is a remarkable result, because the parameters that lead to bone formation are important due to the fact that new bone formation in a healthy individual is one of the limitations of the normal bone repair process.
The discovery and elucidation of the molecular factors that lead to FOP has been an outstanding achievement by providing a better understanding of this disease.Citation29 Furthermore, new therapeutic strategies might be developed,Citation73 with future perspectives never seen before in the history of this clinical pathology. Recently, specific RNA interference (RNAi) has been shown to be able to suppress the expression of a mutant copy of the FOP gene in cells from patients in vitro. It was found that this approach decreased the elevated BMP signaling in FOP cells to levels observed in control cells. Being from a patient with FOP, these cells contained the exact combination of damaged and normal ACVR1/ALK2 receptors found in all classically affected FOP patients.Citation74 These results have raised the hope that RNAi could be of potential therapeutic efficacy for the treatment of FOP. In addition, the molecular mediators observed in FOP could be used to optimize the process of bone repair in several different clinical situations.
In conclusion, FOP is a rare disease that affects multiple body structures, causing heterotopic ossification. The current lack of an effective therapy for FOP is primarily due to the absence of definitive knowledge about the primary genetic defect that causes and rules its complex evolutionary changes. The difficulty in obtaining diagnostic biopsies at defined stages of its evolution, the lack of a suitable animal model for drug testing, the lack of families with multiple affected generations for the study of the natural variability of the disease, and the lack of truly objective clinical studies such as randomized double-blind placebo-controlled trials further hinder efforts to establish a rational therapy for this complex disease with genetic, evolutionary, post-traumatic and autoimmune bases. However, this rare disease raises great interest because the understanding of the genetic mechanism of mature bone formation can encourage research lines related to bone regeneration and the prevention of heterotopic ossification.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank INCT-BIOFABRIS, from the Brazilian government.
References
- Raschke M, Kolbeck S, Bail H, Schmidmaier G, Flyvbjerg A, Lindner T, Dahne M, Roenne IA, Haas N. Homologous growth hormone accelerates healing of segmental bone defects. Bone 2001; 29:368-73; PMID:11595620; http://dx.doi.org/10.1016/S8756-3282(01)00587-7
- Kneser U, Schaefer DJ, Polykandriotis E, Horch RE. Tissue engineering of bone: the reconstructive surgeon's point of view. J Cell Mol Med 2006; 10:7-19; PMID:16563218; http://dx.doi.org/10.1111/j.1582-4934.2006.tb00287.x
- Zhang R, Gao Z, Geng W, Yan X, Chen F, Liu Y. Engineering vascularized bone graft with osteogenic and angiogenic lineage differentiated bone marrow mesenchymal stem cells. Artif Organs 2012; 36:1036-46; PMID:23020776; http://dx.doi.org/10.1111/j.1525-1594.2012.01529.x
- Fernández-Tresguerres-Hernández-Gil I, Alobera-Gracia MA, del-Canto-Pingarrón M, Blanco-Jerez L. Physiological bases of bone regeneration I. Histology and physiology of bone tissue. Med Oral Patol Oral Cir Bucal 2006; 11:E47-51; PMID:16388294
- Shapiro F. Bone development and its relation to fracture repair. The role of mesenchymal osteoblasts and surface osteoblasts. Eur Cell Mater 2008; 15:53-76; PMID:18382990
- Magrini TD, Santos AR Jr., Rodrigues AA, Batista N, Belangero WD, Simas MP, Martinho HS. Analysis by FT-IR of three different bone regions: healthy, endochondral and intramembranous. Proc SPIE 2013; 8577:1
- Hench LL. Biomaterials: a forecast for the future. Biomaterials 1998; 19:1419-23; PMID:9794512; http://dx.doi.org/10.1016/S0142-9612(98)00133-1
- Atala A. Engineering organs. Curr Opin Biotechnol 2009; 20:575-92; PMID:19896823; http://dx.doi.org/10.1016/j.copbio.2009.10.003
- Santos AR Jr. Bioresorbable polymers for tissue engineering. In: Daniel Eberlin (Editor), Tissue engineering. Olajnica: In-Teh, 2010. p. 235-246
- Salter E, Goh B, Hung B, Hutton D, Ghone N, Grayson WL. Bone tissue engineering bioreactors: a role in the clinic? Tissue Eng Part B Rev 2012; 18:62-75; PMID:21902622; http://dx.doi.org/10.1089/ten.teb.2011.0209
- Lucchesi C, Ferreira BMP, Duek EAR, Santos AR Jr., Joazeiro PP. Increased response of Vero cells to PHBV matrices treated by plasma. J Mater Sci Mater Med 2008; 19:635-43; PMID:17619989; http://dx.doi.org/10.1007/s10856-007-0169-3
- Santos AR Jr., Wada MLF. Polímeros biorreabsorvíveis como arcabouços para cultura de células e engenharia tecidual. Polímeros Cien Tecnol 2007; 17:308-17
- Smith R, Athanasou NA, Vipond SE. Fibrodysplasia (myositis) ossificans progressiva: clinicopathological features and natural history. QJM 1996; 89:445-6; PMID:8758048; http://dx.doi.org/10.1093/qjmed/89.6.445
- Delai PLR, Kantanie S, Santili C, Kaplan FS. Fibrodisplasia ossificante progressiva: uma doença hereditária de interesse multidisciplinar. Rev Bras Ortop 2004; 39:205-13
- Romani F, Karam SM. Progressive ossifying fibrodysplasia: case report. Rev Bras Ortop 2011; 46:736-40; http://dx.doi.org/10.1590/S0102-36162011000600019
- Hsu JE. Current Review of Heterotopic Ossification. Uni Pennsyl Orthopaedic J 2010; 20:126-30
- Palhares DB, Leme LM. Miosite ossificante progressiva: uma perspectiva no controle da doença. J Pediatr 2001; 77:431-4
- Rothschild BM, Martin LD, Timm RM. A new spontaneous model of fibrodysplasia ossificans progressive. Brazi Geograph J Geosci Hum Res Medium 2010; 1:228-237
- Araújo CR Jr., Carvalho TN, Costa MAB, Lobo LV, Fonseca CR, Teixeira KS. Fibrodisplasia ossificante progressiva: relato de caso e achados radiográficos. Radiol Bras 2005; 38:69-73; http://dx.doi.org/10.1590/S0100-39842005000100014
- Campos DM, Renck DV, Bebber FE, Marquetto IB, Brum LF, Mendes L. Fibrodisplasia ossificante progressiva: relato de caso. Radiol Bras 2005; 38:393-5; http://dx.doi.org/10.1590/S0100-39842005000500017
- Kartal-Kaess M, Shore EM, Xu M, Schwering L, Uhl M, Korinthenberg R, Niemeyer C, Kaplan FS, Lauten M. Fibrodysplasia ossificans progressiva (FOP): watch the great toes! Eur J Pediatr 2010; 169:1417-21; PMID:20577760; http://dx.doi.org/10.1007/s00431-010-1232-5
- Shehab D, Elgazzar AH, Collier BD. Heterotopic ossification. J Nucl Med 2002; 43:346-53; PMID:11884494
- Sferco A, Naser C, Robledo H, Fili T, Tramunt B. Fibrodysplasia osificante progresiva: pautas para su reconocimiento. Arch Argent Pediatr 2001; 99:249-52
- Kaplan FS, Pignolo RJ, Shore EM, et al. The seventeenth annual report of the fibrodysplasia ossificans progressiva (FOP) collaborative project. FOP Connection 2008;21:1-18, in http://www.tfrd.org.tw/english/news/upload/20080710027828_03.pdf.
- Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, Glaser DL, Goldhamer DJ, Kaplan FS. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surg Am 2009; 91:652-63; PMID:19255227; http://dx.doi.org/10.2106/JBJS.H.01177
- Shaikh N, Arif F. Fibrodysplasia ossificans progressiva. J Pak Med Assoc 2011; 61:397-9; PMID:21465984
- Pignolo RJ, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva: clinical and genetic aspects. Orphanet J Rare Dis 2011; 6:80; PMID:22133093; http://dx.doi.org/10.1186/1750-1172-6-80
- Gonçalves AL, Masruha MR, de Campos CC, Delai PLR, Vilanova LCP. Fibrodysplasia ossificans progressiva: case report. Arq Neuropsiquiatr 2005; 63:1090-3; PMID:16400434; http://dx.doi.org/10.1590/S0004-282×2005000600032
- Kaplan FS, Xu M, Glaser DL, Collins F, Connor M, Kitterman J, Sillence D, Zackai E, Ravitsky V, Zasloff M, et al. Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics 2008; 121:e1295-300; PMID:18450872; http://dx.doi.org/10.1542/peds.2007-1980
- Majmudar DK, Hathila NN, Vaishya KB. Sayani, Trivedi AV, Kalola JG. Fibrodysplasia ossificans progressiva. Ind J Radiol Imag 2005; 15:347-8; http://dx.doi.org/10.4103/0971-3026.29152
- Baysal T, Elmali N, Kutlu R, Baysal O. The stone man: myositis (fibrodysplasia) ossificans progressiva. Eur Radiol 1998; 8:479-81; PMID:9510591; http://dx.doi.org/10.1007/s003300050420
- Hammond P, Suttie M, Hennekam RC, Allanson J, Shore EM, Kaplan FS. The face signature of fibrodysplasia ossificans progressiva. Am J Med Genet A 2012; 158A:1368-80; PMID:22581580; http://dx.doi.org/10.1002/ajmg.a.35346
- Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 2005; 116:e654-61; PMID:16230464; http://dx.doi.org/10.1542/peds.2005-0469
- Jackson WM, Aragon AB, Onodera J, Koehler SM, Ji Y, Bulken-Hoover JD, Vogler JA, Tuan RS, Nesti LJ. Cytokine expression in muscle following traumatic injury. J Orthop Res 2011; 29:1613-20; PMID:21452302; http://dx.doi.org/10.1002/jor.21354
- Nucci A, Queiroz LD, Santos AD, Camargo EE, Moura-Ribeiro MVL. Fibrodysplasia ossificans progressiva: case report. Arq Neuropsiquiatr 2000; 58(2A):342-7; PMID:10849638; http://dx.doi.org/10.1590/S0004-282x2000000200023
- Sendur OF, Gurer G. Severe limitation in jaw movement in a patient with fibrodysplasia ossificans progressiva: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2006; 102:312-7; PMID:16920539; http://dx.doi.org/10.1016/j.tripleo.2005.09.020
- Tonholo-Silva ER, Adachi EA, Tafner MS, Yoshinaga L. Fibrodysplasia ossificans progressiva. Arq Neuropsiquiatr 1994; 52:100-2; PMID:8002796; http://dx.doi.org/10.1590/S0004-282x1994000100020
- Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Goldsby RE, Kitterman JA, Groppe J, Shore EM. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol 2008; 22:191-205; PMID:18328989; http://dx.doi.org/10.1016/j.berh.2007.11.007
- Kaplan FS, Shore EM, Pignolo RJ, Glaser DL. Animal models of fibrodysplasia ossificans progressive. Clin Rev Bone Miner Metabolism 2005; 3:229-34; http://dx.doi.org/10.1385/BMM:3:3-4:229
- Yamaguchi A, Komori T, Suda T. Regulation of osteoblast differentiation mediated by bone morphogenetic proteins, hedgehogs, and Cbfa1. Endocr Rev 2000; 21:393-411; PMID:10950158; http://dx.doi.org/10.1210/edrv.21.4.0403
- Moreira PL, An YH, Santos AR Jr., Genari SC. In vitro analysis of anionic collagen scaffolds for bone repair. J Biomed Mater Res B Appl Biomater 2004; 71:229-37; PMID:15386402; http://dx.doi.org/10.1002/jbm.b.30026
- da Luz Moreira P, Genari SC, Goissis G, Galembeck F, An YH, Santos AR Jr. Bovine osteoblasts cultured on polyanionic collagen scaffolds: an ultrastructural and immunocytochemical study. J Biomed Mater Res B Appl Biomater 2013; 101:18-27; PMID:22987821; http://dx.doi.org/10.1002/jbm.b.32804
- Xiao Y-T, Xiang L-X, Shao J-Z. Bone morphogenetic protein. Biochem Biophys Res Commun 2007; 362:550-3; PMID:17719560; http://dx.doi.org/10.1016/j.bbrc.2007.08.045
- Urist MR. Bone: formation by autoinduction. Science 1965; 150:893-9; PMID:5319761; http://dx.doi.org/10.1126/science.150.3698.893
- Urist MR, Grant TT, Lindholm TS, Mirra JM, Hirano H, Finerman GA. Induction of new-bone formation in the host bed by human bone-tumor transplants in athymic nude mice. J Bone Joint Surg Am 1979; 61:1207-16; PMID:229105
- Santos AA, Miranda CDO, Alves MTS, Faloppa F. The role of bone morphogenetic protein on bone tissue repair. Acta Ortop Bras 2005; 13:194-5
- Chen G, Deng C, Li YP. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 2012; 8:272-88; PMID:22298955; http://dx.doi.org/10.7150/ijbs.2929
- Huang Z, Ren PG, Ma T, Smith RL, Goodman SB. Modulating osteogenesis of mesenchymal stem cells by modifying growth factor availability. Cytokine 2010; 51:305-10; PMID:20580248; http://dx.doi.org/10.1016/j.cyto.2010.06.002
- Gu K, Zhang L, Jin T, Rutherford RB. Identification of potential modifiers of Runx2/Cbfa1 activity in C2C12 cells in response to bone morphogenetic protein-7. Cells Tissues Organs 2004; 176:28-40; PMID:14745233; http://dx.doi.org/10.1159/000075025
- Carvalho DR, Navarro MMM, Martins BJAF, Coelho KEFA, Mello WD, Takata RI, Speck-Martins CE. Mutational screening of ACVR1 gene in Brazilian fibrodysplasia ossificans progressiva patients. Clin Genet 2010; 77:171-6; PMID:19796185; http://dx.doi.org/10.1111/j.1399-0004.2009.01256.x
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006; 38:525-7; PMID:16642017; http://dx.doi.org/10.1038/ng1783
- Mattia S, Mantovani G, Guaral N, Pagano R, Venuta A, Ferrari P. Eteroplasia ossificante progressiva: una nuova mutazione del gene GNAS 1 e review della letteratura. Medico e Bambino 2008; 11
- Blaszczyk M, Majewski S, Brzezinska-Wcislo L, Jablonska S. Fibrodysplasia ossificans progressiva. Eur J Dermatol 2003; 13:234-7; PMID:12804980
- Kaplan FS, Chakkalakal SA, Shore EM. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Dis Model Mech 2012; 5:756-62; PMID:23115204; http://dx.doi.org/10.1242/dmm.010280
- Martelli A, Teixeira LBC, Santos AR Jr. Aspectos histopatológicos e histoquímico de osteossarcomas em cães. Estud Biol 2007; 29:179-89
- Crivelenti LZ, Borin S, Brum AM, Honsho DK. Fibrodysplasia ossificans progressiva-like in a cat. Arq Bras Med Vet Zootec 2012; 64:359-62;; http://dx.doi.org/10.1590/S0102-09352012000200015
- Kaplan FS, Groppe J, Pignolo RJ, Shore EM. Morphogen receptor genes and metamorphogenes: skeleton keys to metamorphosis. Ann N Y Acad Sci 2007; 1116:113-33; PMID:17872396; http://dx.doi.org/10.1196/annals.1402.039
- Ripamonti U, Reddi AH. Periodontal regeneration: potential role of bone morphogenetic proteins. J Periodontal Res 1994; 29:225-35; PMID:7932015; http://dx.doi.org/10.1111/j.1600-0765.1994.tb01216.x
- Hughes FJ, Collyer J, Stanfield M, Goodman SA. The effects of bone morphogenetic protein-2, -4, and -6 on differentiation of rat osteoblast cells in vitro. Endocrinology 1995; 136:2671-7; PMID:7750491
- Kaplan FS, Shen Q, Lounev V, Seemann P, Groppe J, Katagiri T, Pignolo RJ, Shore EM. Skeletal metamorphosis in fibrodysplasia ossificans progressiva (FOP). J Bone Miner Metab 2008; 26:521-30; PMID:18979151; http://dx.doi.org/10.1007/s00774-008-0879-8
- Kaplan FS, Fiori J, DE LA Peña LS, Ahn J, Billings PC, Shore EM. Dysregulation of the BMP-4 signaling pathway in fibrodysplasia ossificans progressiva. Ann N Y Acad Sci 2006; 1068:54-65; PMID:16831905; http://dx.doi.org/10.1196/annals.1346.008
- van der Meij EH, Becking AG, van der Waal I. Fibrodysplasia ossificans progressiva. An unusual cause of restricted mandibular movement. Oral Dis 2006; 12:204-7; PMID:16476045; http://dx.doi.org/10.1111/j.1601-0825.2005.01171.x
- Shore EM, Kaplan FS. Insights from a rare genetic disorder of extra-skeletal bone formation, fibrodysplasia ossificans progressiva (FOP). Bone 2008; 43:427-33; PMID:18590993; http://dx.doi.org/10.1016/j.bone.2008.05.013
- Zhang D, Schwarz EM, Rosier RN, Zuscik MJ, Puzas JE, O’Keefe RJ. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J Bone Miner Res 2003; 18:1593-604; PMID:12968668; http://dx.doi.org/10.1359/jbmr.2003.18.9.1593
- Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 2010; 16:1400-6; PMID:21102460; http://dx.doi.org/10.1038/nm.2252
- Habibovic P, de Groot K. Osteoinductive biomaterials–properties and relevance in bone repair. J Tissue Eng Regen Med 2007; 1:25-32; PMID:18038389; http://dx.doi.org/10.1002/term.5
- Lee SC, Shea M, Battle MA, Kozitza K, Ron E, Turek T, Schaub RG, Hayes WC. Healing of large segmental defects in rat femurs is aided by RhBMP-2 in PLGA matrix. J Biomed Mater Res 1994; 28:1149-56; PMID:7829545; http://dx.doi.org/10.1002/jbm.820281005
- Whang K, Tsai DC, Nam EK, Aitken M, Sprague SM, Patel PK, Healy KE. Ectopic bone formation via rhBMP-2 delivery from porous bioabsorbable polymer scaffolds. J Biomed Mater Res 1998; 42:491-9; PMID:9827671; http://dx.doi.org/10.1002/(SICI)1097-4636(19981215)42:4≤491::AID-JBM3≥3.0.CO;2-F
- Hollinger JO, Schmitt JM, Buck DC, Shannon R, Joh S-P, Zegzula HD, Wozney J. Recombinant human bone morphogenetic protein-2 and collagen for bone regeneration. J Biomed Mater Res 1998; 43:356-64; PMID:9855194; http://dx.doi.org/10.1002/(SICI)1097-4636(199824)43:4≤356::AID-JBM3≥3.0.CO;2-7
- Zhang H, Ahmad M, Gronowicz G. Effects of transforming growth factor-beta 1 (TGF-beta1) on in vitro mineralization of human osteoblasts on implant materials. Biomaterials 2003; 24:2013-20; PMID:12628820; http://dx.doi.org/10.1016/S0142-9612(02)00616-6
- Ripamonti U, Ferretti C, Teare J, Blann L. Transforming growth factor-beta isoforms and the induction of bone formation: implications for reconstructive craniofacial surgery. J Craniofac Surg 2009; 20:1544-55; PMID:19816294; http://dx.doi.org/10.1097/SCS.0b013e3181b09ca6
- Schindeler A, Morse A, Peacock L, Mikulec K, Yu NYC, Liu R, Kijumnuayporn S, McDonald MM, Baldock PA, Ruys AJ, et al. Rapid cell culture and pre-clinical screening of a transforming growth factor-β (TGF-β) inhibitor for orthopaedics. BMC Musculoskelet Disord 2010; 11:105; PMID:20509926; http://dx.doi.org/10.1186/1471-2474-11-105
- Kaplan FS, Groppe J, Shore EM. When one skeleton is enough: approaches and strategies for the treatment of fibrodysplasia ossificans progressiva (FOP). Drug Discov Today Ther Strateg 2008; 5:255-62; PMID:23599718; http://dx.doi.org/10.1016/j.ddstr.2008.11.004
- Kaplan J, Kaplan FS, Shore EM. Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene Ther 2012; 19:786-90; PMID:22011642; http://dx.doi.org/10.1038/gt.2011.152