Abstract
The Wnt/β-catenin signaling pathway, conserved across the animal kingdom, is critical for the development of numerous tissues. Several recent studies have focused on the roles that this pathway plays at different stages of pancreatic organogenesis, including specification, proliferation, differentiation and function. Whereas, during early endoderm development, inhibition of the pathway is required for pancreatic specification, subsequent growth and differentiation of the fetal organ depends on the pathway being active. This appears especially true for exocrine acinar cells, the specification and differentiation of which also depend on β-catenin function. Whether the pathway plays an important role in development or function of endocrine islet cells, including insulin-producing β-cells, remains controversial. This question is particularly important in light of recent studies that implicate a downstream component of the pathway, TCF7L2, in human β-cell function. This review will cover recent work on Wnt/β-catenin signaling in pancreas development, emphasizing those points of controversy that most urgently require further investigation.
Introduction
The mammalian pancreas is anatomically subdivided according to its two major functions: the exocrine pancreas produces enzymes for digestion of food, and the endocrine pancreas regulates glucose metabolism. Digestive enzymes are secreted by acinar cells, located at the termini of a ductal network that drains these secretions into the intestine. Endocrine cells are organized into islets of Langerhans, which are scattered through the core of the organ. The major cell type of the islet, insulin-producing beta (β) cells, is destroyed in type I diabetes, and much interest in the field focuses on generating new β-cells to replace those lost in this disease.
For this reason, and because development of the organ can be followed in vitro, pancreatic organogenesis has been intensely studied for decades. The molecular era has seen considerable success in identifying intrinsic transcriptional regulators of pancreas development.Citation1,Citation2 Older studies of rodent pancreas development in culture highlighted the additional importance of extrinsic signals in this process, particularly for exocrine acinar differentiation.Citation3,Citation4 More recent work has thus focused on understanding the intercellular signaling milieu of the developing pancreas, particularly in the hope that such an understanding will help in directed islet differentiation of stem cells.Citation5 In the past five years, the Wnt signaling pathway has come under particular scrutiny for its potential roles in pancreatic organogenesis, as discussed in this review.
The Wnt/β-catenin signaling pathway is summarized in , and has been well-reviewed elsewhere.Citation6,Citation7 It must be kept in mind that Wnts can also act via alternative, “non-canonical” pathways, which are especially important in tissue morphogenesis.Citation8 Indeed, blocking the function of zebrafish Wnt-5, or its mouse ortholog Wnt5a, both of which appear to act non-canonically, causes defects in islet cell migration and morphogenesis.Citation9 These defects are relatively subtle, however, and do not grossly affect organ specification, proliferation or differentiation. Other studies of Wnt signaling in the pancreas, summarized in , pertain more specifically to the canonical pathway, on which this review will concentrate.
Specification of the Pancreatic Endoderm: Inhibition by Wnt/β-catenin Signaling
Table 1 breaks down pancreas development into stages of organ specification, expansion, differentiation and maintenance; this is a convenient device, but obscures the extent to which these processes coincide during organogenesis. For example, even as the pancreatic epithelium expands dramatically from embryonic day 13.5 (E13.5) in the mouse, considerable numbers of progenitor cells begin to differentiate into β-cell and other islet cell types.Citation10 Similarly, although acinar cell differentiation begins at this time as well, newly-formed acinar cells are not post-mitotic, and in fact are among the most proliferative cells in the embryonic pancreas.Citation11,Citation12
The massive wave of differentiation that occurs between E13.5 and birth in the mouse embryonic pancreas is termed the “secondary transition,” so-called to distinguish it from the “primary transition” that occurs during organ specification, when low levels of pancreas- specific gene products are first detectable.Citation13 The pancreas initially arises from separate dorsal and ventral strips of foregut endoderm, located between the prospective stomach and duodenum and, in the case of the ventral pancreas, adjacent to the forming liver (). During specification, the pancreatic endoderm can be distinguished by its expression of several transcription factors, including Pdx1/Ipf1, which is absolutely required for growth and differentiation of the organ.Citation14,Citation15 Several gain-of-function experiments, including the first to examine Wnt signaling in the pancreas,Citation16 indicate an inhibitory role for this pathway in pancreas specification.
Heller et al.Citation16 were the first to document expression of Wnts and their Frizzled receptors in the developing pancreatic epithelium and surrounding mesenchyme. This group and others have shown that Wnt2b, Wnt4, Wnt5a and Wnt7b are prominently expressed in the developing pancreas;Citation16,Citation17 with the exception of Wnt-5/Wnt5a, noted above, no pancreas defects have yet been described in any Wnt gene mutants. Heller et al.,Citation16 additionally showed that transgenic expression of Wnt1 under control of the Pdx1 promoter, i.e., from the onset of pancreas specification, results in almost complete agenesis of the organ. Precisely what goes wrong in these organs was not determined, but later work by Hebrok and colleagues showed that expression of activated β-catenin in the early pancreas resulted in a hypoplastic organ lacking normal exocrine and endocrine tissue.Citation18 The pancreata of these mutants appeared to be mis-specified as they both downregulated Pdx1 expression and upregulated the hedgehog pathway, which is normally excluded from the pancreas but active elsewhere in the gut. Similarly, global elevation of Wnt signaling in zebrafish, through knockdown of the β-catenin inhibitor Apc (), completely abolishes pancreas development.Citation19 Recent work in Xenopus, by Zorn and colleagues, confirms that repression of canonical Wnt signaling in the foregut is essential for both liver and pancreas specification.Citation20
Together, these studies indicate that Wnt pathway hyperactivation prevents normal pancreas development, likely by inhibiting specification of the organ. In Xenopus, activated β-catenin also causes ectopic intestinal marker expression in the foregut, and blocking β-catenin/TCF function causes at least partial conversion of intestine into liver and pancreas,Citation20 suggesting that canonical Wnt signaling normally acts to specify hindgut fates in the developing endoderm. It is unknown whether a similar respecification to intestine occurs in the mouse gain-of-function studies, or whether loss of Wnt function in the mouse would produce the reciprocal conversion of hindgut to foregut. Interestingly, the timing of the Xenopus and mouse experiments appear to be quite different. ZornCitation20 dissected the critical period during which Wnt pathway manipulation could affect Xenopus pancreas development, showing that it included only gastrula and early somite-forming stages (embryonic stages 11–25, perhaps equivalent to mouse E7.0–E8.5), well before initiation of Pdx1 (also called Xlhbox8 in Xenopus) expression at stage 33.Citation21 The mouse experiments involved Wnt activation after the onset of Pdx1 expression,Citation16,Citation18 at which point Xenopus pancreas specification is refractory to inhibition by Wnt/β-catenin. It is unknown whether these differences reflect quirks of experimental design or a real mechanistic difference between the species. In any event, further work on the role of Wnt/β-catenin signaling in pancreas specification should produce important insights into this process.
Wnt Signals are Essential for Expansion of Pancreatic Epithelium
Following specification, the pancreatic primordia expand into the surrounding mesenchyme, forming paired dorsal and ventral buds by E10.5 in the mouse, and then initiate rapid proliferation and branching morphogenesis. Although early Wnt/β-catenin signaling appears to inhibit pancreatic specification, it is later essential for this growth phase.Citation11,Citation17,Citation22,Citation23 One line of evidence for this comes from a study by Papadopoulou and Edlund, who established transgenic mice expressing a soluble, dominant-negative Frizzled receptor under control of the Pdx1 promoter (Pdx1-Frz8CRD).Citation17 The pancreata of these mice are specified and form buds normally, yet between E11.5 and E15.5 exhibit reduced proliferation. The result is severe pancreatic hypoplasia that affects both endocrine and exocrine cells, without impairing survival or differentiation.Citation17
Is this effect due to inhibition of canonical or non-canonical Wnt signaling? A hallmark of canonical pathway activation is accumulation of unphosphorylated β-catenin protein, which transduces the signal to the nucleus.Citation24 The Edlund study showed that the pancreatic epithelium stains for unphosphorylated β-catenin at E13.5 (a finding confirmed by independent studies that showed unphosphorylated β-catenin in the pancreas from E11.5–E15.5Citation11,Citation23), and that this staining is reduced in Pdx1-Frz8CRD mice.Citation17 This suggests that the endogenous canonical pathway is active during organ growth, and dependent on whatever Wnts are blocked by Frz8CRD. Nonetheless, staining for unphosphorylated β-catenin is not abolished in these mice, suggesting that Frz8CRD expression does not block all Wnt signaling in the pancreas. It also bears noting that this study used stable transgenic mouse lines, which de facto must have been viable; if complete Wnt inhibition in the pancreas were to cause lethality (as found by Lowy and colleagues,Citation23 discussed below), such lines could not have been established. Pdx1-Frz8CRD mice may thus represent Wnt/β-catenin hypomorphs, rather than nulls.
A more complete loss of Wnt/β-catenin signaling should be achievable by conditional deletion of the β-catenin gene, Ctnnb1, yet the lack of a “consensus phenotype” for pancreas-specific β-catenin knockout mice illustrates several pitfalls inherent to conditional knockout studies. Three investigators have attempted to delete β-catenin in the early pancreas, using two separate conditional (“floxed”) alleles of Ctnnb1 and three independent Pdx1-Cre deletor mice, and obtained only partially-overlapping results. A study by Grapin-Botton and colleagues found little or no reduction in organ size in their conditional knockouts, but a small decrease in islet mass and a pancreatitis-like phenotype in late development.Citation22 A study by Melton and colleagues found a dramatic reduction in pancreas size, even more severe than that seen by Edlund, but specifically affecting the exocrine pancreas.Citation11 A study by Lowy and colleagues demonstrated a generally similar exocrine hypoplasia phenotype, along with decreased survival of knockouts after birth, and fibrotic destruction of residual exocrine tissue in surviving adults.Citation23
The different phenotypes of these mice are likely explained by the different Pdx1-Cre transgenic lines used: Grapin-Botton and Lowy obtained dramatically different results with an identical floxed Ctnnb1 allele,Citation22,Citation23 while Melton used another floxed alleleCitation11 and obtained results similar to Lowy. Work by Hebrok and colleagues showed that the Melton deletor line, Pdx1-Creearly, catalyzes widespread recombination in the pancreas by E10.5; that used by Lowy is reportedly similar.Citation18,Citation23 By contrast, the line used by Grapin-Botton, Pdx1-Crelate, produces mosaic recombination only from E11.5 onwards.Citation18 Melton and Lowy observed widely decreased proliferation in their conditional knockout pancreata, consistent with the results of Edlund and indicating that Wnt/β-catenin signaling is essential for efficient proliferation of pancreatic progenitors.Citation11,Citation17,Citation23 The strong requirement for β-catenin in proliferation implies that cells escaping Ctnnb1 deletion, in conditional knockouts, will exhibit a growth advantage and potentially reconstitute the organ. Indeed, careful analysis by Grapin-Botton and colleagues showed that knockout cells are less proliferative than cells in the same organ that retain wildtype β-catenin, and that knockout cells are progressively replaced by wildtype during organ expansion.Citation22
As β-catenin also binds to E-cadherin at cell-cell junctions, its deletion may produce phenotypes independent of roles in Wnt signaling. Nonetheless, β-catenin deficient pancreatic epithelial cells do not exhibit obvious defects in intercellular adhesion,Citation11 and loss of β-catenin leads to decreased expression of two LEF/TCF target genes, cMycCitation11 and NMyc,Citation23 that are themselves important for proliferation in other tissues. Gain-of-function studies further support the conclusion that Wnt/β-catenin signaling promotes proliferation in the pancreas: the Hebrok group used Pdx1-Crelate to drive expression of activated β-catenin in the pancreas, and observed increased postnatal organ size, largely due to increased exocrine cell numbers.Citation18 Also using Pdx1-Crelate, Herrera and colleaguesCitation25 recently deleted Apc, which encodes an inhibitor of canonical Wnt signaling (), and observed similar exocrine hyperplasia. Importantly, this hyperplasia correlated with, and required, cMyc upregulation, confirming the importance of this β-catenin/TCF target gene in pancreas expansion.Citation25
β-catenin Promotes Differentiation and Maintenance of Exocrine Acinar Cells
As noted above, the Melton and Lowy conditional knockout models revealed that acinar cell numbers were especially sensitive to loss of β-catenin, and that endocrine development proceeded essentially normally in conditional knockouts.Citation11,Citation23 One possible explanation for this result is that Wnt/β-catenin signaling specifically regulates proliferation of already-differentiated acinar cells; as noted above, embryonic acinar cells are highly mitotic,Citation11,Citation12 and a specific blockade to their expansion could possibly account for the gross hypoplasia seen in all three conditional knockout models. Nonetheless, both of these groups looked for the appearance of β-catenin-deficient acinar cells and acinar precursors at early stages of the secondary transition (E12.5–E13.5) and were unable to find them, suggesting that β-catenin-deficient progenitor cells are unable to assume an acinar fate; by contrast, knockout cells readily contribute to islet and ductal lineages.Citation11,Citation23 Interestingly, an independent study performed in the embryonic chick found that activated β-catenin could prevent endocrine differentiation induced by misexpression of the bHLH gene Neurogenin3,Citation26 supporting the idea that islet development does not require canonical Wnt signaling, and may in fact be inhibited by the pathway ().
It should be noted that the Lowy group analyzed their knock-outs by microarray and RT-PCR, and found that mutants showed a transient spike of expression, higher than wildtype, of several acinar enzyme genes.Citation23 This occurred at E14.5, before the organ exhibited obvious hypoplasia, and was rapidly followed by almost complete loss of expression of these same genes. This loss of expression was also seen by immunostaining, whereas the transient upregulation could not be detected at the protein level. This result requires further investigation, but would be consistent with a model where Wnt/β-catenin signaling promotes proliferation of acinar precursor cells while inhibiting their differentiation, so that blocking the pathway triggers premature differentiation.Citation23 The Melton study, by contrast, suggested that loss of β-catenin completely prevents entry into the acinar lineage, leaving open the question of what becomes of those cells that would have formed acini in a wildtype background. A recent lineage-tracing study shows that cells expressing acinar markers in the early pancreas (roughly E12–E14) actually represent multipotent progenitors of both endocrine and exocrine lineages;Citation12 it is tempting to hypothesize that Wnt/β-catenin signaling coordinates the expansion and acinar differentiation of these cells.
Regardless of whether β-catenin specifically regulates acinar differentiation, the knockout studies are all consistent with a role for canonical Wnt signaling in proliferation of mature acinar cells.Citation11,Citation22,Citation23 The gain-of-function studiesCitation18,Citation25 also support such a role, as do several older studies by Clarke and colleagues in which spontaneous loss of Apc (in a heterozygous background) was found to promote acinar cell carcinomas.Citation27,Citation28 Mutational activation of the Wnt/β-catenin pathway, through loss of APC (a classic tumor suppressor gene) or activation of CTNNB1, occurs frequently in colon and several other human cancers, but appears to be very rare in pancreatic cancer.Citation7,Citation29 Among the few exceptions, consistent with the Clarke studies, are acinar cell carcinomas, which are rare in humans but a subset of which exhibit mutations in APC or CTNNB1.Citation30 Additional loss-of-function studies are needed to determine if Wnt/β-catenin signaling actually regulates acinar proliferation in the adult, or if its activation in these rare tumors reflects non-physiological effects from reactivating a pathway normally restricted to embryogenesis.
The Grapin-Botton and Lowy β-catenin knockouts also exhibited a pancreatitis-like phenotype in late fetal and postnatal life, respectively, including apparent destruction of residual acinar cells.Citation22,Citation23 Although this might suggest a requirement for β-catenin in survival of differentiated acinar cells, the Melton group specifically deleted β-catenin from acinar cells of adult mice, and observed neither cell death nor pancreatitis.Citation11 This discrepancy may reflect a timing difference: late fetal and neonatal acini are likely to be more proliferative than those of the adult, and therefore may be particularly sensitive to loss of β-catenin (although none of these groups found evidence for apoptosis of β-catenin-deficient cellsCitation11,Citation22,Citation23). Alternatively, some essential function of duct cells could depend on β-catenin, as ductal defects are known to produce similar pancreatitis-like phenotypes in, for example, cystic fibrosis.Citation31 As with other aspects of Wnt/β-catenin signaling in the pancreas, the question of its role in maintenance of normal exocrine function warrants further study.
A Puzzle Still Missing Some Pieces
Despite some phenotypic ambiguities (e.g., moderately reduced islet cell mass in the Edlund and Grapin-Botton loss-of-function studies,Citation17,Citation22 vs. apparently normal islet mass in the work of Melton and LowyCitation11,Citation23), the foregoing studies appear to tell a tidy mechanistic tale, highly congruous with other organ systems. (1) Blocking Wnts in the extracellular milieu causes reduced proliferation.Citation17 (2) Loss of β-catenin also causes reduced proliferation, and activated β-catenin causes hyperplasia, especially of acinar cells.Citation11,Citation18,Citation22,Citation23,Citation25 (3) cMyc and NMyc, known mitogenic transcription factors and β-catenin/TCF target genes, require β-catenin for their expression in the developing pancreas.Citation11,Citation23 (4) Hyperplasia driven by activated β-catenin requires cMyc.Citation25
A major fly in the ointment, however, was introduced by the Grapin-Botton study, which looked at Wnt reporter gene expression in the pancreas.Citation22 Several mouse lines have been developed in which TCF-dependent transcription drives expression of a LacZ transgene, the most reliable of which appears to be the Axin2LacZ mouse.Citation32 Axin2 seems to be activated by β-catenin/TCF in all Wnt-responsive cell types, and the LacZ knock-in thus serves as a readout of canonical Wnt activity. Importantly, Grapin-Botton and colleagues found Axin2LacZ expression neither in developing acinar or duct cells, where Wnt/β-catenin signaling appear to be important, but solely in developing islets, where the pathway appears to be almost completely dispensable!Citation22
Several potential explanations suggest themselves, none of which will be trivial to rule out: (1) Canonical Wnt/β-catenin/TCF signaling is active in developing acinar cells, but is not detectable with Axin2LacZ. This could occur if, for example, the absolute levels of endogenously active β-catenin are insufficient to drive Axin2LacZ expression, but sufficient to drive other target genes such as cMyc. (2) Wnt/β-catenin signaling is active in developing acini, but β-catenin acts through nuclear partners other than LEF/TCF. Such non-TCF partners have been identified, and at least one of these, Nr5a2/Lrh1, is expressed in the embryonic pancreas.Citation33,Citation34 (3) β-catenin plays a Wnt-independent role in proliferation (e.g., via subtle effects on cell adhesion), and the hypoplasia caused by Frz8CRD misexpression reflects a separate role for non-canonical Wnt signaling. In this scenario, hyperplasia driven by activated β-catenin would simply reflect the oncogenic activity of this mutant protein, introduced into a context in which β-catenin/TCF activity is normally silent. Testing these models will be a challenge, but essential to unraveling the question of how, precisely, Wnt/β-catenin signaling acts in the pancreas.
A Recent Wrinkle: Wnts, β-catenin and TCF7L2 in Insulin Secretion and Diabetes
As noted above, none of the loss-of-function mouse studies have found any role for Wnt signaling in maintenance or function of adult islets. Indeed, Edlund, Grapin-Botton and Melton explicitly looked for and did not find dysregulation of glucose homeostasis in their loss-of-function models.Citation11,Citation17,Citation23 By contrast, Sakai and colleaguesCitation35 found that mice heterozygous or homozygous null for Lrp5, encoding a Wnt co-receptor specific to the canonical pathway (), exhibit impaired glucose homeostasis and glucose-stimulated insulin secretion (GSIS). This group also showed that Wnt protein treatment in vitro enhanced GSIS by isolated islets, in an Lrp5-dependent manner.Citation35 Furthermore, Kim and colleaguesCitation36 recently demonstrated that postnatal activation of β-catenin, specifically in β-cells, resulted in enhanced proliferation and increased β-cell mass, and that overexpression of the pathway antagonist Axin caused the opposite phenotype.Citation36 Although these results are somewhat inconsistent with those discussed earlier (in particular, the Hebrok study showed that β-catenin activation by Pdx1-Crelate, which occurs in the majority of β-cells, does not cause β-cell hyperplasiaCitation18), they collectively suggest a possible role for Wnt/β-catenin signaling in islet function, a hypothesis supported by the endocrine-specific expression of Axin2LacZ.Citation22
This hypothesis has recently been invigorated by human genetic studies that implicate the LEF/TCF family member TCF7L2 (also called TCF4) in type II diabetes and β-cell dysfunction. As originally shown by Grant and colleagues,Citation37 and confirmed by many independent studies,Citation38 a common polymorphism in an intron of TCF7L2 is associated with a roughly 1.4-fold increased risk of developing type II diabetes (T2D). T2D is commonly associated with peripheral insulin resistance, but β-cell function is also compromised in this disease, and carriers of the TCF7L2 risk allele exhibit decreased glucose-stimulated insulin secretion (GSIS).Citation38 As mice null for Tcf7l2 fail to develop enteroendocrine cells,Citation39 and enteroendocrine cells indirectly promote GSIS, the human phenotype may be an indirect result of impaired enteroendocrine development or function.Citation38 The risk allele could also affect pancreatic function directly, as the gene is expressed both in the embryonic pancreasCitation11 and in adult human islets.Citation40 Indeed, Lyssenko and colleaguesCitation40 recently demonstrated that islets of risk allele carriers express higher levels of TCF7L2 mRNA than controls, and that forced overexpression of TCF7L2 in cultured human islets resulted in impaired GSIS. It remains unclear whether this situation mimics activation of the Wnt pathway or inhibition (e.g., if higher levels of TCF7L2 protein, in the absence of activated β-catenin, cause target gene repression). What is beyond a doubt, given the extraordinary interest now focused on TCF7L2, is that further studies of its mode and site of action will be soon forthcoming.
Conclusions
The recent interest in Wnt/β-catenin function in pancreatic development began with the assumption that this pathway, so important in other organ systems, would almost certainly play a role in the pancreas. Although this assumption has been amply confirmed, the workings of the pathway in the pancreas may differ subtly from those elsewhere. It is clear that Wnt/β-catenin signaling in the early endoderm inhibits pancreas specification, suggesting that blocking the pathway may be necessary to efficiently derive β-cells from stem cells in vitro. Later in fetal life, the pathway appears to promote proliferation and/or differentiation of acinar cells, and manipulating this process may be useful in alleviating diseases of exocrine pathology such as acute and chronic pancreatitis. Finally, independent studies in mouse and humans implicate Wnt/β-catenin signaling in maintaining adult islet function, although here the fine details are blurriest. Future work on these areas will surely bring these details into sharper focus, and help translate the basic science of pancreatic organogenesis into clinical practice.
Figures and Tables
Figure 1 Canonical Wnt/β-catenin signaling. In the absence of Wnt signals (left), cytoplasmic β-catenin protein (β) is bound by a “destruction complex” of proteins that include APC and Axin, both of which are encoded by tumor suppressor genes, and the serine/threonine-kinase GSK3. Phosphorylation of β-catenin by GSK3 targets it for ubiquitination and degradation. Wnt protein binding to the Frizzled and LRP5/6 co-receptors (right) activates cytoplasmic Disheveled protein (Dsh), which disrupts the destruction complex, possibly by recruitment of Axin, thus permitting accumulation of unphosphorylated β-catenin. As β-catenin levels increase, a fraction of the cytoplasmic pool translocates to the nucleus and associates with DNA-binding proteins of the TCF family, displacing Groucho repressor proteins (Gro, left) and activating expression of downstream target genes such as cMyc. In epithelial cells, β-catenin also associates with cadherin proteins at the cell surface (not shown here), potentially stabilizing cell-cell interactions.Citation6
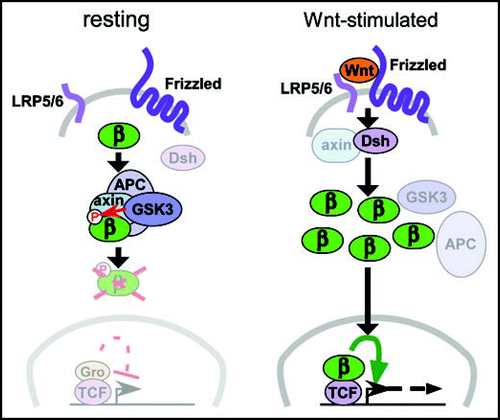
Figure 2 Stages of pancreatic organogenesis. The dorsal and ventral pancreata (left, dp and vp) are specified in the foregut between embryonic days 8.5–9.5 (E8.5–E9.5) in the mouse, near the forming liver (li). Over the next several days, the dorsal and ventral primordia initiate a program of rapid growth and branching morphogenesis (center), eventually producing lobular organs that lie adjacent to the stomach (st) and intestine (in), respectively. A massive wave of differentiation occurs coincident with the later portion of the growth phase (right), producing a branched ductal network (du) terminated by digestive enzyme-secreting acinar cells (ac), with endocrine islets (is) embedded in the interstitial space. As described in the text, Wnt/β-catenin signaling appears to regulate multiple steps in this process, including inhibition of pancreas specification, promotion of growth and acinar differentiation, and possibly inhibition of endocrine development.
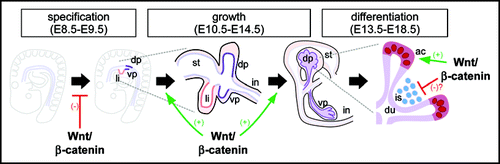
Table 1 Experimental evidence of Wnt/β-catenin function in pancreas development
Acknowledgements
I am grateful to my laboratory and Kristen Kwan for helpful discussions. This work is supported by NIH grant R01DK075072.
References
- Collombat P, Hecksher-Sorensen J, Serup P, Mansouri A. Specifying pancreatic endocrine cell fates. Mech Dev 2006; 123:501 - 512
- Murtaugh LC. Pancreas and beta-cell development: from the actual to the possible. Development 2007; 134:427 - 438
- Golosow N, Grobstein C. Epitheliomesenchymal interaction in pancreatic morphogenesis. Dev Biol 1962; 4:242 - 255
- Wessells NK, Cohen JH. Early pancreatic organogenesis: morphogenesis, tissue interactions and mass effects. Dev Biol 1967; 15:237 - 270
- Spence JR, Wells JM. Translational embryology: Using embryonic principles to generate pancreatic endocrine cells from embryonic stem cells. Dev Dyn 2007; 236:3218 - 3227
- Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004; 303:1483 - 1487
- Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev 2005; 19:877 - 890
- Veeman MT, Axelrod JD, Moon RT. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev Cell 2003; 5:367 - 377
- Kim HJ, Schleiffarth JR, Jessurun J, Sumanas S, Petryk A, Lin S, Ekker SC. Wnt5 signaling in vertebrate pancreas development. BMC Biol 2005; 3:23
- Herrera PL, Huarte J, Sanvito F, Meda P, Orci L, Vassalli JD. Embryogenesis of the murine endocrine pancreas; early expression of pancreatic polypeptide gene. Development 1991; 113:1257 - 1265
- Murtaugh LC, Law AC, Dor Y, Melton DA. Beta-Catenin is essential for pancreatic acinar but not islet development. Development 2005; 132:4663 - 4674
- Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, Melton DA. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell 2007; 13:103 - 114
- Pictet R, Rutter WJ. Steiner D, Freinkel N. Development of the embryonic endocrine pancreas. Handbook of Physiology, Section 7 1972; Baltimore Williams & Williams 25 - 66
- Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 1996; 122:983 - 995
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994; 371:606 - 609
- Heller RS, Dichmann DS, Jensen J, Miller C, Wong G, Madsen OD, Serup P. Expression patterns of Wnts, Frizzleds, sFRPs, and misexpression in transgenic mice suggesting a role for Wnts in pancreas and foregut pattern formation. Dev Dyn 2002; 225:260 - 270
- Papadopoulou S, Edlund H. Attenuated Wnt signaling perturbs pancreatic growth but not pancreatic function. Diabetes 2005; 54:2844 - 2851
- Heiser PW, Lau J, Taketo MM, Herrera PL, Hebrok M. Stabilization of {beta}-catenin impacts pancreas growth. Development 2006; 133:2023 - 2032
- Nadauld LD, Sandoval IT, Chidester S, Yost HJ, Jones DA. Adenomatous polyposis coli control of retinoic acid biosynthesis is critical for zebrafish intestinal development and differentiation. J Biol Chem 2004; 279:51581 - 51589
- McLin VA, Rankin SA, Zorn AM. Repression of Wnt/beta-catenin signaling in the anterior endoderm is essential for liver and pancreas development. Development 2007; 134:2207 - 2217
- Wright CV, Schnegelsberg P, De Robertis EM. XlHbox 8: a novel Xenopus homeo protein restricted to a narrow band of endoderm. Development 1989; 105:787 - 794
- Dessimoz J, Bonnard C, Huelsken J, Grapin-Botton A. Pancreas-specific deletion of beta-catenin reveals Wnt-dependent and Wnt-independent functions during development. Curr Biol 2005; 15:1677 - 1683
- Wells JM, Esni F, Boivin GP, Aronow BJ, Stuart W, Combs C, Sklenka A, Leach SD, Lowy AM. Wnt/beta-catenin signaling is required for development of the exocrine pancreas. BMC Dev Biol 2007; 7:4
- van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wnt signaling controls the phosphorylation status of beta-catenin. J Biol Chem 2002; 277:17901 - 17905
- Strom A, Bonal C, Ashery-Padan R, Hashimoto N, Campos ML, Trumpp A, Noda T, Kido Y, Real FX, Thorel F, Herrera PL. Unique mechanisms of growth regulation and tumor suppression upon Apc inactivation in the pancreas. Development 2007; 134:2719 - 2725
- Pedersen AH, Heller RS. A possible role for the canonical Wnt pathway in endocrine cell development in chicks. Biochem Biophys Res Commun 2005; 333:961 - 968
- Clarke AR, Cummings MC, Harrison DJ. Interaction between murine germline mutations in p53 and APC predisposes to pancreatic neoplasia but not to increased intestinal malignancy. Oncogene 1995; 11:1913 - 1920
- Kongkanuntn R, Bubb VJ, Sansom OJ, Wyllie AH, Harrison DJ, Clarke AR. Dysregulated expression of beta-catenin marks early neoplastic change in Apc mutant mice, but not all lesions arising in Msh2 deficient mice. Oncogene 1999; 18:7219 - 7225
- Gerdes B, Ramaswamy A, Simon B, Pietsch T, Bastian D, Kersting M, Moll R, Bartsch D. Analysis of beta-catenin gene mutations in pancreatic tumors. Digestion 1999; 60:544 - 548
- Abraham SC, Wu TT, Hruban RH, Lee JH, Yeo CJ, Conlon K, Brennan M, Cameron JL, Klimstra DS. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol 2002; 160:953 - 962
- McIntosh I, Cutting GR. Cystic fibrosis transmembrane conductance regulator and the etiology and pathogenesis of cystic fibrosis. FASEB J 1992; 6:2775 - 2782
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol 2002; 22:1184 - 1193
- Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, Schoonjans K. Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Mol Cell 2004; 15:499 - 509
- Annicotte JS, Fayard E, Swift GH, Selander L, Edlund H, Tanaka T, Kodama T, Schoonjans K, Auwerx J. Pancreatic-duodenal homeobox 1 regulates expression of liver receptor homolog 1 during pancreas development. Mol Cell Biol 2003; 23:6713 - 6724
- Fujino T, Asaba H, Kang MJ, Ikeda Y, Sone H, Takada S, Kim DH, Ioka RX, Ono M, Tomoyori H, Okubo M, Murase T, Kamataki A, Yamamoto J, Magoori K, Takahashi S, Miyamoto Y, Oishi H, Nose M, Okazaki M, Usui S, Imaizumi K, Yanagisawa M, Sakai J, Yamamoto TT. Low-density lipoprotein receptor-related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proc Natl Acad Sci USA 2003; 100:229 - 234
- Rulifson IC, Karnik SK, Heiser PW, ten Berge D, Chen H, Gu X, Taketo MM, Nusse R, Hebrok M, Kim SK. Wnt signaling regulates pancreatic beta cell proliferation. Proc Natl Acad Sci USA 2007; 104:6247 - 6252
- Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 2006; 38:320 - 323
- Florez JC. The new type 2 diabetes gene TCF7L2. Curr Opin Clin Nutr Metab Care 2007; 10:391 - 396
- Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 1998; 19:379 - 383
- Lyssenko V, Lupi R, Marchetti P, Del Guerra S, Orho Melander M, Almgren P, Sjogren M, Ling C, Eriksson KF, Lethagen AL, Mancarella R, Berglund G, Tuomi T, Nilsson P, Del Prato S, Groop L. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest 2007; 117:2155 - 2163