Abstract
Tracing cell movements in vivo yields important clues to organogenesis, yet it has been challenging to accurately and reproducibly fluorescently mark single and small groups of cells to build a picture of tissue assembly. In the early embryo, the small size (hundreds of cells) of progenitor cell regions has made it easier to identify and selectively mark superficially located cells by glass needle injection. However, during early organogenesis, subregions of interest may be several millions of cells in volume located deeper within the embryo requiring an alternative approach. Here, we combined (confocal and 2-photon) photoactivation cell labeling and multi-position, multi-time imaging to trace single cell and small subgroups of cells in the developing brain and spinal cord. We compared the photostability and photoefficiency of a photoswitchable fluorescent protein, PSCFP2, with a novel nuclear localized H2B-PSCFP2 protein. We showed that both fluorescent proteins have similar photophysical properties and H2B-PSCFP2 is more effective in single cell identification in dense tissue. To accurately and reproducibly fluorescently trace subregions of cells in a 3D tissue volume, we developed a protocol for multi-position photoactivation and multi-time acquisition in the chick spinal cord in up to 8 tissue sections. We applied our techniques to address the formation of the sympathetic ganglia, a major component of the autonomic nervous system, and showed there are phenotypic differences between early and later emerging neural crest cells and their positions in the developing ganglia. Thus, targeted fluorescent cell marking by confocal or 2-photon multi-position photoactivation and multi-time acquisition offer a more efficient, less invasive technique to trace cell movements in large regions of interest and move us closer towards mapping the cellular events of organogenesis.
Introduction
Resolving the behavior, movement and proliferative activity of every cell within growing tissue holds significant promise to answer complex questions about organ assembly. As regions of interest grow in size from small subgroups of embryonic progenitor cells to developing organs such as the brain and peripheral nervous system, the number of cells to trace becomes significant. In the early mouse embryo, the inner cell mass of the blastocyst that gives rise to most of the fetus consists of ∼100 cells,Citation1,Citation2 yet a developing hindbrain may contain millions of cells.Citation3,Citation4 Tracing cell lineage and cell behaviors in live tissue requires accurate fluorescent cell marking. Consistent results require data from large numbers of embryos and reproducible cell labeling. However, typical methods using focal dye injection and image analysis of one embryo or tissue section at a time can be very time consuming, difficult to reproduce and limited to cells and tissue accessible to glass needle. A new approach that could accurately label cells in a less invasive manner and automate the marking and tracking of multiple tissue sections or embryos would significantly decrease the time and effort to trace cell behaviors during larger morphogenetic events.
Cell labeling techniques have ranged from gold particlesCitation5 to lipophilic dyes microinjected or iontophoretically delivered into cells and tissueCitation6,Citation7 and single cell electroporation of fluorescent protein constructs.Citation8 However, these techniques deliver dyes by glass needle microinjection, and are fairly limited to superficially located cells or cells deeper in tissue but visually distinguishable due to sparsely populated tissue. The recent explosion of photoactivatable fluorescent proteins has helped to overcome this roadblock and offers a less invasive cell marking approach.Citation9 When photoactivation of GFP or its variants is combined with 2-photon microscopy, this allows for optical selection of single cells deep within an embryo or tissue, since 2-photon excitation can be confined to the focal plane of interest. There are now several photoactivatable fluorescent proteins with a wide range of excitation and emission schemes that offer photoactivation by 405 nm excitationCitation10 or 488 nm excitationCitation11 and possess distinct photoefficiencies and photostability for various uses in embryos.Citation13,Citation14 Thus, photoactivation has become an important tool for accurate cell tracing and has allowed the analysis of cell behaviors in vitroCitation15 and in vivo during morphogenetic events in avian neural crest migration,Citation16,Citation17 Xenopus developmentCitation18 and Drosophila mesoderm migration.Citation19
How do we harness advances in photoactivation cell marking and automated microscopy to develop techniques to trace events in organogenesis? As an example, we consider the assembly of the peripheral nervous system (PNS). The PNS, including sensory, sympathetic, enteric and parasympathetic divisions of the embryo is entirely derived from a multipotent, highly invasive cell population in the embryo called the neural crest.Citation20,Citation21 An important question in development is how a common pool of progenitor neural crest cells that follow stereotypical trunk migratory pathways become patterned into many diverse components of the PNS. Previous lineage studies, using vital dyes to label individual premigratory trunk neural crest cells have revealed that cells follow two distinct migratory pathwaysCitation12,Citation22,Citation23 and give rise to diverse derivatives.Citation24,Citation25 Though these studies have been extremely insightful in understanding neural crest cell lineage, vital dyes are typically applied to only superficially located cells at a single time point, making it difficult to trace entire cell subpopulations and the relationship between their order of emergence and position within a target site. This is especially challenging for trunk neural crest cells that may travel deep within the embryo. With the advent of new photoactivatable proteins localized to the cytoplasm, in particular KikGR,Citation14,Citation17,Citation26 we can selectively label small subpopulations of pre-migratory neural crest cells, thus overcoming the cell labeling roadblocks and further address PNS development. Additionally, advances in optical imaging (hardware and software) now allow multi-position (mark and find) and multi-time image acquisition for widefield and confocal microscopy.Citation27,Citation28 Thus, the timing is right for development of automated cell labeling and tracking methods to study cell behaviors during organogenesis.
Here, we have determined the optical parameters and constraints of confocal and 2-photon in vivo photoactivation using a photoswitchable fluorescent protein, PSCFP2. We compared the in vivo photophysical properties of PSCFP2 with a novel nuclear localized version, H2B-PSCFP2. To develop tools for cell tracing during organogenesis, we combined the strengths of photoactivation cell labeling with multi-time, multi-position image acquisition. We then applied this approach to study cell behaviors during chick spinal cord and sympathetic ganglia development (in a quasi-automated manner) in multiple tissue sections in a throughput manner. We specifically addressed whether different waves of migratory trunk neural crest cells selectively give rise to neural or glial populations within the developing sympathetic ganglia. This approach allowed us to overcome limitations of less invasive cell labeling and large-scale cell tracing towards monitoring morphogenetic events during organogenesis.
Results
In vivo single cell photoactivation: separating the individuals from the herd.
The use of photoactivation to trace single cell behaviors during organ development will require deep tissue, confined (local) excitation and observation. This suggests that typical confocal photoactivation must adapt to using 2-photon microscopy in order to overcome out-of-plane photoactivation, an ability to selectively mark a cell(s) deep within a thick tissue section or embryo, and in densely populated cells. We have previously shown that in comparison to other popular photoactivatable fluorescent proteins (PAGFP, KikGR, and Kaede), PSCFP2 was resistant to photobleaching and remained visually distinguishable longer, up to 48 hours, after 405 nm confocal laser photoactivation in cells of the avian embryo hindbrain.Citation14 Here, we determined the optical parameters and constraints of 2-photon in vivo photoactivation () using the embryonic avian brain as our model system and a photoswitchable cyan fluorescent protein, PSCFP2.Citation29 The early brain consists of a pseudo-stratified cellular organization within a developing neural tube of densely populated cells. Each cell within the neural tube extends cellular processes to contact both the apical and basal sides of the neural tube. This made it nearly impossible to visually distinguish an individual fluorescently labeled cell by typical widefield or confocal microscopy due to the overlap of cellular processes and tissue density. We found that in ovo 405 nm confocal laser scanning excitation of PSCFP2-labeled neural tube cells caused out of plane photoactivation of neighboring cells due to the cone of the focused laser beam (). After photoactivation with a continuous wave laser, X—Z scanning clearly revealed the out-of-plane photoactivation (, arrowhead; B′, bottom cell). Using a 2-photon laser, we were able to selectively photoactivate a single neural tube cell at 770 nm excitation without out of plane photoactivation of neighboring cells (, arrow; B′, top cell). An individual 2-photon photoactivated cell within the neural tube was clearly identified by its nucleus and long, thin extended processes (. arrow; B′, top cell). Thus, by using 2-photon photoactivation we were able to confine fluorescent marking to single cells and limit out of plane photoactivation.
To selectively mark single cells within the densely populated avian hindbrain with increased accuracy for identification and cell tracking, we needed to develop a localized fluorescent photoactivatable protein. To accomplish this, we developed an H2B-PSCFP2 plasmid that localized the fluorescent protein to the cell nucleus and more precisely defined (identified) single cell photoactivation (). To test the applicability and in vivo photophysical properties of H2B-PSCFP2, we directly targeted cell nuclei within the neural tube that had been labeled with standard chick electroporation delivery techniques.Citation30 By targeting only the nuclei, we found that H2B-PSCFP2 photoactivation with 405 nm confocal laser light removed the possibility of accidental photoactivation to neighboring cells by indirect laser light in cells that were sparsely labeled ( and F). Measurements of the photoefficiency of the PSCFP2 unbound and H2B-PSCFP2 localized to the cell nucleus showed that the photophysical properties remained intact (). The photoefficiency (a measure of photaostability and efficiency of photoactivation) showed a steady increase in fluorescence intensity in cells after continuous excitation at 405 nm (). Thus, by utilizing a nuclear localized photoactivatable fluorescent protein, we were able to more selectively fluorescently mark and identify cells within dense tissue.
Automated cell tracing by photoactivation.
The developing spinal cord consists of many distinct tissue subregions of homogeneous progenitor cells that give rise to a specific neuronal architecture.Citation31 Axonal projections from the spinal cord to the periphery and between the spinal cord and brain complete the neural circuits of the peripheral nervous system.Citation32 Cell tracing of progenitors within the spinal cord would yield clues to the assembly of a complex neuroarchitecture. However, the 3D volume of the spinal cord for a particular axial level (e.g., forelimb, hindlimb, etc.) makes it challenging to perform traditional single embryo, single injection labeling and 2-photon imaging since the number of embryos required to build an accurate picture of this morphometric event becomes significantly large and the ventral portion of the spinal cord is too deep to image with a light microscope. For example, to trace the lineage of single cells within a 3D volume of the spinal cord between the fore- and hind-limbs of day 4 chick embryos would take a student approximately 13.8 yrs! (assuming a spinal cord of length 800 um, half-width of 60 um, height of 340 um [total volume = π × (half-height) × (half-width) × (length)], with each cell volume of 3,375 cubic um; 15 cells (embryos) marked per day and n = 10 at each position for redundancy). Thus, in order to build a detailed picture of cell behaviors within a subregion of the developing spinal cord in a reasonable time, we needed to develop an automated manner to fluorescently mark subgroups of cells within the spinal cord and monitor cell behaviors over time in tissue sections that could be rendered into a 3D volume.
To approach this, we electroporated PS-CFP2 into HH St.10 chick embryos to label one-side of the developing spinal cord. Embryos were re-incubated for 72 hours and we generated transverse sections from the volume between the fore- and hind-limbs. Using the spinal cord from successive transverse sections from the same embryo, we defined regions of interest (for photoactivation cell labeling) by measuring the average dorsoventral length and medioventral width of the spinal cord (). In a typical transverse section through the E4 chick trunk, we found the average spinal cord height (dorsal-ventral) to be 341.6 ± 30.1 um and average width (medial-lateral) to be 119.4 ± 9.5 um. We sectioned the trunk region of the embryo into 200 um rostral to caudal sections to cover the region between the fore- and hind-limbs. Typically, the number of sections averaged n = 6 from each embryo. The regions for photoactivation can vary depending on the cellular resolution needed, i.e., single or small groups of cells. We chose to trace groups of cells on one side of the spinal cord in approximately 50 um dorsal to ventral length increments, starting at the dorsal spinal cord and continuing ventrally to the floor plate (), as a potential application to map zones of neurogenesis. In each tissue section we manually recorded x–y position, rotated the field of view to align the spinal cord to vertical, set the z-stack settings, and marked the photoactivation regions of interest. We executed the multi-time macro to proceed with automated photoactivation at 405 nm excitation at each predefined region of interest and collected z-stack data in each slice. For a typical set of spinal cord sections the progressive regions of interest covered nearly one-side of the spinal cord (). We calculated the average time required to set up (align each tissue section and mark its location in a dish) and acquire each confocal image (). Interestingly, for n = 6 tissue sections, we calculated the average setup time to be 24 min (4 minutes per slice; ). The initial scan time (pre-photoactivation) was approximately 1 min (0.2 min/slice) and photoactivation time was approximately 7 min (1.2 min/slice). The z-sectioning through the slices was 3 minutes (0.5 min/slice). For a typical imaging session, our total setup and acquisition time for n = 6 sections was 35 min (). In this scenario, the setup was the most significant time requirement, with approximately 68% of the total time (). Thus, using automated multi-position photoactivation allowed us to perform cell marking on a large number of tissue sections in significantly less time (decreased by a factor of at least six) than with typical methods.
To extend our approach to multi-time acquisition for time-lapse imaging, we prepared the embryo and tissue slices in the same manner as mentioned above and drawn schematically (). After the multi-position photoactivation, a confocal z-stack of images was collected at set time points, for each tissue section (). This resulted in 4D (3D + time) data generated for multiple locations for on average 8–10 hrs (). Photoactivated cell movements were visually observed from time-lapse recordings, such as changes in cell direction, without loss in fluorescence (; lower, cells of interest marked by arrow and arrowhead), interkinetic nuclear migration and axonal projections (data not shown).
Towards cell tracing during organogenesis.
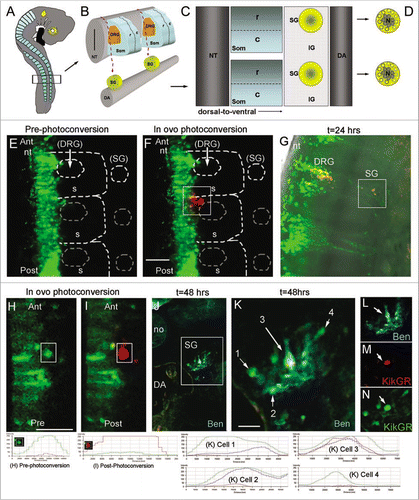
Sympathetic ganglia (SG) of the autonomic nervous system form deep within the embryo from a common pool of premigratory trunk neural crest cells (). Early emerging trunk neural crest cells travel long distances along a medio-to-ventral migratory pathwayCitation12,Citation22,Citation23 to the dorsal aorta and very early in their development sort into neural (core) and glial (perimeter) subpopulations.Citation33 It is still unclear how the sympathetic ganglia is assembled and how and when neural crest cells are influenced to sort into these divergent lineages. We used this model system with photoactivation to initially determine whether the spatial order of emergence from the trunk neural tube specified them to be a neural or glial cell. Our lab has previously used photoactivatable constructs to map certain waves of cranial neural crest cell migration and have now extended this to mapping single and small numbers of migrating cells.Citation17 We used KikGR for these experiments, as we previously determined it was one of the most photoefficient PFP, as measured by the time to reach maximum photoconversion.Citation14 This was due to the rapid exponential decrease of green fluorescence and increase in red fluorescence during the photoconversion process at 2% laser power, which is optimal for in ovo photoconversion at later stages after the heart has begun to beat. Premigratory trunk neural crest cells were electroporated in ovo (n = 7) with KikGR. When lead trunk neural crest cells emerged after 24 hours of electroporation of KikGR, we photoactivated, in ovo, the initial subgroup of neural crest cells, including cells that migrated into the rostral somite, but excluding cells in the neural tube (, F, H, I). After re-incubation to allow for SG development, we assessed the final location of the photoactivated cells both in whole mount and sectioned embryos. Photoactivated red cells were discernible from green, un-photoactivated, cells both visually and by analysis of the red to green fluorescence intensity ratio (measured by line scan intensities) immediately after photoactivation and after 24 and 48 hrs of re-incubation (, J). Photoactivated embryos, re-incubated for 48 hrs, were transverse sectioned and stained with a neural marker (Ben) to identify the core (neural population) of the SG (). We found that the majority, 87% (s.d. = 5.7, n = 20 cells) of photoactivated lead neural crest cells were found within the core of the ganglia, suggesting that the order of neural crest cell emergence predicted the cell position within the developing SG.
Discussion
Tracing and mapping single cells during organogenesis holds much importance for understanding complex cellular behavior and tissue assembly. Thus far, it has been technically difficult and time consuming to label single and small subgroups of cells in dense tissue and trace cells in large 3D volumes with current techniques. In this paper, we used a novel, nuclear localized H2B-PSCFP2 photoactivatable fluorescent protein that allowed us to label single cells in densely populated tissue (), thus overcoming the difficulty to selectively illuminate cells in dense tissue. We compared the photophysical properties of H2B-PSCFP2 and PSCFP2 and showed that H2B-PSCFP2 had comparable photo-stability and photoefficiency parameters, making it useful for cell tracing studies. To address large scale cell tracing in 3D tissue volumes, we developed a protocol for multi-position photoactivation and multi-time acquisition of multiple tissue slices cut from 3D volumes within whole chick embryos. This overcame significant time and effort involved in typical single embryo, single site microinjection studies. As an application of our techniques to the development of tissue structures and organs, we applied photoactivation to the formation of sympathetic ganglia of the peripheral nervous system, initially revealing that the order of trunk neural crest cell emergence from the neural tube may predict locations within sympathetic ganglia. This showed by example our efforts could be applied to address cell tracing during organogenesis.
The advantages of using a 2-photon laser for single cell photoactivation were specificity, depth of tissue, and out-of-plane photoactivation. However, 2-photon photoactivation had some disadvantages. The amount of power that a 2-photon laser had at 770 nm can be more than 50–100 times that compared to a 405 nm laser. This amount of power within the photoactivation wave-length was more likely to photobleach cells of interest than the excitation wavelength from prolonged 405 nm laser light exposure. The region of interest (ROI) to create a single photoactivated cell had to be within the cell of interest or multiple cells were photoactivated. We found that limiting tissue movement or drift during 2-photon excitation minimized error in single cell marking. Therefore, we suggest that 2-photon photoactivation holds significant advantages over single-photon for accurate single cell labeling, however, care should be taken to monitor photoefficiency and photobleaching during the cell marking process.
Alignment of tissue sections and marking of regions of interest were the most time consuming of the multi-position photoactivation and multi-time acquisition process (). With large tissue structures and homogeneous cell labeling, it was difficult to determine any prospective overlap of regions of interest between consecutive tissue sections. We anticipate this issue would become more problematic with increasing complexity of 3D tissue structures. We decided on regions of interest of an appropriate size (50 um∼60 um) to label small subgroups of cells within the spinal cord, however depending on the size desired, this could also complicate the issue of overlapping regions of interest. Further efforts to automate the tissue alignment and region of interest marking, perhaps by identification of tissue boundaries and calculation of non-overlapping regions of interest to cover a desired area or volume would significantly decrease the user interface and time to data acquisition.
Automated multi-position photoactivation and multi-time acquisition significantly decreased the time and effort for typical cell tracing studies. In typical E4 chick embryos, the spinal cord had an average dorsal-to-ventral height of ∼340 um and half width of ∼60 um. We subdivided the half spinal cord into 6–7 regions of interest, approximately 50 um in height and the half width of the spinal cord. The entire anterior-to-posterior length of the spinal cord between the fore- and hind-limbs was approximately 1,200 um (sectioned into 150–200 um widths); this translated into approximately 56 regions of interest. In each time-lapse imaging session, we were able to image approximately 6–7 tissue sections simultaneously, for up to 24 hrs (). To build a complete picture of cell behaviors of the E4 half spinal cord, we estimated it would take approximately 1.5 yrs (56 regions of interest and 10 time-lapse sessions per region of interest for redundancy, 1 time-lapse session per day; 560/365 days = 1.53 yrs). Thus, the multi-position acquisition of 7 regions of interest per time-lapse session would decrease the total time by a factor of 7 to approximately 80 days or ∼12 weeks. In terms of cost savings for microscopy time for this set of experiments, if each time-lapse session cost $960 (24 hrs at $40/hr), then multi-position acquisition would cost $76,800 versus $537,600 for the standard imaging approach or a savings of $460,800.
Tracing photoactivated cells that migrated deep within the intact embryo posed an additional layer of complexity. After 24 hours in culture, KikGR-photoactivated cells were easy to visually identify, as the protein localized to the cytoplasm of the cell and the entire cell body was detectable. Analysis of the line scan intensity for both the red and green channels prior to photoconversion should be set so the green channel is approximately 250 AFU and the red channel is barely visible and less than 50 AFU. Immediately after photoconversion, the green channel should drop down to baseline (near zero AFU) and the red channel reach above 250 AFU for complete photoconversion (). After 24 hours in culture, we found red, photoactivated cells, in both the DRG and SG. Cells from the trunk neural tube follow the same migratory pathways to populate the DRG and SG. Specifically analyzing the contribution of photoconverted cells in the SG, the red channel maintained its intensity at approximately 250 AFU, but the green channel had increased to approximately 100 AFU (slightly less than half the intensity of the red channel). Tracing photoactivated cells after 48 hours, the red channel intensity maintained between 200 and 250 AFU, but the green channel intensity increased to 250 AFU (). This increase in green intensity on un-photoactivated cells was expected as each cell continued to produce the green fluorescence form of the protein, even after photoactivation. However, beyond 24 hours, a photoactivated cell was identified by comparing the red channel intensity; those cells that were not photoactivated maintained a baseline close to 0 (between 0 and 50) AFU in the red channel (). This technique provided better resolution over vital dye labeling of small numbers of cells as the vital dyes often become diluted in the cell after successive cell divisions and it is more difficult to positively identify a punctate labeled cell deep within the embryo.
Overall, by optimizing and balancing the above considerations, in ovo photoactivation of single and small cell numbers, combined with a multi-position photoactivation and multi-acquisition platform offered a means to study both cell-cell interactions and trace cell behaviors during organogenesis with increased accuracy and decreased time and effort. These techniques, when combined with experimental manipulations and gene expression studies will offer a powerful approach to help us better understand cell behaviors and tissue assembly during organogenesis.
Methods
Embryos.
Fertilized White Leghorn chicken eggs (Ozark Hatchery, Meosho, MO) were placed in a rocking incubator at 37°C (Kuhl, Flemington, NJ). Eggs were rinsed with 70% alcohol and 3 mL albumin was removed. Eggs were windowed and embryos staged according to Hamburger and HamiltonCitation34 (HH). Embryos at HH St.10 were injected with photoactivatable-photoconversion plasmids, KikGR, to label premigratory neural crest cells located in the dorsal neural tube. Fast Green FCF (Sigma, F-7252) at 10 ug/mL, was added 1:5 to the injection needle to visualize injection of the construct in ovo. Plasmids were micro-injected into the lumen of the neural tube using a borosilicate glass capillary pulled needle (World Precision Instruments, MTW100-4) until the region of the neural tube between the forelimbs and hindlimbs was filled. Constructs were electroporated into premigratory neural crest cells using gold coated Genetrode electrodes (Fisher, BTX512) and an electroporator (BTX ECM830, Genetronics, San Diego, CA). Eggs were resealed with adhesive tape and incubated at 38°C for 2–3 days. After this incubation period we evaluated each embryo prior to manipulation for normal development.
Injected embryos were re-incubated for 24 hours, after trunk neural crest migration has started. Embryos were mounted on an upright confocal microscope stage. Eggs were windowed and covered with a Teflon membrane to allow for visualization and photoactivation of the embryo in ovo. The initial migratory wave of NCCs that have migrated away from the neural tube were selected (visually identified when NCCs have reached approximately 50 um from the neural tube) and photoactivated (measured as a green-to-red fluorescence conversion) using a 405 nm laser line in ovo. Premigratory NCCs within the neural tube will not be photoactivated and thus will remain green. Eggs were sealed and reincubated for an additional 36 hours, to allow for the formation of the primary sympathetic ganglia, harvested and images whole mount on glass slides.
Photoactivation.
We obtained KikGR (kind gift from Prof. Atsushi Miyawaki) and PSCFP2 (from Evrogen, PS-CFP2-N vector, #FP802, Moscow, Russia). Each construct was grown up to 5 ug/uL. The general photoactivation process, including cell target acquisition and selection for photoactivation, followed a previous protocol developed for PAGFPCitation16 and adapted for each PFP. For the comparison of PFPs, we used constant imaging parameters, including laser power at 2% (measured at 44 uW at the sample and the back aperture using a handheld optical power meter (Model #840, Newport)) in some experiments. All photoactivation was performed in ovo on intact chick embryos. A teflon membrane (Fisher Scientific, LLC) was stretched across an acrylic ring (∼2.2 cm i.d.* 2.6 cm o.d.* 0.5 cm) and secured into a window in the eggshell with beeswax (Fisher Scientific, LLC) according to a previous protocol.Citation35 All imaging was performed on an upright LSM 5 PASCAL microscope (Carl Zeiss, Inc., Thornwood, NY) using a Plan-NeoFluor 10X/0.3, 40X/0.75, or C-Apochormat 40X W/1.20 (Zeiss). Collected images were saved using the AIM Software (Zeiss) and processed (histogram stretch only) in Adobe Photoshop CS2 (Adobe Systems, Inc., San Jose, CA).
Photoefficiency.
Under the same imaging parameters, a time-lapse of each construct expressed by neural crest or neural tube cells was compared. The cells were continuously photoactivated for †9 minutes and 75 confocal scans. We took the intensity mean ratio of the photoactivated fluorescence (blue) over the non-photoactivated fluorescence (green) of cells from each construct to create the photoefficiency graph.
Immunohistochemistry.
Embryos were collected in PBS and fixed in 4% PFA for 4 hours. Embryos for cryostat sectioning were rinsed in PBS and run up in a sucrose gradient (5%, 15%, 30% in PBS), embedded in OCT and stored at −80°C. Sagittal cryostat sections were obtained at 15 um thickness. Slides with sectioned embryos for immunohistochemistry were removed from −80°C and rehydrated in TBS for 20 minutes. Slides were washed with TBS + 0.5% triton x-100 and blocked with TBS + 0.5% triton x-100 + 20% goat serum and 0.01 M glucose for 1 hour at room temperature. Primary antibody, Ben (Developmental Studies Hybridoma Bank), was applied in blocking solution at 4°C overnight. Slides were then washed and incubated for 1 hour in block at room temperature. Secondary antibody, Alexa Fluor 633 (Invitrogen), was applied for 1 hour in block at room temperature. Slides were washed and coverslipped using the ProLong Antifade reagent kit (Molecular Probes, P-7481).
Transverse slice culture.
HH St. 10 embryos were injected with EGFP control, photoactivatable constructs (described above), or unlabeled (for DiI labeling experiments) and embryos were re-incubated for 2–3 hrs. Embryos were harvested in Ringers solution, membranes removed. Using a tungsten needle, the trunk region (between the forelimbs and hindlimbs was excised). The trunk region was then transferred to a piece of Parafilm and with a Kimwipe, excess Ringers solution was removed from the tissue. Using forceps, the trunk tissue was embedded in a solution of 7% low melting agarose (Sigma; made in Ringer's solution). The agarose was allowed to cool and solidify and a block was cut containing the trunk tissue and mounted for vibratome sectioning. Transverse sections were vibratome sectioned at 50–75 um. Transverse sections were then transferred to Millipore culture plate inserts (Millipore, PICMORG50) coated with 20 ug/ml fibronectin (Gibco, 33016-015). Culture insert and transverse sections were then transferred to a glass bottom 35 mm Petri dish (MatTek Corp., P35G-0-14-C). Neuralbasal medium (Gibco, 21103-049), supplemented with B27 (Gibco, 17504-044) was added to the bottom of the glass bottom petri dish. The petri dish was sealed with parafilm and imaged on an inverted Confocal LSM5 Zeiss Pascal microscope.
Static imaging.
After the desired reincubation period (24–48 hours), each embryo was prepared for confocal imaging as described in Teddy and Kulesa.Citation36 Briefly, the embryo was cut from the egg with a pair of scissors (Fine Science Tools, 14060-10) and placed in a dish of Ringer's solution. Since all of the fluorescent labeling was focused in the head region of the embryo, all of the undesired tissues were cut away including portions of the tail and entire heart. On a glass slide (VWR, 48312-024), a circle of vacuum grease (Dow corning, 79810-99) was placed to create a well and the embryo was transferred in the circle with the left side facing down. All liquid was removed from around the embryo and a no. 1, 25 mm circle coverslip (VWR, 48380-080) was carefully placed over the entire grease ring. Confocal imaging was performed using the same configuration settings per PFP described above.
Multi-position photoactivation and multi-time acquisition.
By combining photoactivation and multitime acquisition, we were able to photoactivate a different subregion in each tissue section cut from a subregion of the same embryo. In this way, we were able to mark and image cells in the same embryo in a semi-automated manner and acquired several different samples from the same embryo, within the same experiment. The setup for the automation was time consuming. Each position required time to configure the microscope for automated mark and find and sub-segment acquisition of confocal z-stacks in regions of interest in several different samples. To setup for automated data acquisition we rotated each image to align the neural tube to a dorsoventral axis to a vertical position. We coordinated x-y positions from each tissue section from transmitted light images. We calculated confocal z-stack settings and photoactivation regions of interest for each fluorescently labeled neural tube half and saved the settings for each position (typically, the right hand side). The microscope (LSM 510 META, Carl Zeiss Microimaging, Thornwood, NY, USA) was configured for PS-CFP2 fluorescent label detection and photoactivation using 488 nm excitation to locate potential cell targets and 405 nm excitation for photoactivation. PS-CFP2 photoactivation is described in more detail in Stark and Kulesa, 2007. After all the desired parameters were set, acquisition started (using a 10x/0.45 Plan Apochromat) with an initial scan at every marked location to establish background intensity before photoactivation for all locations. We used the multi-time macro (Zeiss) zoomed to produce approximately 50 pixels/um2 at the predefined locations. We used 405 nm laser activation (20%) to photoactivate all PS-CFP2 labeled cells within the regions of interest at all defined locations. When the photoactivation was completed, a z-stack at every location was collected. Collected images were processed using the AIM software (Zeiss) and visualized for analysis in Imaris (Bitplane, Saint Paul, MN, USA).
Figures and Tables
Figure 1 In ovo single cell photoactivation. (A) An E3.5 chick embryo 16 hours post in ovo microinjection and electroporation with H2B-PSCFP2 in the hindbrain and fixed for 1 hour before mounting on a glass coverslip. The black box outlines the unphotoactivated fluorescence (pink) and photoactivated (green) cells in the neural tube and migrating neural crest cells from rhombomere 6 (r6) and r7 regions. The photoactivated fluorescence (green) was created using a 405 nm laser, 10% laser power. (B) Single cell photoactivation can be challenging if the protein is diffuse throughout the cytoplasm and if the location of the cell is in a densely labeled population. Two cells with PSCFP2 have been photoactivated (green) and unphotoactivated cells are blue. The top cell (arrow) was photoactivated with 2-photon photoactivation at 770 nm, 7% laser power. The bottom cell (arrowhead) was photoactivated with confocal laser excitation at 405 nm. (B′) The X–Z scan, zoomed in from the white box, show a single cell photoactivated with a 2-photon laser (arrow in B) and 2–3 cells photoactivated with a 405 nm (arrowhead in B) inside the neural tube. (C) The line profile through the two photoactivated cells of the X–Z scan in (B′) compares the fluorescence intensity of the 2-photon (arrow in B) and the 405 nm (arrowhead in B) photoactivated cells by recording of the log (base10) ratio of the mean fluorescence intensity ratio (I/Io) of photoactivated to non-photoactivated fluorescence. (D) An area of cells expressing H2B-PSCFP2 pre-photoactivation (pink) and (D′) after the exposure of a 405 nm laser the single cell in the center becomes photoactivated (green). (E) The dense tissue of the neural tube is labeled with H2B-PSCFP2 with both unphotoactivated (blue) and photoactivated (green) cells. The localization in the nucleus helps distinguish individual cells for targeted photoactivation. (F) The X–Z projection of the photoactivated nuclei shown in isosurface reveals several nuclei surrounding the targeted cells. (G) The photoefficiency of PSCFP2 is maintained after localizing to the nucleus using 10% laser power of a 25 mW 405 nm laser over 9 minutes of scanning. The curves are an average of n = 4 mean intensities of cells for PSCFP2 (blue line) and H2B-PSCFP2 (green line).
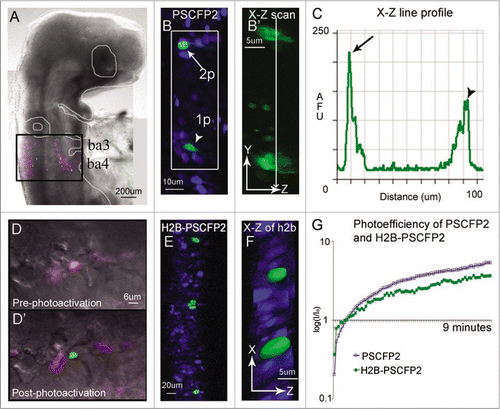
Figure 2 Multi-position photoactivation set up. (A) Transverse section of E4 trunk tissue with neural tube outlined in white. Dorsoventral length (L) and one-half of medioventral width (W) of the neural tube were measured to calculate the approximate ROI size necessary to cover the entire length of half the neural tube. (B) Multi-position mark and find alignment schematic of ROI's, placed in consecutive sections with an increasing position along the dorsoventral axis, green rectangular boxes. (C–H) Collapsed z-stacks of each slice (6 slices from one embryo), with and without brightfield for clarity, after the ROI's were photoactivated. Slices shown based on the placement of the photoactivated regions in serial sections. (I) Chronological breakdown of the time requirements are necessary for set-up, photoactivation and z-stack in one position. Dor, dorsal;Ven, ventral; NT, neural tube; L, length;W, width; ROI's, regions of interest. The scale bar in (C) is 100 um.
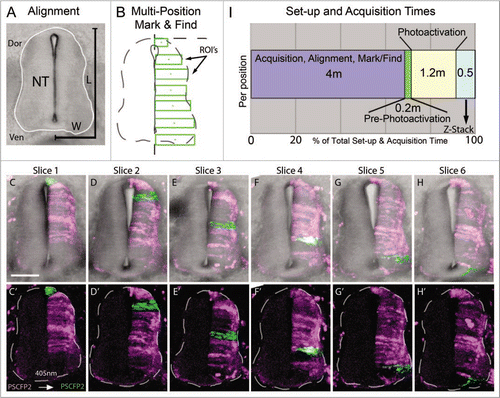
Figure 3 Multi-position photoactivation and multi-time acquisition. (A) A typical fluorescently labeled (PSCFP2) chick embryo at day 4 with (B) the spinal cord removed and (C) cut into slice preparations that (D) are laid onto a culture insert. (E) Within each tissue slice, a region of interest is photoactivated and set up for time-lapse acquisition in a sequential manner, with three typical slice data shown at 0.5 hr, 4.5 hr and 8.5 hr intervals. Individual photoactivated cells are identified by arrows (to show maintenance of photoactivated fluorescence signal) and by arrowheads (to show changes in cell direction). Each photoactivated region of interest is approximately 50 um in height.
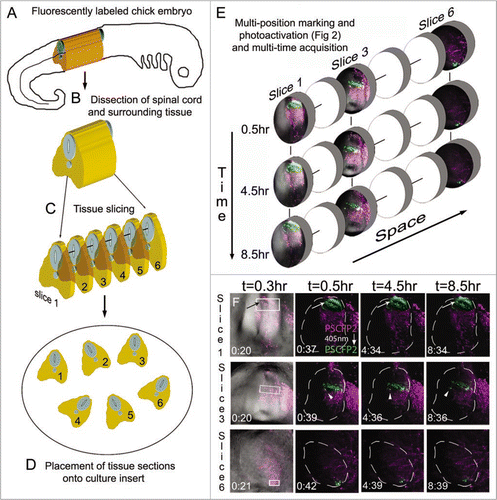
Figure 4 Towards cell tracing during organogenesis: determining the fates of early versus late migrating sympathetic precursor cells. (A) Schematic of E3 chick embryo with boxed region indicating region of interest for photoactivation cell labeling of premigratory neural crest cells. (B) Schematic of typical trunk neural crest cell medial to ventral migratory pathway through the presumptive dorsal root ganglia (DRG) to reach the sympathetic ganglia (SG) adjacent to the dorsal aorta (DA). (C) Sagital view of the trunk structures including the rostral (R) and caudal (C) somite and the SG and inter-ganglionic region (IG). (D) Schematic of the SG that form into a neuronal core (N), grey shaded area, and progenitor neural crest cells, yellow area. (E) In ovo image of KikgR unphotoactivated (green) construct expressed by premigratory neural crest cells in the neural tube, and beginning to emerge into the rostral somite. (F) In ovo image of post-photoactivation (red) of neural crest cells within the highlighted box, showing a group of cells that recently emerged from the neural tube beginning their medio-ventral migration path to populate either the dorsal root or sympathetic ganglia. Unphotoactivated cells remained green. (G) Whole embryo explant image of where the photoactivated cells migrated and localized. From the initial group of photoactivated cell, a portion stopped in the dorsal root ganglia and some continued ventral to give rise to the sympathetic ganglia. (H and I) pre- and post-in ovo photoactivation of early neural crest cells emerging from the neural tube (within box). (J) Embryo after 48 hours from (I), transverse sectioned and stained with Ben (blue). (K–N) Higher magnification of boxed region in (J). Line scan intensities for cells in part (H, I and K) are shown at the bottom of the figure. Pre-photoconversion levels show GFP intensity above 250 AFU and RFP channel below 50 AFU. Post-photoconversion shows an increase in RFP intensity above 250 AFU and a decrease in GRF intensity to below 50 AFU. Cells from 1 K include cell 1 = unphotoconverted non-neural cell (green only), cell 2 = unphotoconverted neural cell (Green and Blue), cell 3 = photoactivated neural cells (Red, Blue and Green-this cell has green fluorescence after 72 hours post-photoconversion because the PSCFP will continue to be made in cells that express it whether it is photoconverted or not), cell 4 = unphotoconverted non-neural cell (green only). nt, neural tube; DRG, dorsal root ganglia; SG, sympathetic ganglia; DA, dorsal aorta; no, notochord.
Acknowledgements
P.M.K. would like to thank the Stowers Institute for their generous support. This work was funded by NIH grant 1R01HD057922.
References
- Rossant J, Tam PP. Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development 2009; 136:701 - 713
- Nishioka N, Inoue K, Adachi K, Kiyonari H, Ota M, Ralston A, et al. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell 2009; 16:398 - 410
- Trainor PA, Krumlauf R. Hox genes, neural crest cells and branchial arch patterning. Curr Opin Cell Biol 2001; 13:698 - 705
- Rosa-Molinar E, Krumlauf R, Pritz MB. Hindbrain development and evolution: past, present and future. Brain Behav Evol 2005; 66:219 - 221
- Weston JA. A radioautographic analysis of the migration and localization of trunk neural crest cells in the chick. Dev Biol 1963; 6:279 - 310
- Fraser SE. Iontophoretic dye labeling of embryonic cells. Methods Cell Biol 1996; 51:147 - 160
- Bhattacharyya S, Kulesa PM, Fraser SE. Vital labeling of embryonic cells using fluorescent dyes and proteins. Meth Cell Biol 2008; 87:187 - 210
- Haas K, Jensen K, Sin WC, Foa L, Cline HT. Targeted electroporation in Xenopus tadpoles in vivo-from single cells to the entire brain. Differentiation 2002; 70:148 - 154
- Lippincott-Schwartz J, Patterson GH. Fluorescent proteins for photoactivation experiments. Methods Cell Biol 2008; 85:45 - 61
- Patterson GH. Photoactivation and imaging of photoactivatable fluorescent proteins. Curr Protoc Cell Biol 2008; 21:21 - 26
- Chudakov DM, Lukyanov S, Lukyanov KA. Using photoactivatable fluorescent protein Dendra2 to track protein movement. Biotechniques 2007; 42:553 - 563
- Raible DW, Wood A, Hodson W, Henion PD, Weston JA, Eisen JS. Segregation and early dispersal of neural crest cells in the embryonic zebrafish. Dev Dyn 1992; 195:29 - 42
- Lukyanov KA, Chudakov DM, Lukyanov S, Verkhusha VV. Innovation: Photoactivatable fluorescent proteins. Nat Rev Mol Cell Biol 2005; 6:885 - 891
- Stark DA, Kulesa PM. An in vivo comparison of photoactivatable fluorescent proteins in an avian embryo model. Dev Dyn 2007; 236:1583 - 1594
- Miyawaki A, Schnitzer MJ. New technologies for neuroscience. Curr Opin Neurobiol 2007; 17:565 - 566
- Stark DA, Kulesa PM. Photoactivatable green fluorescent protein as a single-cell marker in living embryos. Dev Dyn 2005; 233:983 - 992
- Kulesa PM, Teddy JM, Stark DA, Smith SE, McLennan R. Neural crest invasion is a spatially-ordered progression into the head with higher cell proliferation at the migratory front as revealed by the photoactivatable protein, KikGR. Dev Biol 2008; 316:275 - 287
- Wacker SA, Oswald F, Wiedenmann J, Knöchel W. A green to red photoconvertible protein as an analyzing tool for early vertebrate development. Dev Dyn 2007; 236:473 - 480
- Murray MJ, Saint R. Photoactivatable GFP resolves Drosophila mesoderm migration behaviour. Development 2007; 134:3975 - 3983
- Huber K. The sympathoadrenal cell lineage: specification, diversification, and new perspectives. Dev Biol 2006; 298:335 - 343
- LeDouarin NM, Smith J. Development of the peripheral nervous system from the neural crest. Annu Rev Cell Biol 1988; 4:375 - 404
- Serbedzija GN, Bronner-Fraser M, Fraser SE. A vital dye analysis of the timing and pathways of avian trunk neural crest cell migration. Development 1989; 106:809 - 816
- Serbedzija GN, Fraser SE, Bronner-Fraser M. Pathways of trunk neural crest cell migration in the mouse embryo as revealed by vital dye labeling. Development 1990; 108:605 - 612
- Bronner-Fraser M, Fraser SE. Cell lineage analysis reveals multipotency of some avian neural crest cells. Nature 1988; 335:161 - 164
- Henion PD, Weston JA. Timing and pattern of cell fate restrictions in the neural crest lineage. Dev 1997; 124:4351 - 4359
- Tsutsui H, Karasawa S, Shimizu H, Nukina N, Miyawaki A. Semi-rational engineering of a coral fluorescent protein into an efficient highlighter. EMBO Rep 2005; 6:233 - 238
- O'Brien GS, Rieger S, Martin SM, Cavanaugh AM, Portera-Cailliau C, Sagasti A. Two-photon axotomy and time-lapse confocal imaging in love zebrafish embryos. J Vis Exp 2009; 24:1129
- Hagen GM, Caarls W, Lidke KA, De Vries AH, Fritsch C, Barisas BG, et al. Fluorescence recovery after photobleaching and photoconversion in multiple arbitrary regions of interest using a programmable array microscope. Microsc Res Tech 2009; 72:431 - 440
- Chudakov DM, Verkhusha VV, Staroverov DB, Souslova EA, Lukyanov S, Lukyanov KA. Photoswitchable cyan fluorescent protein for protein tracking. Nat Biotechnol 2004; 22:1435 - 1439
- Itasaki N, Bel-Vialar S, Krumlauf R. ‘Shocking’ developments in chick embryology: electroporation and in ovo gene expression. Nat Cell Biol 1999; 1:203 - 207
- Jessell TM, Sanes JR. Development. The decade of the developing brain. Curr Opin Neurobiol 2000; 10:599 - 611
- Glebova NO, Ginty DD. Growth and survival signals controlling sympathetic nervous system development. Annu Rev Neurosci 2005; 28:191 - 222
- Tsarovina K, Schellenberger J, Schneider C, Rohrer H. Progenitor cell maintenance and neurogenesis in sympathetic ganglia involves Notch signaling. Mol Cell Neurosci 2008; 37:20 - 31
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morph 1951; 88:49 - 92
- Kulesa PM, Fraser SE. In ovo time-lapse analysis of chick hindbrain neural crest cell migration shows cell interactions during migration to the branchial arches. Development 2000; 127:1161 - 1172
- Teddy JM, Kulesa PM. In vivo evidence for short- and long-range cell communication in cranial neural crest cells. Development 2004; 131:6141 - 6151