Abstract
Urinary tract obstruction leads to obstructive nephropathy, which in turn, frequently results in renal failure. Congenital urinary tract obstruction can be traced back to errors during the organogenesis of the urinary system. A fundamental understanding of the causes of urinary tract obstruction and the developmental processes involved are critical for improving the diagnostic and therapeutic strategies for this disease. A number of laboratories, including ours, have been using genetically engineered and spontaneously occurring mouse models to study the primary causes and the pathogenesis of urinary tract obstruction. These studies have shown that urinary tract obstruction is a very heterogeneous disease that can be caused by a diverse set of factors targeting multiple levels of the urinary system. Accumulating evidence also indicates that the development of the urinary tract requires the integration of progenitor cells of diverse embryonic origins, leading to the formation of multiple junctions prone to developmental errors. In addition, the high sensitivity of the pyeloureteral peristaltic machinery to disturbance affecting the structural or functional integrity of its components also contributes to the high incidence rate of urinary tract obstruction.
Introduction
Congenital obstructive nephropathy is the principal cause of renal failure in the pediatric population. Compared to adults, urinary tract obstruction in children is particularly damaging due to the permanent impairment inflicted on the maturing kidneys. If left untreated, congenital obstructive nephropathy can lead to renal failure and even death.Citation1 Congenital obstructive nephropathy is not only a devastating disease, but also a very common one. Hydronephrosis is found in antenatal screening at 1:100 with about 1 in 500 being clinically significant, making it the leading cause for renal failure in the pediatric population.Citation2 There are different types of obstructive nephropathy based on the location of the obstruction and/or the cause of the obstruction. Obstruction at the junction between the ureter and the renal pelvis is called Ureteropelvic Junction (UPJ) obstruction, which usually presents as hydronephrosis without obvious dilatation of the ureter (hydroureter). UPJ obstruction is estimated to occur in one out of 1,000–1,500 new births. Obstruction at other levels, such as ureterovesical junction (UVJ) and bladder outlet are also common. Obstruction in the urinary tract, in a strict sense, describes the presence of a physical blockage to the urine flow. However, the word “obstruction” is also used to describe the failure of urine transport from the kidney to the external environment regardless of the causes. The word “obstruction” is used in this manuscript in its broader sense to describe the physical blockage (physical obstruction) of the urinary tract and functional failure in urine transport (functional obstruction).
Experimental surgical animal models for obstructive nephropathy have been very useful in determining the pathologic impacts of the obstruction but less informative about the molecular and cellular lesions causing the disease.Citation3–Citation12 Despite this progress, the genetic or environmental determinants in most human patients with congenital defects in the kidney and the urinary tract are not known. In addition to familial cases, strong genetic determinants in congenital kidney and urinary tract defects were reported by a number of studies in seemingly sporadic cases.Citation13–Citation15 For example, Feather et al. found that there is a 30–50 fold increased incidence of vesicoureteral reflux in first-degree relatives of probands, compared with the general population.Citation14 The strong genetic component in congenital kidney and urinary tract defects demands more studies in the genetic factors and pathways involved in the pathogenesis of these devastating diseases. Mouse models, both genetically engineered and naturally occurring, have been making and will continue to make major contributions to these studies. Pathological consequences of congenital obstructive nephropathy have been described in detail from both human and animal studies for decades.Citation16–Citation21 The molecular and cellular lesions causing congenital urinary tract obstruction only started to come to light in recent years. Results from both genetically engineered and spontaneously occurring mouse models have revealed a high degree of heterogeneity in the origins of obstructive nephropathy and the developmental processes where most of the disease-causing anomalies occur.Citation22 Continuing advances in mouse genetics and genomics will no doubt speed up the identification of the genetic factors and pathways involved in the pathogenesis of obstructive nephropathy.
Loss of Calcineurin in the Metanephric and Ureteric Mesenchyme causes Congenital Obstructive Nephropathy
Our interest in congenital obstructive nephropathy originated from the study of the calcineurin mutant mice. Calcineurin is a Ca2+-dependent serine/threonine phosphatase composed of a catalytic subunit, CnA and a regulatory subunit, CnB (). CnB has a ubiquitously expressed isoform (Cnb1) and a testis-specific isoform (Cnb2). The regulatory subunit of calcineurin is indispensible for calcineurin activity.Citation23,Citation24 Thus inactivation of Cnb1 in non-testis cells will render those cells deficient for calcineurin function.Citation25 The NFATc (Nuclear Factor of Activated T Cells) transcription factors are the most extensively studied calcineurin substrates. They appear to be the major regulator of transcription downstream of Ca2+/Calcineurin signals.Citation24 NFATc proteins reside in the cytoplasm in resting cells. Upon activation by an increase in intracellular Ca2+, calcineurin dephosphorylates the NFATc proteins. This dephosphorylation exposes the concealed nuclear localization signals of the NFATc proteins, leading to translocation from the cytoplasm to the nucleus. Once in the nucleus, the NFATc proteins form NFAT transcription complexes with their nuclear partners to control the transcription of target genes.
In an earlier study, we used a Pax3-Cre transgene to direct the deletion of a loxP-flanked Cnb1 allele and serendipitously created a model for congenital obstructive nephropathy.Citation26 The Pax3-Cre transgene (Pax3-Cre) was originally generated to direct the recombination of loxP sites in neural crest cells and other tissues.Citation27 Our analyses of mice carrying the Pax3-Cre transgene and the ROSA26RLacZ (ROSA) alleleCitation28 revealed that, within the urinary system, this transgene is expressed specifically in the metanephric mesenchyme (MM) and ureteric mesenchyme (UM) derived tissues (), but not in ureteric bud (UB) derived tissues. Especially, the smooth muscle cells of the renal pelvis and ureter (mesenchymal derivatives) have strong Cre expression as evidenced by the strong β-galactosidase activity in these cells (). We unexpectedly discovered severe hydronephrosis in mutant mice (Pax3-CreT/+; Cnb1lox/lox) with Cnb1 deleted in the MM and UM (). The blood urea nitrogen (BUN) level in the mutants is on average more than 10 times higher than that of the controls. All mutants died of renal failure within 3 weeks after birth.
Calcineurin is Required for the Development of the Pyeloureteral Peristaltic Machinery
We further showed by polymer molding that the mutant urinary tract is not physically obstructed. Instead, the pyeloureteral peristalsis is defective in the Pax3-CreT/+; Cnb1lox/lox mutants, leading to failure of urine transfer and functional obstruction.Citation26,Citation29 A key reason for the functional obstruction is the defective development of the renal pelvis and smooth muscle along the urinary tract. The mutants fail to form the funnel-shaped muscular structure of the renal pelvis that is important for normal peristalsis ( and B). The defective development of the renal pelvis is, in turn, caused by reduced proliferation of the smooth muscle cells and their progenitors lining the developing renal pelvis. Ki67 staining revealed a reduction of proliferation in the mesenchymal cells with calcineurin deficiency but not in the epithelium that retains calcineurin activity ( and D). The renal pelvis is critical for normal urine transfer in at least three ways. First, constriction of the junction between the renal pelvis and the ureter (ureteropelvic junction, UPJ) can lead to obstruction to the urine flow. Second, it has been proposed that the pacemakers for pyeloureteral peristalsis reside in the renal pelvis.Citation30 These pacemaker cells are thought to be specialized smooth muscle cells that control the initial contraction of the smooth muscles lining the urinary tract. Third, the contraction of the funnel-shaped muscular structure of the renal pelvis provides the strong initial thrust that effectively propels urine down the ureter from the pelvicocaliceal space inside the kidney. Spontaneous peristalsis was observed in pyeloureteral complexes removed from the Pax3-CreT/+; Cnb1lox/lox mutants, suggesting that the pacemaker cells were still present in the mutants for the initiation of peristalsis. As revealed by polymer molding of the urinary tract in the mutants, physical constriction of the UPJ was not observed. Thus defective development of the “powertrain” for peristalsis (the renal pelvis and smooth muscle along the urinary tract) in the Pax3-CreT/+; Cnb1lox/lox is the major cause for the ineffective urine transfer and functional obstruction of the urinary tract.Citation26
Interaction of Signaling Pathways in Urinary Tract Development
Since not all tissues and cells with Cnb1 deletion have reduced proliferation, we believe that calcineurin is not simply serving as a generic proliferation regulator in the MM and UM. Instead, it functions as a key factor in the mesenchyme for the interpretation of signals that originate in the UB-derived epithelium. A number of factors have important functions in the UB derivatives for urinary tract development.Citation22 Among these factors, Shh communicates with its receptor Ptch in the mesenchyme to regulate smooth muscle differentiation.Citation31 The deletion of Shh in the UB derivatives leads to defective smooth muscle development and obstructive nephropathy. In addition, Dlgh1 is expressed strongly in the epithelium, though it also has expression in the mesenchyme. Its inactivation results in a range of urogenital defects, including urinary tract smooth muscle defects and obstructive nephropathy.Citation32,Citation33 Since many signaling pathways directly or indirectly induce calcium increase in the target cells, it will not be surprising to see that the calcineurin signaling is embedded in a more complex signaling network and is constantly being modified by other interacting signals. The Renin-Angiotensin system (RAS) is known to be involved in renal injury in congenital obstructive nephropathy. RAS, however, has a constructive role during urinary tract development.Citation34,Citation35 Mutations in a number of RAS component genes cause congenital anomalies of the kidney and urinary tract (CAKUT).Citation36–Citation42 Although the broad effect of RAS in blood pressure control and regulation of the ability to concentrate urine may contribute to the observed renal pathological changes, evidence suggests that direct function of RAS in the ureteral SM differentiation plays an important role.Citation38 In fact, a number of studies suggest that RAS can induce activation of calcineurin. Our recent studies indicate that transgenic activation of Nfatc1 in the urinary tract also leads to severe developmental anomalies including hydroureters and hydronephrosis (Wang and Chen F, unpublished data). It is possible that within the urinary system, RAS activates calcineurin to regulate Nfat-mediated transcription for proper urinary tract development and differentiation.
Genetic Mutations causing Congenital Obstructive Nephropathy
Besides genetically engineered mice, the identification of unknown causative mutations in mice with congenital obstructive nephropathy is another way to reveal the genetic determinants in this disease. One example of such forward genetic studies is the identification of the genetic mutation in the cph (congenital progressive hydronephrosis) mice.Citation43 cph was first discovered as a spontaneous, autosomal recessive trait in the 1970s.Citation44,Citation45 Since these mutants were thought to have renal cysts, they were named jpk for Juvenile Polycystic Kidney until a later study found that the pathologic changes were cystic renal dysplasia caused by progressive hydronephrosis secondary to UPJ obstruction.Citation45 We performed a thorough analysis of the urinary system in the cph mutants and found that besides UPJ obstruction, some of them also have UVJ obstruction and hydroureter.Citation43 Based on polymer molding, there is no physical blockage of the urinary tract, suggesting another example of functional obstruction. Early postnatal hydronephrosis usually progresses rapidly, leading to loss of renal function and death (). To identify the causative mutation, we performed genetic mapping that located the cph mutation to a 0.7 Mb chromosomal region on mouse chromosome 15 (). There were still approximately 30 known genes and predicted open reading frames in this interval.Citation43
After studying the genes within the suspected chromosomal interval, we discovered that the distribution of the water channel protein Aqp2 was altered in the mutants. Aqp2 functions as a water channel to reabsorb water from the collecting duct lumen back to the interstitium. While Aqp2 was predominantly distributed on the apical membrane of the principal cells in the collecting ducts in control mice, its apical accumulation was completely lost at all levels of the collecting duct in all homozygous mutants ( and B). At that point, we could not be certain if the observed difference in distribution was merely a pathological change due to obstructive nephropathy or was in fact the cause for the renal defects. We thus examined the distribution of Aqp2 in the principal cells of the Pax3CreT/+; Cnb1lox/lox mutants that also have severe congenital obstructive nephropathy.Citation26 The distribution of Aqp2 was not significantly changed in the Pax3CreT/+; Cnb1lox/lox mutants despite overt collecting duct dilatation (data not shown), suggesting that absence of Aqp2 apical accumulation was not simply a secondary effect of the congenital obstructive nephropathy.
By sequencing the Aqp2 gene, we discovered a mutation in nucleotide 767 in the fourth exon of Aqp2 in the mutants (). The C-T change in codon 256 led to a Ser to Leu substitution in the protein. This S256 is actually a phosphorylation site and is highly conserved in all vertebrate species. The loss of this key phosphorylation due to the cph mutation results in the loss of apical trafficking of the Aqp2 protein. The direct physiological effect of this trafficking defect is in urine concentration. As a consequence, the cph mutants produce a high volume of hypotonic urine and show classic characteristics of Nephrogenic Diabetes Insipidus, including insensitivity to vasopressin.Citation43 The cph mutants were first described as having polycystic kidney disease, before it was later found to have progressive hydronephrosis and obstructive nephropathy instead. Our study shows that the underlying cause is in Aqp2 distribution and urine concentration. They actually have Nephrogenic Diabetes Insipidus and the drastic increase of urine output overwhelms the urine transfer machinery, secondarily causing hydronephrosis. Many diseases share similar structural anomalies and present with similar symptoms, but accurate diagnosis can only be made by revealing the underlying molecular and cellular lesions.
Diverse causes of Obstructive Nephropathy
Our ongoing research involves other urinary tract disease models that are either spontaneously occurring murine mutants, transgenic mice with alterations in specific genes, or transgenic mice with specific ablation of progenitor cells involved in the development of the urinary system. As we learn from our own studies and studies from many other laboratories, obstructive nephropathy can be caused by a diverse set of factors targeting different parts of the urinary system.Citation22,Citation46 Most of the known causes affect the development of the lower urinary tract. These include the underdevelopment of the pelvis and the malformation of the ureteric smooth muscle as we see in the calcineurin mutants. Defects in the ureteric epithelium can directly or indirectly affect the pyeloureteral peristalsis, leading to obstruction. The joining of the ureter to the bladder is a complex process where errors are prone to occur.Citation47–Citation49 As seen in the cph mutants, in addition to problems in the lower urinary tract, anomalies in the kidney proper can also lead to physiological changes that eventually break down the urine transfer machinery, causing obstruction. The essential pyeloureteral peristalsis is a process that requires high structural and functional integrity of all of its components, making it sensitive to disturbances caused by genetic and environmental factors. Perhaps this sensitivity of pyeloureteral peristalsis to such disturbances and the complexity in the formation of the junctional complexes along the urinary conduit set the stage for the high incidence of congenital urinary tract obstruction ().
A Systematic View of the Development of Urinary Tract Obstruction
Development of the urinary tract requires the integration of progenitor populations with diverse embryonic origins (). The majority of the nephron components are from the MM within the intermediate mesoderm. The UB contributes to the ureteric epithelium and the collecting duct epithelium.Citation50,Citation51 The tailbud-derived mesenchyme gives rise to the UM which, in turn, produces the smooth muscle and other mesenchymal derivatives along the ureter and along the ureter.Citation52 While the vesical (bladder) epithelium is from the hindgut endoderm, the vesical mesenchyme appears to arise from tailbud-derived mesenchyme.Citation52 The lineage origin of the urethral mesenchyme is not entirely clear, but the urethral epithelium is also derived from the same hindgut endoderm that gives rise to the vesical epithelium.Citation53 The temporal and spatial integration of these diverse progenitor populations requires precise control by a number of signaling pathways. Any disruption of these signaling pathways could affect the integration, leading to a wide range of renal defects, including agenesis, hypoplasia, supernumerary ureter, hydroureter, hydronephrosis and others. Presence of multiple types of defects in the kidney and the urinary tract is described as CAKUT (congenital anomalies of the kidney and urinary tract).Citation54,Citation55 On one hand, the term CAKUT reflects the fact that defects in the urinary system can have very different phenotypes. It may also encourage urologists and nephrologists to work together to tackle these complex diseases that tend to affect both the kidney and the lower urinary tract. Some investigators, however, dislike the term CAKUT because of the perceived risk of blurring the pathological distinction of distinct disease subtypes. In any case, it is clear that disturbance during embryogenesis can affect multiple interconnecting processes for the development of the urinary system, causing a wide spectrum of defects in the kidney and urinary tract. Even if the primary molecular and cellular lesions initially affect only part of the system, the structural and functional integration of the system eventually leads to disruption of the development and integrity of other parts of the urinary system. Thus, a systematic view of the development of the urinary system is necessary even when a particular defect, such as urinary tract obstruction, is being studied.
Phenotypic Variations in Congenital Urinary Tract Defects
It is common to see phenotypic variations in human patients and animal models with urinary tract defects. We have seen variations among affected individuals of the same genotype or even between the left and right sides of the same individual. Variations are seen in any organ systems, but some unique characters of urinary system may have made it more prone to extreme variations. I will use two hypothetical examples here to better illustrate this point (). In the first example, a genetic defect slows UB budding to a degree that the UB barely reaches the MM just in time to prevent its apoptosis. Other problems associated with this mutation then lead to the development of urinary tract obstruction. However, any additional disturbance, including intrinsic variations or random extrinsic factors, may cause an additional minor delay in UB outgrowth, causing the failure of the UB to reach MM before its degeneration and leading to renal agenesis. This is similar to a scenario where you are running late to catch a train. You may be lucky to get on it at the last second, or your taxi to the station may hit one red light too many and you miss it all together. The outcomes are very different (a train journey or no train journey), but result from what could be considered a minor difference in timing (one red light too many resulting in a delay of seconds). By analogy, in kidney and urinary tract development, what could be considered a minor quantitative difference (slower UB outgrowth, for example) may be translated into qualitatively distinct outcomes (renal agenesis or a kidney with urinary tract obstruction) in affected individuals. In another hypothetical example, abnormal intermediate mesoderm patterning causes the MM from both sides to position closer to the middle than normal. This sets up an unstable state that any slight variation may cause fusion of the MM from both sides and, subsequently, the formation of horseshoe kidneys. The ascent of the horseshoe kidneys tends to be impeded by crossing blood vessels, causing subsequent problems such as hydronephrosis and hydroureter. However, if no fusion occurs, the left and right kidneys may develop relatively normally despite their unusually close proximity to each other. Therefore, very slight and random variations may separate a rather healthy state and a potentially very morbid state.
Conclusions
Genetically engineered and naturally occurring murine models are powerful tools in the study of congenital obstructive nephropathy. With the advance of mouse genetics and genomics, the use of these tools will certainly expand. While we now have a much better understanding of urinary tract development and the causes of congenital urinary tract obstruction than a decade ago, identifying the cause and determining the most suitable treatment for a particular patient remain a challenge for basic researchers and clinicians to tackle for years to come.
Conference Questions and Answers
When do the cph mutants die?
Usually they die within the first month.
Are they normal at birth?
Yes, at birth they look normal. The hydronephrosis usually becomes obvious within the first week of life.
You indicate that polyuria will induce breakdown of the peristaltic machinery and that produces renal insufficiency. Most patients with diabetes insidious do not have a decrease in renal function. Why is it different in the mice?
Patinents with diabetes insipidus can have hydronephrosis and hydroureter, if they don't take medication and drink too much. Mice drink as much as they want and are not medicated. It is possible that polyuric mice are more prone to hydronephrosis than polyuric humans. In any case, I think the breakdown of the peristaltic machinery induced by polyuria is the most likely explanation for the functional obstruction in the cph mice.
I would agree with you because we see clinically that pediatric patients with polyuria can overwhelm their bladder capacity. This results in a functional obstruction that can cause renal scarring. In many of those kids, we recommend over night Foley drainage to decompress the bladder and prevent elevated pressure from the functional obstruction.
Right. Thanks.
You showed that a mutation in Aqp2 disrupting its trafficking causes urine concentration defect and obstructive nephropathy. Did you have the opportunity to study the osmolality of the medulla of those animals. If you don't have a hypertonic medulla, even if you have a working aquaporin, you cannot concentrate the urine, and you get polyuria. You need both a functional aquaporin and you need hypertonic medulla.
That is a good point. We did not measure the osmolality of the mutant medulla. However, one would expect that the Aqp2 defect would affect the medullary gradient.
Does the lower urinary tract have transport function?
As you know, aquaporin 2 is expressed in the collecting duct principal cells and serves as a channel for water transport. However, in the lower urinary tract, namely the ureter and the bladder, the expression of Aqp2 is insignificant, if there is any at all. In fact, we looked for Aqp2 expression in the ureter and bladder using immunehistochemistry and found no positive staining. We doubt that aquaporin 2 has any significant water reabsorptive function in the lower urinary tract.
You haven't talked about the innervations of the urinary tract. It has been speculated that nervous system abnormalities might be a cryptic cause of obstruction. Can you talk about the neural control of peristalsis? The reason I ask is because calcineurin has important functions in the nervous system and is a target of a class of drugs.
That is a good question. One of the first things we did when we began the calcineurin was to examine the ureters from mutants and controls histologically looking for differences in neural innervation. We saw no structural differences. However, it is much harder to test functionally. Certainly, the nerves are there and must be doing something. On the other hand there are experiments showing that the peristaltic process is primarily a myogenic. In the movie I showed in the talk, pyeloureteral peristalsis happened in the isolated urinary system without any innervation in the dish. In fact, you can add neural blockers to the dish to block nerve activity without affecting peristalsis.
There is no innervation following kidney and ureter transplantation, at least not in the initial stage. However, the ureter still functions.
Abbreviations
| UB | = | ureteric bud |
| MM | = | metanephric mesenchyme |
| UM | = | ureteric mesenchyme |
| UPJ | = | ureteropelvic junction |
| UVJ | = | ureterovesical junction |
| CAKUT | = | congenital anomalies of the kidney and urinary tract |
| RAS | = | renin-angiotensin system |
Figures and Tables
Figure 1 The Calcineurin-NFAT signaling pathway. Calcineurin is a calcium-dependent serine-threonine phosphatase. Calcineurin has a catalytic subunit “A” and a regulatory subunit “B”. Both of these subunits are indispensible for calcineurin activity. The activity of calcineurin can be inhibited by a number of intrinsic and pharmacological inhibitors, such as Cyclosporin A (CsA) and FK506. The increase in cytoplasmic calcium level can be triggered by many factors, including the activation of a diverse set of cell surface receptors, ion channels and even gap junctions. Intracellular calcium increase leads to the activation of calcineurin. Activated calcineurin dephosphorylates its substrates, including the NFATc transcription factors. The dephosphorylated NFATc proteins translocate from the cytoplasm to the nucleus to regulate the transcription of their target genes. The regulation of transcription by NFATc proteins requires nuclear partners (NFATn). These nuclear partners can be different for different target genes and in different cells. These nuclear partners are usually activated by a different set of receptors on the cell surface.
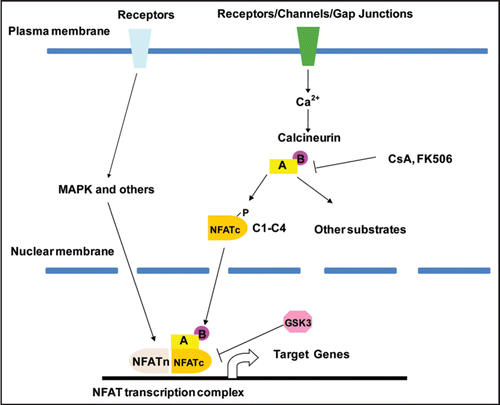
Figure 2 Inactivation of Cnb1 in the metanephric and ureteric mesenchyme causes congenital obstructive nephropathy. (A–E) are samples from P1 Pax3-CreT/+, ROSAT/+ mice. In the kidney proper, LacZ expression (blue, reflecting the Cre expression) is evident in the glomeruli and tubules that are MM-derived but not in the collecting duct system originated from the UB (A). The black arrow in (B) points to the developing glomeruli. The open arrow points to one of the UB branches. In the developing renal pelvic region, the connective tissues, the smooth muscle layers (SM) and the adventitia (AD) of the urinary tract express LacZ while the UB-derived urothelium (UT) remains LacZ-negative (C–E). U: ureter; PP: Papilla. The SM layers in the developing renal pelvic wall are illustrated by αSMA staining on a wild-type newborn sample of the same area (D). LacZ is also selectively expressed in the SM layers in the ureter but not in the stratified transitional epithelium (E). Mutants (Pax3-CreT/+, Cnb1lox/lox) have severe hydronephrosis and erosion of the kidney parenchyma (G and I compared with F and H. Samples G and H were collected at P12). Modified from and from ref. Citation26 with permission.
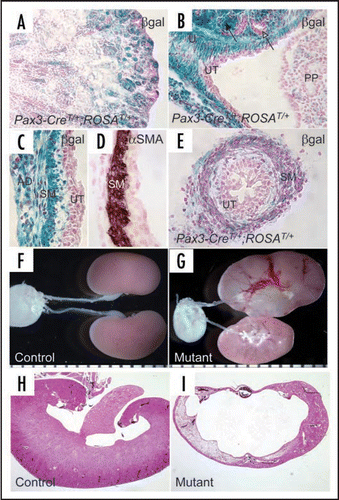
Figure 3 Inactivation of Cnb1 in the metanephric and ureteric mesenchyme disrupts the development of the pyeloureteral peristaltic machinery. (A and B) are hemi-sected control (A) and mutant (B) kidneys. The white arrows point to the UPJ. (C and D) are sections of the developing renal pelvic wall of control (C) and mutant (D) immunostained with a Ki67 antibody. The controls have significantly more proliferating mesenchymal cells along the developing renal pelvic wall. PP: papilla. M: mesenchymal derivatives. UT: urothelium. Modified from and from ref. Citation26 with permission.
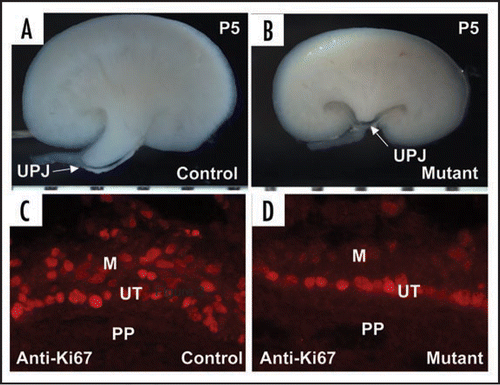
Figure 4 Genetic mapping of the cph mutants with congenital obstructive nephropathy. (A–C) The mutants have unilateral or bilateral hydronephrosis and hydroureter (arrow). (D) The BUN level in the mutants (P5–P16) is dramatically increased. (E and F) Kidney sections from P14 control and mutant littermates stained with H&E. (G) Physical map of relevant portions of mouse chromosome 15. Our genetic mapping efforts locate the cph locus to the chromosomal interval of about 0.7 Mbp, defined by the microsatellite markers C15LD6 and C15LD5. The black triangle indicates the chromosomal location of Aqp2. Modified from and from ref. Citation43 with permission.
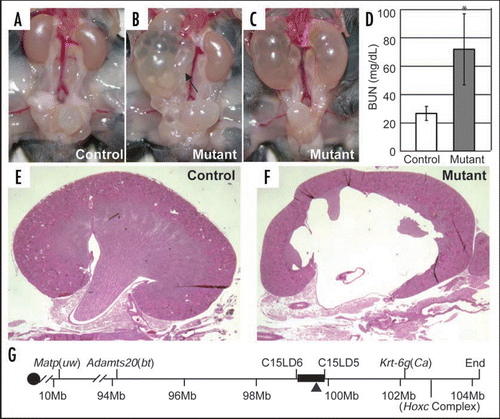
Figure 5 A mutation in Aqp2 disrupting its trafficking causes urine concentration defect and obstructive nephropathy. (A and B) Immunostaining of the collecting ducts in the outer medulla shows apical accumulation of Aqp2 in the controls (A) but a diffuse staining with no apical accumulation in the mutant (B). (C) Sequence of the fourth exon of Aqp2 reveals the C-T substitution at nucleotide 767 in the homozygous mutants, while the heterozygotes have both C and T represented at position 767. This substitution results in a Ser to Leu change at amino acid 256 in the cytoplasmic tail of the Aqp2 protein. +, wild-type allele; c, cph mutant allele. Modified from from ref. Citation43 with permission.
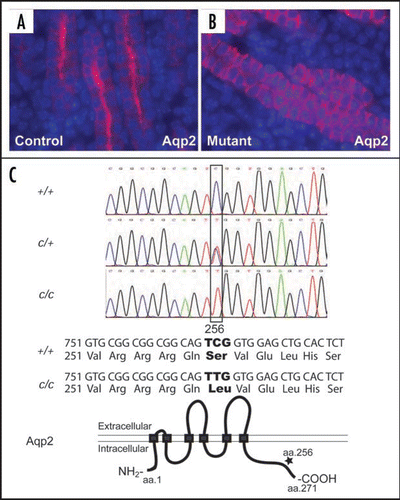
Figure 6 Urinary tract development requires the integration of diverse progenitor cell populations and the formation of multiple junctions. Nephron components, excluding the collecting ducts, are derived from the MM within the intermediate mesoderm. The collecting duct and the ureteric epithelium are derived from the UB. The tailbud-derived mesenchyme gives rise to the UM that in turn produces the smooth muscles and other mesenchymal derivatives along the ureter and along the ureter. While the vesical (bladder) epithelium is from the hindgut endoderm, the vesical mesenchyme appears to arise from tailbud-derived mesenchyme. The lineage origin of the urethral mesenchyme is not entirely clear, but the urethral epithelium is also derived from the same hindgut endoderm that gives rise to the vesical epithelium. The origin of the renal capsule is less clear but appears to be distinct from the other structures within the urinary system. The use of different colors is for the distinction of tissues with different embryonic origins. The different colors used for the UM and vesical mesenchyme (both from the tailbud-derived mesenchyme) reflect the fact that they develop as separate structures before being joint together later in development. The entire urinary tract is also highly innervated. The integration of these diverse progenitor populations also involves the formation of junctional complexes, especially at the UPJ and UVJ. Any disruption of the regulation of the integration could lead to urinary defects, including urinary tract obstruction.
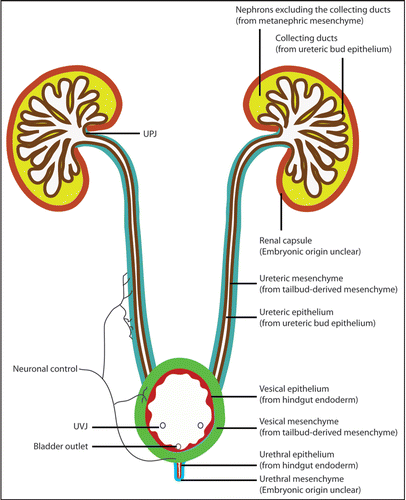
Figure 7 Quantitative differences can be translated into qualitatively distinct outcomes in urinary tract development, contributing the phenotypic variations. (A) In a hypothetical scenario, a genetic defect affects UB budding to a degree that the UB barely reaches the MM just in time to prevent its apoptosis. Other problems associated with this mutation then lead to the development of urinary tract obstruction. Any additional disturbance may delay the UB outgrowth further, causing the failure of the UB to reach MM before its degeneration and leading to renal agenesis. This is similar to a scenario (depicted in B) where you are running late to catch a train. You may be lucky to get on it at the last second, or your taxi to the station may hit one red light too many and you miss it all together. In another hypothetical example (C), abnormal intermediate mesoderm patterning causes the MM from both sides to position closer to the midline than normal. The left and right kidneys may develop relatively normally despite their unusual positions. However, the close proximity of the MM from both sides sets up an unstable state that any slight variation may cause MM fusion and the occurrence of horseshoe kidneys that tend to be obstructed. Therefore, very slight and random variations may separate a rather healthy state and a potentially very morbid state.
Acknowledgements
The author wishes to thank Drs. Helen Liapis, and Qiusha Guo for critical reading of the manuscript. F.C. has been supported in part by a NIH grant (DK067386) and the George M. O'Brien Washington University Center for Kidney Disease Research (NIHP30DK079333).
Note
Edited transcripts of research conferences sponsored by Organogenesis and the Washington University George M. O'Brien Center for Kidney Disease Research (P30 DK079333) are published in Organogenesis. These conferences cover organogenesis in all multicellular organisms including research into tissue engineering, artificial organs and organ substitutes and are participated by faculty at Washington University School of Medicine, St. Louis, Missouri.
References
- Chevalier RL. Pathophysiology of obstructive nephropathy in the newborn. Semin Nephrol 1998; 18:585 - 593
- Grasso M, Gitlin J. Ureteropelvic Junction Obstruction. eMedicine 2001; Online
- Guo G, Morrissey J, McCracken R, Tolley T, Liapis H, Klahr S. Contributions of angiotensin II and tumor necrosis factor-alpha to the development of renal fibrosis. Am J Physiol Renal Physiol 2001; 280:777 - 785
- Hruska KA, Guo G, Wozniak M, et al. Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am J Physiol Renal Physiol 2000; 279:130 - 143
- Liapis H, Barent B, Steinhardt GF. Extracellular matrix in fetal kidney after experimental obstruction. J Urol 2001; 166:1433 - 1438
- Liapis H, Yu H, Steinhardt GF. Cell proliferation, apoptosis, Bcl-2 and Bax expression in obstructed opossum early metanephroi. J Urol 2000; 164:511 - 517
- Orsola A, Adam RM, Peters CA, Freeman MR. The decision to undergo DNA or protein synthesis is determined by the degree of mechanical deformation in human bladder muscle cells. Urology 2002; 59:779 - 783
- Miyajima A, Chen J, Poppas DP, Vaughan ED Jr, Felsen D. Role of nitric oxide in renal tubular apoptosis of unilateral ureteral obstruction. Kidney Int 2001; 59:1290 - 1303
- Miyajima A, Chen J, Lawrence C, et al. Antibody to transforming growth factor-beta ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int 2000; 58:2301 - 2313
- Felsen D, Loo MH, Marion DN, Vaughan ED Jr. Involvement of platelet activating factor and thromboxane A2 in the renal response to unilateral ureteral obstruction. J Urol 1990; 144:141 - 145
- Park JM, Yang T, Arend LJ, et al. Obstruction stimulates COX-2 expression in bladder smooth muscle cells via increased mechanical stretch. Am J Physiol 1999; 276:129 - 136
- Morrissey J, Guo G, Moridaira K, et al. Transforming Growth Factor-beta Induces Renal Epithelial Jagged-1 Expression in Fibrotic Disease. J Am Soc Nephrol 2002; 13:1499 - 1508
- Kenda RB, Kenig T, Budihna N. Detecting vesico-ureteral reflux in asymptomatic siblings of children with reflux by direct radionuclide cystography. Eur J Pediatr 1991; 150:735 - 737
- Feather SA, Malcolm S, Woolf AS, et al. Primary, nonsyndromic vesicoureteric reflux and its nephropathy is genetically heterogeneous, with a locus on chromosome 1. Am J Hum Genet 2000; 66:1420 - 1425
- Queisser-Luft A, Stolz G, Wiesel A, Schlaefer K, Spranger J. Malformations in newborn: results based on 30,940 infants and fetuses from the Mainz congenital birth defect monitoring system (1990–1998). Arch Gynecol Obstet 2002; 266:163 - 167
- Bascands JL, Schanstra JP. Obstructive nephropathy: insights from genetically engineered animals. Kidney Int 2005; 68:925 - 937
- Chevalier RL. Molecular and cellular pathophysiology of obstructive nephropathy. Pediatr Nephrol 1999; 13:612 - 619
- Chevalier RL. Promise for gene therapy in obstructive nephropathy. Kidney Int 2004; 66:1709 - 1710
- Peters CA. Congenital obstructive nephropathy: is the fog lifting?. Kidney Int 2005; 67:371 - 372
- Liapis H. Biology of congenital obstructive nephropathy. Nephron Exp Nephrol 2003; 93:87 - 91
- Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol 2002; 283:861 - 875
- Chen F. Genetic and developmental basis for urinary tract obstruction. Pediatric Nephrology 2009; In press
- Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem 1998; 273:13367 - 13370
- Crabtree GR. Calcium, calcineurin and the control of transcription. J Biol Chem 2001; 276:2313 - 2316
- Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 2001; 105:863 - 875
- Chang CP, McDill BW, Neilson JR, et al. Calcineurin is required in urinary tract mesenchyme for the development of the pyeloureteral peristaltic machinery. J Clin Invest 2004; 113:1051 - 1058
- Li J, Chen F, Epstein JA. Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis 2000; 26:162 - 164
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature Genetics 1999; 21:70 - 71
- Mendelsohn C. Functional obstruction: the renal pelvis rules. J Clin Invest 2004; 113:957 - 959
- Santicioli P, Maggi CA. Myogenic and neurogenic factors in the control of pyeloureteral motility and ureteral peristalsis. Pharmacological Reviews 1998; 50:683 - 721
- Yu J, Carroll TJ, McMahon AP. Sonic hedgehog regulates proliferation and differentiation of mesenchymal cells in the mouse metanephric kidney. Development 2002; 129:5301 - 5312
- Mahoney ZX, Sammut B, Xavier RJ, et al. Discs-large homolog 1 regulates smooth muscle orientation in the mouse ureter. Proc Natl Acad Sci USA 2006; 103:19872 - 19877
- Iizuka-Kogo A, Ishidao T, Akiyama T, Senda T. Abnormal development of urogenital organs in Dlgh1-deficient mice. Development 2007; 134:1799 - 1807
- Fujinaka H, Miyazaki Y, Matsusaka T, et al. Salutary role for angiotensin in partial urinary tract obstruction. Kidney Int 2000; 58:2018 - 2027
- Yosypiv IV, El-Dahr SS. Role of the renin-angiotensin system in the development of the ureteric bud and renal collecting system. Pediatr Nephrol 2005; 20:1219 - 1229
- Oshima K, Miyazaki Y, Brock JW 3rd, Adams MC, Ichikawa I, Pope JCt. Angiotensin type II receptor expression and ureteral budding. J Urol 2001; 166:1848 - 1852
- Nishimura H, Yerkes E, Hohenfellner K, et al. Role of the angiotensin type 2 receptor gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Mol Cell 1999; 3:1 - 10
- Miyazaki Y, Tsuchida S, Nishimura H, et al. Angiotensin induces the urinary peristaltic machinery during the perinatal period. J Clin Invest 1998; 102:1489 - 1497
- Oliverio MI, Kim HS, Ito M, et al. Reduced growth, abnormal kidney structure and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci USA 1998; 95:15496 - 15501
- Esther C Jr, Howard TE, Marino EM, Goddard JM, Capecchi MR, Bernstein KE. Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology and reduced male fertility. Lab Invest 1996; 74:953 - 965
- Niimura F, Labosky PA, Kakuchi J, et al. Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. J Clin Invest 1995; 96:2947 - 2954
- Nagata M, Tanimoto K, Fukamizu A, et al. Nephrogenesis and renovascular development in angiotensinogen-deficient mice. Lab Invest 1996; 75:745 - 753
- McDill BW, Li SZ, Kovach PA, Ding L, Chen F. Congenital progressive hydronephrosis (cph) is caused by an S256L mutation in aquaporin-2 that affects its phosphorylation and apical membrane accumulation. Proc Natl Acad Sci USA 2006; 103:6952 - 6957
- Fox S, Eicher E. Juvenile polycystic kidneys—jpk. Mouse News Letter 1978; 58:47
- Horton CE Jr, Davisson MT, Jacobs JB, Bernstein GT, Retik AB, Mandell J. Congenital progressive hydronephrosis in mice: a new recessive mutation. J Urol 1988; 140:1310 - 1315
- Chevalier RL, Peters CA. Congenital urinary tract obstruction: Proceedings of the State-Of-The-Art Strategic Planning Workshop-National Institutes of Health, Bethesda, Maryland USA, 11–12 March 2002. Pediatr Nephrol 2003; 18:576 - 606
- Batourina E, Choi C, Paragas N, et al. Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nat Genet 2002; 32:109 - 115
- Batourina E, Tsai S, Lambert S, et al. Apoptosis induced by vitamin A signaling is crucial for connecting the ureters to the bladder. Nat Genet 2005; 37:1082 - 1089
- Viana R, Batourina E, Huang H, et al. The development of the bladder trigone, the center of the anti-reflux mechanism. Development 2007; 134:3763 - 3769
- Dressler GR. The cellular basis of kidney development. Annu Rev Cell Dev Biol 2006; 22:509 - 529
- Schedl A. Renal abnormalities and their developmental origin. Nature Reviews genetics 2007; 8:791 - 802
- Brenner-Anantharam A, Cebrian C, Guillaume R, Hurtado R, Sun TT, Herzlinger D. Tailbud-derived mesenchyme promotes urinary tract segmentation via BMP4 signaling. Development 2007; 134:1967 - 1975
- Seifert AW, Harfe BD, Cohn MJ. Cell lineage analysis demonstrates an endodermal origin of the distal urethra and perineum. Dev Biol 2008; 318:143 - 152
- Miyazaki Y, Ichikawa I. Ontogeny of congenital anomalies of the kidney and urinary tract, CAKUT. Pediatr Int 2003; 45:598 - 604
- Pope JCt, Brock JW 3rd, Adams MC, Stephens FD, Ichikawa I. How they begin and how they end: classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J Am Soc Nephrol 1999; 10:2018 - 2028