Abstract
We previously reported that some cattle affected by bovine spongiform encephalopathy (BSE) showed distinct molecular features of the protease-resistant pion protein (PrPres ) in Western blot, with a 1-2 kDa higher apparent molecular mass of the unglycosylated PrPres associated with labelling by antibodies against the 86-107 region of the bovine PrP protein (H-type BSE). By Western blot analyses of PrPres, we now showed that the essential features initially described in cattle were observed with a panel of different antibodies and were maintained after transmission of the disease in C57Bl/6 mice. In addition, antibodies against the C-terminal region of PrP revealed a second, more C-terminally cleaved, form of PrPres (PrPres #2), which, in unglycosylated form, migrated as a ≈ 14 kDa fragment. Furthermore, a PrPres fragment of ≈ 7kDa, which was not labelled by C-terminus-specific antibodies and was thus presumed to be a product of cleavage at both N- and C-terminal sides of PrP protein, was also detected. Both PrPres #2 and ≈ 7 kDa PrPres were detected in cattle and in C57Bl/6 infected mice. These complex molecular features are reminiscent of findings reported in human prion diseases. This raises questions regarding the respective origins and pathogenic mechanisms in prion diseases of animals and humans.
Introduction
Transmissible spongiform encephalopathies (TSE) are fatal neurodegenerative diseases, affecting both humans (Creutzfeldt-Jakob disease; CJD) and animals, such as sheep and goats (scrapie), deer and elk (chronic wasting disease; CWD) and cattle (bovine spongiform encephalopathy; BSE). Although the nature of the infectious agent causing these diseases still remains controversial,Citation1–Citation3 a central event in their pathogenesis is the accumulation of an abnormal form of the host-encoded prion protein, in infected tissues. Under conditions where the normal cellular protein (PrP C) is fully sensitive to proteases, the disease-associated prion protein (PrPd) is only degraded to a partly resistant fragment (PrPres) by removal of the amino-terminal end of PrPd. The sizes of PrPres fragments and their N-terminal amino acid residues of PrPres may vary between different prion strains.Citation4–Citation10
Experimental transmissions of these diseases revealed a biological diversity, reminiscent of strains of classical infectious agents such as viruses.Citation11 This has mainly been described in genetically defined inbred wild-type mice by the different features of the disease, which include differences in incubation periods and in the distribution of brain lesions.Citation12 Different and specific features of the PrPres protein have also been found in mice or hamsters infected with different biological strains of prion diseases, suggesting that biological properties of the infectious agent might be enciphered in the conformation of the protein.Citation4–Citation10
It was believed until recently that TSEs in cattle were caused by a single strain of infectious agent identified as responsible for the food-borne BSE epidemic. Characterization of the infectious agent associated with BSE indeed showed unique features, with well-defined incubation periods and distribution of brain lesions following transmission of the disease to wild-type mice, not only directly from cattle, but also, after natural or experimentally-induced cross-species transmissions.Citation13,Citation14 The surprisingly uniform features of the disease in cattle were also illustrated by analysing the distribution of neurodegenerative brain lesions in affected cattle, at different places and periods of the BSE epidemic.Citation15,Citation16 However, twenty years after identification of the first cases in cattle, the origin of the actual BSE agent remains controversial.Citation17,Citation18 The possible recycling of an infectious agent derived from prion diseases found naturally in other species, such as scrapie in sheep and goats, has been the most famous hypothesis, but the possible existence of a previously unidentified “sporadic” form of prion disease in cattle, as are most cases of Creutzfeldt-Jakob disease in humans, has also been proposed.Citation19
Western blot analyses of PrPres accumulating in the brains of BSE-infected animals and humans have demonstrated specific molecular features, compared to most other forms of prion diseases, with a low molecular mass of the unglycosylated PrPres moiety and high proportions of diglycosylated PrPres.Citation8 Recent studies however have reported cases of prion abnormalities in cattle with distinctly deviant PrPres features.Citation20,Citation21 We previously described three French cattle isolates essentially characterized by a higher apparent molecular mass of unglycosylated PrPres (later called H-type isolates) and decreased levels of diglycosylated PrPres, in comparison to BSE.Citation20 Also, only PrPres from H-type isolates was labelled by monoclonal antibody P4 with defined PrPres N-terminus epitope specificity, in contrast with the PrPres from BSE isolates, suggesting a different cleavage of the protein by proteinase K (PK).Citation20 We recently demonstrated that such H-type isolates of bovine prion diseases were transmissible to wild-type mice, and that these specific features initially described in cattle as distinct from typical BSE, were maintained in mice.Citation22 Similar results were also obtained after transmission in bovine and ovine transgenic mice, favouring the hypothesis that a distinct strain of prion should be involved in H-type BSE.Citation23
We now further describe the existence of complex molecular features of these H-type cases in cattle, by Western blot analyses of PrPres using a panel of anti-PrP antibodies, and demonstrate the transmissibility of these features after transmission of the disease in C57Bl/6 mice. These data are discussed with respect to their similarities with different prion diseases in humans.
Materials and Methods
Cattle TSE isolates.
The H-type BSE isolates used in the mouse transmission experiments in this study included two cases which showed, as previously described with RB1 polyclonal antibody, a higher apparent molecular mass of unglycosylated PrPres, as well as labelling by P4 monoclonal antibody.Citation20 A typical BSE case was used in comparison in both sets of mouse transmission experiments. Main features of these three cases, identified from active surveillance at rendering plants, were previously reported.Citation21
Mouse inoculations and follow-up.
Four to six weeks old female C57Bl/6 mice (Charles Rivers, L'Arbresle, France) were inoculated intracerebrally, following anaesthesia, with 20 ml per mouse of a 10% (wt/vol) homogenate prepared from cattle brain stem samples in glucose (5%) using disposable blenders. Mice were cared for and housed according to the European guidelines (directive 86/609/EEC) and of the French Ethical Committee (decree 87–848), and were supplied with food and drink ad libitum. Following experimental challenge, experiments were conducted in the Biohazard prevention area (A3) of the author's institution with the approval of the Rhône-Alpes Ethical Committee for Animal Experiments. The mice were then checked twice a week for the presence of clinical signs, such as tremors, plastic tail, coarse coat, abnormal gait, hind limb paralysis, clasping feet, hunched posture, bradykinesia or leanness. The animals were sacrificed by anaesthetic solution overdose when they exhibited any signs of distress or confirmed evolution of clinical signs of prion disease; in a few cases they were found dead. The whole brain of every second mouse was frozen and stored at -80°C before Western Blot analysis and the other brains were fixed in paraformaldehyde 4% for histopathological studies.
Western blot analysis.
(i) Extraction of PrPres. PrPres was extracted from bovine brain stem samples using the TeSeE® Western blot Bio-Rad kit (Ref 355 1169) following the manufacturer's instructions. Briefly, 250µl of 20% brain homogenate were incubated with an equal volume of reconstituted proteinase K solution (reagent A + PK) at 37°C for 10 min. After adding 250 µl of reagent B, samples were centrifuged at 15,000 x g for 7 min. The pellets were resuspended and heated for 5 min at 100°C in 50 µl of denaturing buffer (TD4215)(4% sodium dodecyl sulfate, 2% β-mercaptoethanol, 192 mM glycine, 25 mM Tris and 5% sucrose) and centrifuged at 12,000 g for 15 min. Finally, the pellets were discarded and the supernatants were run on a sodium dodecyl sulphate polyacrylamide gel. Mouse brain tissues representing half of the whole brain after sagittal section were homogenized in a 5% glucose solution (10% wt/vol). The homogenates were forced through a 0.4-mm diameter needle before incubation for 1h at 37°C with proteinase K (PK) (10 µg/100 mg of brain tissue). Samples were then incubated for 15 min with N-lauroylsarkosyl (final concentration 10%) and centrifuged at 100,000 rpm for 2h on a 10% sucrose cushion (Beckman TL100 ultracentrifuge). The pellets were resuspended and heated in denaturing buffer as previously described, before SDS-PAGE. (ii) Western Blotting procedures. Proteins were separated by SDS-PAGE on 15% polyacrylamide gels and transferred to 45 µm nitrocellulose membranes (Amersham) in a 25 mM Tris, 192 mM glycine, 10% isopropanol buffer at 400 mA constant for 1h. For immunoblotting, the membranes were blocked for 1h with fat milk 5% (12B2, SAF84), TeSeE® Western blot Bio-Rad kit Blocking Solution (Sha31) or 3% bovine serum albumin in PBS-Tween 20 (0.1%) (PBST)(other antibodies). They were then incubated for 30 min at room temperature with antibodies against different PrP domains, as summarised in . After washes in PBST, the membranes were incubated 30 min at room temperature with peroxidase-labelled conjugates diluted in PBST against mouse IgG (TeSeE® Western blot Bio-Rad conjugate (1/10) for Sha31 antibody or anti-mouse IgG (H+L) from Clinisciences (ref 1010-05)(1:2500) for other monoclonal antibodies) or against rabbit IgG (1:2500)(Clinisciences ref 41010-05) for polyclonal antibody RB1. Streptavidin (5 ng/ml)(S5512, Sigma) was added to the conjugate solution. The membranes were finally washed three times in PBST and once in PBS, and bound antibodies were detected by enhanced chemiluminescence ECL (Amersham) or Supersignal (Pierce) and visualized either on film (Biomax, Kodak) or directly in an image analysis system for quantitative measurements (Versadoc, Biorad). (iii) Analyses. For quantitative studies of the glycoform ratios, chemiluminescent signals corresponding to the three glycoforms of the protein were measured by using Quantity One (Biorad) software. Glycoform ratios were expressed as mean percentages (± standard deviations) of the total signal from at least three separate gel runs. Apparent molecular masses (Mr) of PrPres glycoforms were determined as the average of the centre positions of the bands from at least 3 repeated runs, measured in comparison with a biotinylated marker (B2787, Sigma) run on each gel. In experiments for quantitative measures of the apparent molecular masses and glycoform ratios, PrPres from C57Bl/6-infected mice infected with C506M3 scrapie strain was also used as a control.Citation4 Immunological reactivities of the most N-terminus-specific antibodies (P4, 12B2, SAF32, 4F2) on samples were assessed by visual examination and quantitative image analysis, in comparison with analyses run in parallel on the same samples using antibody Sha31.
PNGase treatment.
Deglycosylation was performed using PNGase F (kit P07043, BioLabs). Denatured samples of PrPres in TD4215 buffer (1–2 µl) were mixed with denaturing buffer from the PNGase kit, G7 buffer, NP40 and PNGase according to the manufacturer's instructions. After incubation at 37°C for 1 h, samples were ready for Western blot analysis following appropriate dilution in TD4215 buffer.
Results
PrPres characterization in cattle TSE isolates.
We characterized the H-type cattle BSE isolates, in comparison with typical BSE, using a panel of antibodies against different regions of the PrP protein (). Results showed a quite different Western blot pattern according to the antibodies used ().
The first group of antibodies (group A), which include antibodies recognizing the most N-terminal region of the PrPres (amino-acids 62–107), exhibited an unusual three band pattern at ≈28, 23, and 19 kDa in H-type BSE, whereas the staining of PrPres in typical BSE, in contrast, was very poor (). In addition, low levels of a small ≈7 kDa PrPres fragment could be detected following overexposure of the membranes (arrowhead in inset of ).
A second group of antibodies (group B), against epitopes in the 110–164 amino-acids region, recognized the same PrPres pattern with almost the same apparent molecular masses (Mr) as the group A antibodies. The group B antibodies however did also stain the three PrPres bands in typical BSE. The PrPres pattern recognized by group B antibodies in the H-type isolates was however characterized by a 1–2 kDa higher Mr of each of the three PrPres bands in comparison to those of the PrPres in typical BSE ().
A third group of antibodies (group C), against the more C-terminal part of the PrP protein (154–236), showed a strikingly different pattern in H-type isolates from that obtained with the group A and B antibodies (). This pattern was characterized by (i) the presence of four bands at 28, 23, 19 and 14 kDa (respectively referred as bands I to IV), of which those at 23 (II) and 19 (III) kDa are quite broad (ii) the predominance of the band II, in contrast to the typical BSE isolate that showed a clear predominance of the diglycosylated PrPres band (see bracket in ), and (iii) the width of the band III (see arrow in ), with diffuse labelling below the position of the well-defined 19 kDa band as recognised by groups A or B antibodies. Taken together, in H-type BSE, the group A and group B antibodies show similar PrPres binding to a triple-band PrPres population in the Mr 19–30 kDa range, whereas the group C antibodies reveal the presence of more complex pattern with four main bands, including also a lower band at 14 kDa. Such an unusual pattern with the additional ≈14 kDa band was not observed using SAF84 (group C) antibody in any of more than 200 cases otherwise characterized, after analysis using 6H4 (group B) antibody, by PrPres molecular weights of typical BSE.
Analysis of PrPres after deglycosylation by PNGase treatment first confirmed the higher PrPres apparent molecular mass (≈19 kDa) in H-type BSE compared to typical BSE (), as well as the more intense labelling using group A antibodies (). In addition the group C antibodies specifically recognized an additional ≈14 kDa in H-type BSE (). This strongly suggests that PrPres from H-type isolates consists of a mixture of two populations - PrPres#1 (producing the ≈ 19 kDa unglycosylated band) and PrPres#2 (producing the ≈ 14 kDa unglycosylated band). Regarding PrPres#2, (i) a diglycosylated form derived from the ≈ 14 kDa band would overlap with the monoglycosylated band of PrPres #1, and (ii) a monoglycosylated form derived from the ≈ 14 kDa band could lead to the more diffuse labelling observed below the position of the ≈ 19 kDa unglycosylated band of PrPres#1. As shown in (), this interpretation is consistent with the unusual and complex Western profiles specifically observed with group C antibodies prior to deglycosylation.
PrPres characterization in C57Bl/6 mice infected with H-type and typical BSE isolates.
The molecular features associated with H-type isolates from cattle were further studied in brain from wild-type mice (C57Bl/6) inoculated with either of the two bovine H-type isolates, in comparison with a typical BSE isolate. Most C57Bl/6 mice were positive for PrPres by Western blot. However, the PrPres levels were significantly lower in mice infected with H-type isolates than in BSE-infected mice since, when group B antibodies were used, four times as much brain tissue from H-type infected mice was needed per lane to obtain equivalent levels of PrPres signals to those found in mice infected with typical BSE (). In these latter conditions, and in relation to the recognition of an additional PrPres form (PrPres #2) with SAF84, the detected PrPres signal was strongly increased in H-type infected mice compared to BSE-infected mice ().
PrPres from Western blot-positive mice was further characterized for comparison with the molecular features previously described in cattle, by analysing the PrPres staining patterns with antibodies representative of the 3 above-defined groups. Using the same immunological reagents as in studies with bovine tissues, analysis of the ≈14 kDa region of the gel was hampered by an unspecific immunoreactivity in this region, which was also detected in uninfected mouse brain (). This was not linked to PrP since it is also detected in the absence of anti-PrP primary antibody () and from PrP 0/0 mouse brain tissue (data not shown).
With group A antibody 12B2, the H-type mouse PrPres was stained, but that of typical BSE only very poorly () and when the group B antibody Sha31 was used, the PrPres in C57Bl/6 mice inoculated with H-type isolates revealed a 1–2 kDa higher Mr for the three PrPres bands compared to those observed in BSE-infected mice ( and ). The differences in Mr of the PrPres bands observed with group B antibodies were maintained after deglycosylation by PNGase (). These data are consistent with a PrPres #1 population as observed in cattle with group A and B antibodies, and show that the PK resistance of the PrP region recognized by group A antibodies was specifically preserved in mice infected with H-type isolates, whereas it was largely removed from BSE-infected mice. In addition to the triple band pattern of PrPres, a conspicuous ≈7 kDa band was also identified using 12B2 and even more strongly with Sha31 (), which could also be identified after deglycosylation (). The labelling in the ≈14 kDa region that was clearly apparent with 12B2 antibody remained consistent with the unspecific labelling found in normal mice, even in the absence of anti-PrP primary antibody.
Using group C antibodies (SAF84 and R524) the same distinct 4-band pattern was observed in mice as described in cattle (compare with ). Although the ≈ 14 kDa in mice might at least in part result from unspecific labelling, this pattern was characterized by the two other salient features of the SAF84 WB profile in H-type BSE in cattle, namely (i) the predominance of band II (≈40% of signal of bands I–IV together, compared to ≈30% for band I) () (see bracket in ) and (ii) the width and diffuse aspect of band III (see arrow in ). All together these data are consistent with the presence of PrPres #2 in mouse brains, as described in cattle. Accordingly, an ≈14 kDa PrPres form, corresponding to unglycosylated PrPres #2, was indeed clearly detected with group C antibody SAF84 after PNGase deglycosylation, in addition to the unglycosylated PrPres #1 at ≈19 kDa ().
Glycoform analyses with Sha31 antibody showed comparable proportions (≈40% of the 19–30 kDa signal) of the diglycosylated and monoglycosylated bands in H-type BSE (), the ratios of which were closer to those observed in a mouse-adapted scrapie strain (C506M3) than in mice infected with typical BSE (≈55% of diglycosylated band).
Discussion
Our study provides new information regarding the molecular characterization of one of two deviant phenotypes described in cattle diagnosed with BSE.Citation20,Citation21 In contrast with the previously described uniformity of BSE in cattle, this phenotype was initially revealed by the higher apparent molecular mass of the unglycosylated PrPres, compared to that of typical BSE. This molecular mass difference is determined by an extended N-terminus of PrPres that only occurs in the deviant phenotype, as evidenced by the presence of epitope 101-WGQGGTH-107 of bovine PrP.Citation4,Citation20,Citation22,Citation24,Citation25 This higher apparent molecular mass of PrPres has now been confirmed using 11 other antibodies (groups B and C) which recognize different epitopes along the central or C-terminal part of the PrP protein, and with three other antibodies (group A) against its more N-terminal end, all of which showed a prominent binding to the PrPres from H-type isolates.
Some of the antibodies used during the study (group C), revealed an unexpected feature of H-type isolates leading to the identification of a second, previously unrecognised form of PrPres (14–24 kDa)(PrPres #2), in addition to the previously described PrPres (19–30 kDa)(PrPres #1). This appears as a major new molecular feature that distinguishes H-type form typical BSE. Although both the size of PrPres #2 and its specific labelling only with the most C-terminus-specific antibodies suggest that it differs from PrPres #1 by a more C-terminal cleavage, the data also imply the presence of glycosyl-groups. The coexistence of different PrPres cleavage products, including different glycosylated fragments, was also recently reported in the recently described form of atypical scrapie or Nor98, which is characterized by very unusual PrPres Western blot features.Citation26 Although N-terminal sequencing of PrPres was not considered in this study, epitope mapping suggests a cleavage of the PrP region around amino-acid 154–163. This situation is similar to that recently described in several forms of Creutzfeldt-Jakob disease, including sporadic,Citation27 iatrogenicCitation28 and geneticCitation29 CJD in which the presence of C-terminally cleaved fragments (11–13 kDa) were revealed by using C-terminus-specific antibodies. A similar finding was also reported in cases of human Gerstmann-Sträussler-Sc heinker (GSS) syndrome but only in those patients in which the ≈21–29 kDa PrPres was also detected.Citation30
Beside the two isolates here described that were transmitted to C57Bl/6 mice, similar molecular features were also obtained in five other H-type isolates identified in France at abattoir or at slaughterhouse, but also in similar cases recently identified from active surveillance in other countries (Netherlands, Germany, Poland, Sweden, USA, Switzerland)(1 case in each of these countries).Citation31–Citation33 Interestingly the case identified in Switzerland had been identified in a miniature zebu (Bos indicus) kept in a zoological park, which allowed detailed investigations of the case in very good sampling conditions.Citation33 Detailed study of this case also showed, in agreement with Western blot results, that PrPd in the central nervous system was labelled by immunohistochemistry using P4 antibody, which was not the case in a typical BSE case under the conditions used. All together our data also clearly show that the presence of lower molecular weights PrPres forms is not linked to the quality of the cattle brain samples, which in some cases may be autolysed, since similar features were found in all the samples examined in the natural host (13 cases), including some samples of very good quality.
Furthermore, we then used selected antibodies to analyse the PrPres features following transmission of the disease in C57BL/6 mice, and showed that the salient features previously recognized in cattle were faithfully maintained in mice. We have already reported the higher apparent molecular mass of PrPres #1 in C57Bl/6 mice, associated with presence of the WGQGG epitope.Citation22 We now report the additional recognition in these mice of the PrPres #2 form, as identified in cattle, by group C C-terminal antibodies. As in H-type infected cattle, this resulted in a stronger detection of total PrPres by SAF84 antibody in H-type BSE than in typical BSE. Western blot analyses in mice also showed the presence of an additional ≈7 kDa band recognised by antibodies 12B2 and Sha31 (groups A and B respectively), i.e., corresponding to the most N-terminal region in comparison to those recognized by group C antibodies. A similar fragment could also be detected in the H-type cattle brain stems, but clearly at lower levels (compare left and right lanes in inset of . It is presently unknown whether this is associated with the bovine-to-mouse transmission. It should also be emphasized that the studies of bovine brains were limited to the brain stem, whereas the PrPres analyses in mice were performed on the whole brain. The precise mechanisms involved in PrPres formation in such cases remain to be elucidated, and a proposed interpretation of the PrPres cleavage is shown in . Such a ≈7 kDa PrPres fragment, non-reactive with group C C-terminus-specific antibodies, might potentially represent an N-terminal fragment of PrPres #2 which would roughly correspond to the region 85–156 of bovine PrP. However, the identification of low molecular weight PrPres fragments cleaved at both C- and N-terminal ends of the prion protein is reminiscent of a major feature of Gerstmann-Sträussler-Scheinker (GSS) syndrome in humans, in which this may even be the only detected PrPres fragment in the absence of PrPres 27–30.Citation30,Citation34,Citation35 Sequencing of the open reading frame in the H-type isolates in cattle has already shown that such cases are not necessarily associated with changes in the PrP sequenceCitation20,Citation31–Citation33 as is the case in human GSS. Interestingly, it should be noted that recently the identification of a proteinase K resistant N- and C-terminally truncated form of PrPres has also been reported in atypical scrapie.Citation26,Citation37 Although this PrPres fragment appears to be a signature of this recently described form of prion disease in sheep,Citation38 it is not associated with a specific genetic change.Citation26,Citation39
It has been stated that the abnormal PrP deposits detected by immunohistochemistry in C57Bl/6 mice infected with H-type isolates are only present as amyloid plaques, in contrast to BSE in such mice in which only diffuse deposits are found.Citation22,Citation40 PrP amyloids are also consistently detected in human GSS, in contrast to most other forms of Creutzfeldt-Jakob disease.Citation41 Unfortunately, due to the lack of fixed material, histopathological studies could not be considered in these initial cattle samples. It is thus unknown whether these remarkable features in mice reflect a property of the pathological process also present in the original host, since amyloid formation was shown to be strongly influenced by the primary structure of PrP.Citation42,Citation43 In GSS, the presence of a C-terminally cleaved 8 kDa PrPres fragment was associated with the presence of multicentric amyloid deposits, with which it colocalizes.Citation29,Citation44 The high propensity of synthetic PrP peptides corresponding to N- and C-terminally cleaved PrP, such as the human 85–148 sequence, to form amyloid fibrils was also demonstrated in vitro.Citation45,Citation46
Similarities of PrPres in H-type BSE in cattle with that found in different human prion diseases, including both CJD in its different forms and GSS, raise crucial questions regarding the molecular mechanisms involved in PrPres accumulation in such cases in cattle. Among human prion diseases, both human GSS and genetic CJD could be transmitted experimentally, in some cases, to animals such as rodents or primates expressing only a wild-type prion protein.Citation47–Citation50 These latter data clearly indicate that a host expressing a normal form of the prion protein can develop such forms of prion diseases, although molecular analyses of the specific features associated with these forms of human diseases have been poorly described in experimental models.
Our data obtained from one of the deviant phenotypes of prion diseases in cattle (H-type) showed some remarkable molecular similarities with human prion diseases. Although the origin of such cases is still unknown, these findings raise important questionsCitation17–Citation19 regarding the respective origins and pathogenic mechanisms of natural prion diseases in humans and in animals prion diseases.
Abbreviations
| BSE | = | bovine spongiform encephalopathy |
| CJD | = | Creutzfeldt-Jakob disease |
| CWD | = | chronic wasting disease |
| GSS | = | Gerstmann-Sträussler-Scheinker |
| H-type BSE | = | high type bovine spongiform encephalopathy |
| PK | = | proteinase K |
| PNGase | = | peptide N-glycosidase F |
| PrPd | = | disease-associated prion protein |
| PrPres | = | protease-resistant prion protein |
| PrPSc | = | scrapie prion protein |
| TSE | = | transmissible spongiform encephalopathy |
Figures and Tables
Figure 1 Molecular discrimination of H-type and typical BSE in cattle and in C57Bl/6 infected mice. Western blot detection of PrPres in cattle (A–C) or C57Bl/6 infected mice (E–G), from H-type isolate (right lane in each panel) or typical BSE (left lane in each panel), using 12B2 (A and E), Sha31 (B and F) or SAF84 (C and G) monoclonal antibodies. In each Western blot, typical BSE is shown in the left lane and H-type BSE in the right lane. Brain equivalent quantities loaded per lane were 100 µg (left lanes) and 250 µg (right lanes) for cattle samples and 1.2 mg (left lanes) and 4.8 mg (right lanes) for mouse samples. Inset of (A) shows the N-terminal PrPres fragment (≈7 kDa) in cattle (left lane) and mouse (right lane). (D) Interpretation of the Western blot profile observed with SAF84 monoclonal antibody. (H) Unspecific immunoreactivity of anti-mouse Ig conjugate on samples prepared from normal brain of C57Bl/6 mouse.
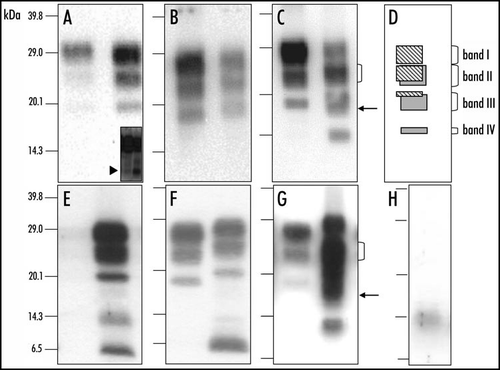
Figure 2 Molecular analysis of PrPres after deglycosylation in C57Bl/6 infected mice. Western blot analysis of PrPres from cattle (A–C) or C57Bl/6 mice (D–F) infected with H-type (lanes 3 and 4) or typical BSE (lanes 1 and 2), after deglycosylation by PNGase treatment. Monoclonal antibodies used for PrPres detection were 12B2 (A and D), Sha31 (B and E) or SAF84 (C and F). Brain equivalent quantities loaded per lane were (i) from cattle, for typical BSE, 50 (lane 1) or 2,6 (lane 2) mg, and for H-type BSE, 50 (lane 3) or 6 (lane 4) mg, and (ii) from mice, for typical BSE, 0.5 (D and F) or 0.3 (E) mg, and for H-type BSE, 2.5 (D and F) or 1.3 mg (E).
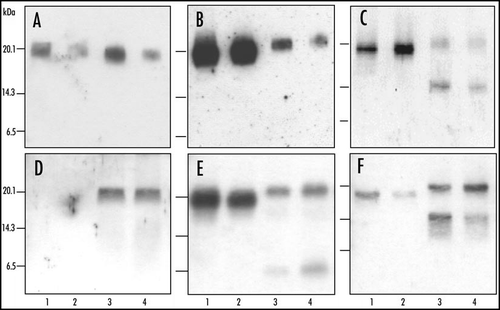
Figure 3 Quantitative molecular features of PrPres in C57Bl/6 infected mice. Molecular masses (A) and glycoform ratios (B) (means ± standard deviations) of PrPres detected with Sha31 antibody, from C57Bl/6 mice infected with H-type or typical BSE isolates. PrPres of mice infected with an experimental scrapie (C506M3) strainCitation4 was used as a control.
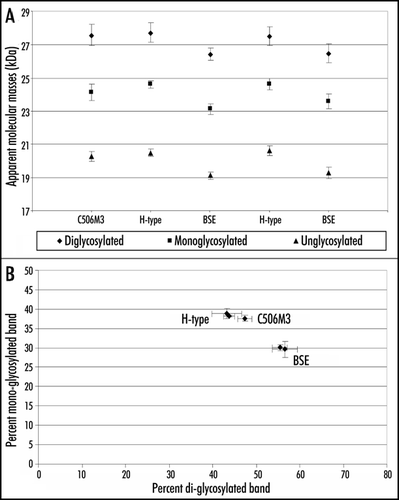
Figure 4 Schematic representation of proteinase K-resistant PrP fragments identified from H-type or typical BSE. Putative regions for proteinase K cleavage are indicated, as assessed by immunoreactivities with anti-PrP antibodies, and categories of epitope locations (groups A, B and C).
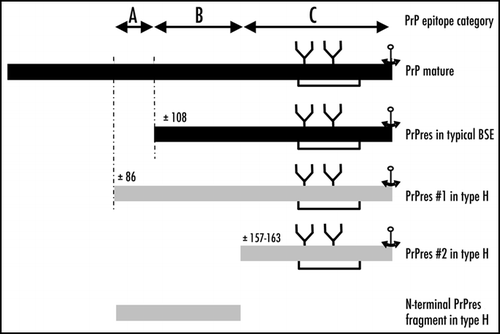
Table 1 Antibodies used to characterize H-type and typical BSE
Acknowledgments
We greatly acknowledge the technical assistance of Dominique Canal, Jérémy Verchère and Johann Vulin for the Western blot experiments and of Emilie Antier and Clément Lavigne for the follow-up of animal experiments, as well as Diana Warwick for careful reading of the manuscript. We also acknowledge collaborative work on H-type cases with A. Buschmann and M. Groschup (FLI, Germany), M. Polak (NVRI, Poland), D. Gavier-Widen (SVA, Sweden), J. Richt (USDA, USA) and T. Seuberlich (NC, Switzerland). This work was supported by grants from the Neuroprion European network of Excellence (FOOD-CT-2004-506579 EUROSTRAINS project) and from GIS ”Infections à prions.“
References
- Chesebro B. Prion protein and the transmissible spongiform encephalopathy diseases. Neuron 1999; 24:503 - 506
- Lasmézas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, Fournier JG, Hauw JJ, Rossier J, Dormont D. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 1997; 275:402 - 405
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Baron TGM, Biacabe AG. Molecular analysis of the abnormal prion protein during coinfection of mice by bovine spongiform encephalopathy and a scrapie agent. J Virol 2001; 75:107 - 114
- Baron T, Crozet C, Biacabe AG, Philippe S, Verchère J, Bencsik A, Madec JY, Calavas D, Calavas D. Molecular analysis of the protease-resistant prion protein in scrapie and bovine spongiform encephalopathy transmitted to ovine transgenic and wild-type mice. J Virol 2004; 78:6243 - 6251
- Kuczius T, Haist I, Groschup MH. Molecular analysis of bovine spongiform encephalopathy and scrapie strain variation. J Infect Dis 1998; 178:693 - 699
- Somerville RA, Chong A, Mulqueen OU, Birkett CR, Wood SCER, Hope J. Biochemical typing of scrapie strains. Nature 1997; 386:564 (with response from Collinge J, et al.).
- Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996; 383:685 - 690
- Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Nongenetic propagation of strain-specific properties of scrapie prion protein. Nature 1995; 375:698 - 700
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathological isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274:2079 - 2082
- Bruce ME, Dickinson AG. Biological evidence that scrapie agent has an independent genome. J Gen Virol 1987; 68:79 - 89
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 1968; 78:301 - 311
- Fraser H, Bruce ME, Chree A, McConnell I, Wells GA. Transmission of bovine spongiform encephalopathy and scrapie to mice. J Gen Virol 1992; 73:1891 - 1897
- Green R, Horrocks C, Wilkinson A, Hawkins SAC, Ryder SJ. Primary isolation of the bovine spongiform encephalopathy agent in mice: Agent definition based on a review of 150 transmissions. J Comp Pathol 2004; 40:1 - 15
- Simmons MM, Harris P, Jeffrey M, Meek SC, Blamire IWH, Wells GAH. BSE in Great Britain: Consistency of the neurohistopathological findings in two random annual samples of clinically suspect cases. Vet Rec 1996; 138:175 - 177
- Orge L, Simas JP, Fernandes AC, Ramos M, Galo A. Similarity of the lesion profile of BSE in Portuguese cattle to that described in British cattle. Vet Rec 2000; 147:486 - 488
- Colchester CF, Colchester NTH. The origin of bovine spongiform encephalopathy: The human prion disease hypothesis. Lancet 2005; 366:856 - 861
- Baron T, Biacabe AG. Origin of bovine spongiform encephalopathy. Lancet 2006; 367:297 - 298
- Commission E. Opinion on: Hypotheses on the origin and transmission of BSE. Brussels: EC Health and Consumer Protection Directorate General 2001; 1 - 67
- Biacabe AG, Laplanche JL, Ryder SJ, Baron T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep 2004; 5:110 - 114
- Casalone C, Zanusso G, Acutis P, Ferrari S, Cappuci L, Tagliavini F, Monaco S, Caramelli M. Identification of a second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA 2004; 101:3065 - 3070
- Baron T, Biacabe AG, Bencsik A, Langeveld JPM. Transmission of new bovine prion to mice. Emerg Infect Dis 2006; 12:1125 - 1128
- Béringue V, Bencsik A, Le Dur A, Reine F, Laï TL, Chenais N, Tilly G, Biacabe AG, Baron T, Vilotte JL, Laude H. Isolation from cattle of a prion strain distinct from that causing bovine spongiform ncephalopathy. PLoS Pathog 2006; 2 (doi:10.1371/journal.ppat.0020112).
- Thuring CMA, van Keulen LJM, Langeveld JPM, Vromans MEW, van Zijderveld FG, Sweeney T. Immunohistochemical distinction between preclinical bovine spongiform encephalopathy and scrapie infection in sheep. J Comp Pathol 2005; 132:59 - 69
- Stack J, Chaplin MJ, Clark J. Differentiation of prion protein glycoforms from naturally occurring sheep scrapie sheep-passaged scrapie strains (CH1641 and SSBP1) bovine spongiform encephalopathy (BSE) cases and Romney and Cheviot breed sheep experimentally inoculated with BSE using two monoclonal antibodies. Acta Neuropathol 2002; 104:279 - 286
- Arsac JN, Andreoletti O, Bilheude JM, Lacroux C, Benestad S, Baron T. Similar biochemical signatures and prion protein genotypes in atypical scrapie and Nor98 cases, France And Norway. Emerg Infect Dis 2007; 13:58 - 65
- Zou WQ, Capellari S, Parchi P, Sy MS, Gambetti P, Chen SG. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J Biol Chem 2003; 278:40429 - 40436
- Satoh K, Muramoto T, Tanaka T, Kitamoto N, Ironside JW, Nagashima K, Yamada M, Sato T, Mohrio S, Kitamoto T. Association of an 11–12 kDa protease-resistant prion protein fragment with subtype of dura graft-associated Creutzfeldt-Jakob disease and other prion diseases. J Gen Virol 2003; 84:2885 - 2893
- Capellari S, Parchi P, Russo CM, Sanford J, Sy MS, Gambetti P, Petersen RB. Effect of the E200K mutation on prion protein metabolism: Comparative study of a cell model and human brain. Am J Pathol 2000; 157:613 - 622
- Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, Gajdusek GC, Gambetti P. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc Natl Acad Sci USA 1998; 95:8322 - 8327
- Buschmann A, Gretzschel A, Biacabe AG, Schiebel K, Corona C, Hoffmann C, Eiden M, Baron T, Casalone C, Groschup MH. Atypical BSE in Germany - Proof of transmissibility and biochemical characterization. Vet Microbiol 2006; 117:103 - 116
- Richt JA, Kunkle RA, Alt D, Nicholson EM, Hamir AN, Czub S, Kluge J, Davis AJ, Hall SM. Identification and characterization of two bovine spongiform encephalopathy cases diagnosed in the United States. J Vet Diagn Invest In press.
- Seuberlich T, Botteron C, Wenker C, Café-Marçal V, Oevermann A, Haase B, Leeb T, Heim D, Zurbriggen A. Spongiform encephalopathy in a miniature zebu. Emerg Infect Dis 2006; 12:1950 - 1953
- Piccardo P, Seiler C, Dlouhy SR, Young K, Farlow MR, Prelli F, Frangione B, Bugiani O, Tagliavini F, Ghetti B. Proteinase-K-resistant prion protein isoforms in Gerstmann-Sträussl er-Scheinker disease (Indiana kindred). J Neuropath Exp Neurol 1996; 55:1157 - 1163
- Piccardo P, Liepnieks JJ, William A, Dlouhy SR, Farlow MR, Young K, Nochlin D, Bird TD, Nixon RR, Ball MJ, DeCarli C, Bugiani O, Tagliavini F, Benson MD, Ghetti B. Prion proteins with different conformations accumulate in Gerstmann-Straussler-Scheinker disease caused by A117V and F198S mutations. Am J Pathol 2001; 158:2201 - 2207
- Buschmann A, Gretzschel A, Biacabe AG, Schiebel K, Corono C, Hoffmann C, Eiden M, Baron T, Casalone C, Groschup MH. Atypical BSE in Germany: Proof of transmissibility and biochemical characterisation. Vet Microbiol 2006; 117:103 - 116
- Klingeborn M, Wik L, Simonsson M, Renström LHM, Ottinger T, Linné T. Characterization of proteinase K-resistant N- and C-terminally truncated PrP in Nor98 atypical scrapie. J Gen Virol 2006; 87:1751 - 1760
- Benestad SL, Sarradin P, Thu B, Schonheit J, Tranulis MA, Bratberg B. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet Rec 2003; 153:202 - 208
- Moum T, Olsaker I, Hopp P, Moldal T, Valheim M, Benestad SL. Polymorphisms at codons 141 and 154 in the ovine prion protein gene are associated with scrapie Nor98 cases. J Gen Virol 2005; 86:231 - 235
- Brown DA, Bruce M, Fraser JR. Comparison of the neuropathological characteristics of bovine spongiform encephalopathy (BSE) and variant Creutzfeldt-Jakob (vCJD) in mice. Neuropath Appl Neuro 2003; 29:262 - 272
- Tagliavini F, Prelli F, Porro M, Rossi G, Giaccone G, Farlow MR, Dlouhy SR, Ghetti B, Bugiani O, Frangione B. Amyloid fibrils in Gerstmann-Sträussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell 1994; 79:695 - 703
- Scott M, Foster D, Mirenda C, Serban D, Coufal F, Walchli M, Torchia M, Groth D, Carlson G, DeArmond SJ, Westaway D, Prusiner SB. Transgenic mice expressing hamster prion protein produce species- specific scrapie infectivity and amyloid plaques. Cell 1989; 59:847 - 857
- Scott M, Groth D, Foster D, Torchia M, Yang SL, DeArmond SJ, Prusiner SB. Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell 1993; 73:979 - 988
- Piccardo P, Dlouhy SR, Lievens PMJ, Young K, Dlouhy SR, Bird TD, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IR, Kish SJ, Ang LC, De Carli C, Pocchiari M, Brown P, Gibbs CJ, Gajdusek DC, Bugiani O, Ironside J, Tagliavini F, Ghetti B. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropath Exp Neurol 1998; 57:979 - 988
- Tagliavini F, Prelli F, Verga L, Giaccone G, Sarma R, Gorevic P, Ghetti B, Passerini F, Ghibaudi E, Forloni G, Salmona M, Bugiani O, Frangione B. Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc Natl Acad Sci USA 1993; 90:9678 - 9682
- Tagliavini F, Lievens PMJ, Tranchant C, Warter JM, Mohr M, Giaccone G, Perini F, Rossi G, Salmona M, Piccardo P, Ghetti B, Beavis RC, Bugiani O, Frangione B, Prelli F. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Straussler-Scheinker disease A117V. J Biol Chem 2001; 276:6009 - 6015
- Tateishi J, Kitamoto T. Inherited prion diseases and transmission to rodents. Brain Pathol 1995; 5:53 - 59
- Tateishi J, Kitamoto T, Hoque MZ, Furukawa H. Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology 1996; 46:532 - 537
- Brown P, Gibbs CJJ, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 1994; 35:513 - 529
- Baker HF, Duchen LW, Jacobs JM, Ridley RM. Spongiform encephalopathy transmitted from Creutzfeldt-Jakob and familial Gerstmann-Sträussler-Scheinker diseases. Brain 1990; 113:1891 - 1909
- Feraudet C, Morel N, Simon S, Volland H, Frobert C, Creminon D, Vilette S, Lehmann S, Grassi J. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem 2005; 280:11247 - 11258
- Harmeyer S, Pfaff E, Groschup MH. Synthetic peptide vaccines yield monoclonal antibodies to cellular and pathological prion proteins of ruminants. J Gen Virol 1998; 79:937 - 945
- Madec JY, Groschup MH, Buschmann A, Belli P, Calavas D, Baron T. Sensitivity of the Western blot detection of prion protein PrPres in natural sheep scrapie. J Virol Methods 1998; 75:169 - 177
- O'Rourke KI, Baszler TV, Besser TE, Miller JM, Cutlip RC, Wells GAH, Ryder SJ, Parish SM, Hamir SM, Cockett NE, Jenny A, Knowles DP. Preclinical diagnosis of scrapie by immunohistochemistry of third eyelid lymphoid tissue. J Clin Microbiol 2000; 38:3254 - 3259
- Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R, Schultz-Schaeffer W, Kretzschmar H, Raeber A, Braun U, Ehrensperger F, Hornemann S, Glockshuber R, Riek R, Billeter M, Wuthrich K, Oesch B. Prion (PrPsc)-specific epitope defined by a monoclonal antibody. Nature 1997; 390:74 - 77
- Garssen GJ, Van Keulen LJM, Farquhar CF, Smits MA, Jacobs JG, Bossers A, Meloen RH, Langeveld JM. Applicability of three anti-PrP peptide sera including staining of tonsils and brainstem of sheep with scrapie. Microsc Res Techniq 2000; 50:32 - 39
- Thuring CM, Erkens JHF, Jacobs JG, Bossers A, van Keulen LJM, Garssen GJ, van Zijderveld FG, Ryder SJ, Groschup MH, Sweeney T, Langeveld JPM. Discrimination between scrapie and bovine spongiform encephalopathy in sheep by molecular size immunoreactivity and glycoprofile of prion protein. J Clin Microbiol 2004; 42:972 - 980
- Langeveld JPM, Jacobs JG, Erkens JHF, Bossers A, Van Zijderveld FG, Van Keulen LJM. Rapid and discriminatory diagnosis of scrapie and BSE in retro-pharyngeal lymph nodes of sheep. BMC Vet Res 2006; 2:19