Abstract
There is strong evidence that the oligomers of key proteins, formed during the early steps of aggregation, could be the primary toxic species associated with human neurodegenerative diseases, such as Alzheimer’s and prion diseases. Here, we review recent progress in the development of computational approaches in order to understand the structures, dynamics and free energy surfaces of oligomers. We also discuss possible research directions for the coming years.
Introduction
More than 20 human diseases are associated with the aggregation of key proteins. For instance, the 40- and 42-residue amyloid β-protein (Aβ),Citation1 99-residue microglobulin (β2m) protein,Citation2 and 210-residue PrP prion protein,Citation3 are linked to Alzheimer's disease, dialysis-related amyloidosis, and prion diseases, respectively. All these proteins have different amino acid sequences and biophysical properties in solution: Aβ is random coil,Citation4 β2m has an all-β topology while PrP is essentially α-helical,Citation5 however, they all misfold and aggregate into amyloid fibrils with a cross-β structure, characterized by β-sheets perpendicular to the fibril axis.Citation6–Citation10
Although the amyloid plaques are one of the hallmarks of the diseases, there is strong evidence that early oligomers during the aggregation process may be the primary toxic species.Citation11–Citation15 The initial assemblies of oligomers are difficult to characterize at an atomic level of detail using biophysical methods, because they are transient and in dynamic equilibrium between dimers, trimers, tetramers etc. The experimental signature of the rate-limiting step is the presence of a lag phase in polymer growth, which varies with protein concentration, salt and agitation. Once a nucleus is present, maturation into fibrils is rapid.Citation16–Citation18 It follows that we have very limited information on both the secondary and tertiary structures of these early oligomers, although an α-helix signal has been detected by circular dichroism,Citation19,Citation20 and based on antibody experiments,Citation21 they may share a common topology, different from the fibrils, independently of the amino acid sequence.
In this context, computer simulations of protein aggregation have provided insights into the early steps of amyloid formation. Such theoretical investigation is made possible by the fact that peptides of four to seven amino acids can form fibrils indistinguishable from those formed by proteins. For example, a reptation mechanism first identified numerically,Citation22,Citation23 seems to be dominant in the rearrangement of amyloid chains.Citation24 These results, and others presented below, show that simulations can complement and even guide experimental measurements in the study of amyloid aggregation. In this review, we limit ourselves to coarse-grained protein aggregation simulations taken largely from our work. Excellent reviews on the use of all-atom molecular dynamicsCitation25 or discontinuous molecular dynamics simulationsCitation26 to understand protein aggregation or bioinformaticsCitation27 to predict regions promoting amyloid fibril formation can be found elsewhere.
In the first part, we report the progress in the development of computational approaches to monitor amyloid fibril formation with their strengths and weaknesses. Then, we present the main results that have been extracted from coarse-grained protein simulations in order to understand the structures, dynamics and free energy surfaces of oligomers. Finally, we list three directions that others and we will probably follow in the field.
In Silico Approaches to Simulate Amyloid Aggregation
Molecular dynamics (MD), which integrates directly Newton's equation of motion, offers the most detailed picture at the atomic level, providing both dynamical and thermodynamic information. Because of the integration 2 fs timestep, all-atom MD in explicit solvent is limited to trajectories of 1 µs—typically about 100 ns. Such a time scale might be sufficient to study the stability of preformed structuresCitation28–Citation34 such as the cross-beta-spine steric zipper of the Sup-35 prion fragment,Citation7,Citation35 the very early events in the dynamics of AβCitation36–Citation40 or the docking of unstructured monomer on preformed structured oligomers,Citation41 but other methods of various degrees of efficiency and accuracy are needed to span the aggregation regime from monomers to fibrils, which requires several days in vitro.
A first approach is the replica exchange procedure, initially proposed by Swendsen and WangCitation42 and then reformulated with a molecular dynamical scheme by Sugita and Okamoto.Citation43 The replica exchange molecular dynamics (REMD) mixes a series of MD trajectories (or replica) run in parallel at different temperatures through Monte Carlo accept/reject moves. The probability of exchanging two conformations i and j run at Th and Tl, respectively, is given by min {1,exp[(βh - βl)(Ej - Ei)]} where β is 1/kT. Because REMD allows conformations to move between various T, one expects a more extensive sampling of the low-energy structures than with standard MD, providing a better description of the thermodynamic properties of the system, at the cost of the loss of dynamical information. However, even at high temperature, the decorrelation time can be longer than the simulations in slow systems. This is the case, for example, when explicit solvent is used or when large assemblies of full-length proteins are studied.Citation44 It follows that this limitation directly affects the quality of the results. Another severe limitation of REMD is that the number of replicas increases rapidly with the size of the system, limiting its applicability to small all-atom systems in solvent such as the monomer of full-length Aβ, and dimers and trimers of short fragments.Citation45–Citation48
The use of implicit solvent and coarse-grained models lifts some of the limitations of both MD and REMD on two counts. First, by removing some of the dampening due to the collision with the solvent, they accelerate the sampling of the phase space by as much as two orders of magnitude.Citation49 Second, the simplification of the potential decreases the computational cost allowing longer simulations.Citation50–Citation52 For example, Jang and Shin could generate multiple 150-ns MD trajectories for a trimer of Aβ(10–35) using a Born-generalized implicit solvent.Citation50 Similarly, Paci et al could reach a total time of 0.5 µs on tetramers of TTR(105–115).Citation53 The use of implicit solvent and coarse-grained models do not affect all motions uniformly, however. This is not important for thermodynamics, but these modifications can affect the details of the aggregation dynamics.Citation54
Implicit solvent protein models also allow the use of a wider range of methods. Activated approaches, such as the activation-relaxation technique (ART nouveau) for example,Citation55,Citation56 explores the space of conformations, through well-defined transition states. Coupled with the implicit solvent coarse-grained protein OPEP force field,Citation57,Citation58 this method has provided the best fit with the NMR data of the fragment Aβ(21–30) in solutionCitation59 and has allowed to monitor the aggregation of various amyloid peptides with 4 to 15 amino-acids, in settings ranging from dimer to dodecamer.Citation22,Citation23,Citation60–Citation66 Because it lacks detailed balance ART cannot provide solid thermodynamic information. Trajectories are physically-based, however, and are qualitatively correct, based on comparisons with other approaches using a 16-residue b-hairpin model.Citation67
Other approaches have also been used to characterize the first steps of aggregation. Irbäck et al uses a simplified off-lattice potential with a Monte Carlo approach based on two elementary backbone moves. This reduction in motion speeds up the simulations, making it possible to follow aggregation of hexamers of Aβ(16–22).Citation68 This is also the case of discrete molecular dynamics (DMD), which requires a much-simplified force field with square-well interactions in order to evolve the time based on collisions. Such an approach would be unworkable in the presence of explicit solvent, but with implicit solvent DMD generates trajectories corresponding to seconds or more, treating up to 100 chains or so.Citation69–Citation71 It remains to be determined whether the very simple force field used provides the correct dynamics or even the proper thermodynamics. More characterization on a wide range of sequences and structures is clearly necessary at this moment.
Free Energy Surfaces of Dimers
It is well established that the early steps of amyloid-fibril formation are characterized by the formation of low molecular weight oligomers consisting of a mixture of dimers, trimers, tetramers, and more in rapid equilibrium.Citation17,Citation18 Because Aβ exists as a stable dimer in solution at low concentrations,Citation72,Citation73 and dimers may act as seeds for larger oligomers, there has been considerable theoretical efforts to characterize the free energy surface and the dynamics of dimer formation using various protein models.Citation22,Citation44,Citation74–Citation78
Here, we probe the free energy surface of the Aβ(16–22) dimer, resulting from a 50-ns REMD-OPEPCitation79 simulation in a sphere of 70 Å diameter, starting from two chains in random orientation. The integration timestep is 1 fs and T is controlled by the Berendsen's bathCitation80 with a coupling constant of 0.1 ps. We use eight replicas with T varying between 287 and 500 K with exponential distribution and an exchange time between neighboring replicas of 20 ps, leading to an acceptance ratio between 30–40%.
shows the free energy surface of Aβ(16–22) dimer at 310 K. The two reaction coordinates are the cosine of the angle between the two chains and the extended status of the chains. The extended status is the product of the end-to-end distance of the chains divided by the product of the end-to-end distance for two ideal β-strands. We see multiple free energy minima. These are in-register and out-of-register parallel strands (structures A, B and D), parallel chains (structure C), cross chains (structure E), antiparallel loops (structure F), and antiparallel strands (structures G and H). The Boltzmann probabilities of structures A–H at 310 K are: 1, 1, 6, 5, 3, 21, 10 and 12%. The calculated percentage of β-strand content is 36% at 310 K. Taken together, these results indicate that the antiparallel arrangement of the chains is preferred over the parallel arrangement, in agreement with previous reports.Citation22,Citation77 The in-register and out-of-register antiparallel β-structures (structures G and H) are similar in free energies, helping explain the experimental dependency of β-sheet registry on pH conditions.Citation81 It also follows that this free energy surface generated by REMD-OPEP is very similar in character to that generated by all-atom explicit solvent REMD simulations.Citation44 This finding is interesting for two reasons. First, it shows that cross conformations (structure E, ) are also populated using a coarse-grained protein model with implicit solvent if thermal fluctuations are considered. Second, it opens the door to the study of the free energy surfaces for many peptides in dimers or higher-order species using reasonable computer resources. We find, for example, that the free energy surface of the human β2m(83–89) dimer is very similar to that shown in , although the 7-residue sequences differ: KLVFFAE for Aβ(16–22) vs. NHVTLSQ for β2m(83–89).
Structures and Dynamics of Transient Oligomeric Species
While simulations of dimers and trimers in solution provide insights into the populated structures,Citation23,Citation41,Citation49,Citation48 larger systems are needed to better understand the early events of aggregation and the dynamics leading to ordered structures. Experimentally, we often observe amorphous and annular aggregates prior to the formation of protofibrils.Citation82,Citation83
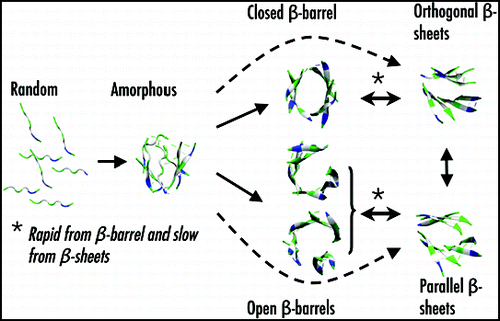
Using ART and MD simulations coupled to OPEP, we study the aggregation of a number of peptides of increasing length (from 4 to 15 amino acids) and oligomer size (from trimer to dodecamer), starting from random orientations of the chains and random coil conformations of each chain. While we observe a high percentage of ordered β-sheet structures for trimers and tetramers of Aβ(16–22) and KFFE,Citation23,Citation63 tetramers and heptamers of β2m(83–89),Citation84,Citation85 and tetramers to octamers of KFFE,Citation60,Citation61,Citation63,Citation64 self-assembly to fibrillar structures is more problematic for a tetramer of Aβ(11–25)Citation62 and a dodecamer of NFGAIL.Citation66 shows a generic aggregation picture leading to fibrillar states, emerging from all these simulations. Starting from an initial state with all chains randomly placed, the peptides first come together to form amorphous aggregates with two-stranded or three-stranded β-sheet rapidly in place. Then, the outcome of both ART and MD simulations using OPEP varies according to the chain length and the oligomer size.
Small oligomer size and small chain length.
For a system containing less than 8 chains of small lengths, the amorphous aggregates evolve either directly to fully ordered structures (orthogonal or parallel β-sheets) or indirectly through closed or open β-barrels. The generated parallel ordered structures display the cross-β characteristics observed experimentally,Citation7 with Cα..Cα distances of 5.0 Å between the strands and around 10.0 Å between the layers. In contrast, orthogonal β-sheets only display the meridional 5.0 Å reflection. Both structures have been observed by MC simulations of six Aβ(16–22) chainsCitation68 and DMD simulations of polyalaninesCitation69 and prion fragments.Citation70 Similarly, orthogonal layers have been observed by all-atom MD simulations starting from parallel or antiparallel layers.Citation31,Citation63,Citation64 The amorphous aggregates, the bilayer β-sheet and the β-barrel structures are all in dynamic equilibrium.Citation64,Citation65 Using MD-OPEP on seven β2m(83–89) chains, we find that the transition is more rapid from β-barrel to β-sheets than from β-sheets to β-barrel, and the estimated time scale for both reactions is on the order of the ms range in explicit solvent.Citation85 Of course, this time will increase with the oligomeric size, and the shape of the β-barrel will also vary with the oligomer size and the length of the peptides. Seven or eight chains of KFFE can only form open β-barrel,Citation61,Citation64 while seven chains of β2m(83–89) are sufficient to stabilize into a closed β-barrel with hydrogen-bonding interactions between all chains.
We identify two important mechanisms in all ART simulations, and more recently in MD simulations of four Aβ(16–22) chains.Citation52 The first one is the reptation move of the chains. This motion allows the chains to rearrange their H-bond networks without having to fully detach, decreasing significantly the free-energy cost of realignment.Citation22,Citation52,Citation62 This move has recently been observed experimentally in oligomers formed by Aβ(16–22) using isotope-edited IR spectroscopy.Citation24 The second mechanism we find refers to as the two-stage dock-lock mechanism for adding a monomer on a preformed oligomer.Citation41,Citation66 Both mechanisms are likely general processes for error-correction in the early steps of protein aggregation, but also in amyloid growth by monomer addition.Citation60,Citation66,Citation86
Large oligomer size or long chain length.
When the number of chains is twelve or more, or when the chain length increases to 15 amino acids, aggregation to ordered β-sheet structures is very rare, and the peptides form amorphous aggregates that are in dynamic equilibrium. Among a total of 12 ART simulations of twelve NFGAIL chains, one run locates highly ordered β-sheet structure within 12,000 events.Citation66 Similarly, multiple simulations of four Aβ(11–25) chains fail to explore a four-stranded β-sheet within 30,000 events.Citation62 In both cases, the elongation process is difficult, as the amorphous oligomer is dense, even when detachment/reattachment of the peptides and reptations moves of the chains are considered. This implies that, just like in the case of monomeric protein folding, there are many kinetic traps and the acquisition of a β-sheet oligomeric structure requires minimal frustration and a low conformational entropy.Citation66
Future Directions
Simulating protein aggregation is a challenging problem that pushes the limits of current methods. Progress will likely be achieved by combining these various methods to take advantages of their particular strengths while minimizing their weaknesses. Thus far, most simulations of oligomers have been performed in solution at various pH and concentration conditions. While they have already provided insights into self-assembly pathways and structures of oligomers, three open questions remain to be addressed.
First, it is essential that these simulations are repeated in more realistic cellular environment, with full treatment of metallic ions (especially copper) and membrane. Interactions with copperCitation87 and membraneCitation37 have already been investigated by short MD simulations, but they only explore local fluctuations around the starting structures. In the case of Aβ, we may go one step beyond and incorporate the effect of cholesterol, and even apolipoprotein E and its allele A4, but the latter system is problematic because there is no structure available.Citation88
Second, there have been many reports suggesting constrained pathways leading to fibrils, but very few solid verifications. Several theoretical studies have suggested intermediate states with α-helixCitation89 and even α-sheet character.Citation90 Similarly, an α-helical signal has been detected by circular dichroism in the late steps of Aβ aggregation, but there is no evidence that these intermediates are on-pathways.Citation17 Rather, our simulations on Aβ(16–22), NFGAIL, Aβ(11–25) and β2m(83–89)Citation23,Citation62,Citation66,Citation85 suggest several pathways for self-assembly, in agreement with experimental data, and transient sampling of species including amorphous aggregates, α-helical intermediates with a very low population, and β-barrels. This β-barrel differs from the disorganized annular prototype seen by DMDCitation91 and microscopy measurements.Citation83 While we do not know if the β-barrel is accessible to full-length Aβ, its structural characteristics makes it an ideal system to create pores within the membranes, which in turn could contribute to the enhanced toxicity of the oligomeric intermediates.Citation83
Third, there has been no theoretical attempt to target the interaction sites between current inhibitors and oligomers at an atomic level of detail. These compounds reported to block amyloid aggregation are based either on D-peptides, N-methylated peptides, peptides containing proline substitutions.Citation92–Citation94 For instance, Meredith et al find that the N-methylated Aβ(16–22) peptide at positions 17, 19 and 21 inhibits the fibrillogenesis of full-length Aβ.Citation95 Similarly, Yang et al showed that curcumin inhibits formation of Aβ oligomers and fibrils, and reduces amyloid in vivo.Citation96 This is a very difficult numerical problem, but free energy surfaces of oligomers-inhibitors using coarse-grained models should tell us where inhibitors bind, as this would help rational drug design.
Figures and Tables
Figure 1 Free energy surface of Aβ(16–22) dimer at 310 K obtained from REMD-OPEP simulation. The two reaction coordinates used are the cosine of the angle between the two chains and the extended status of the two chains. The structures A–H of low free energy (in kcal/mol) are shown.
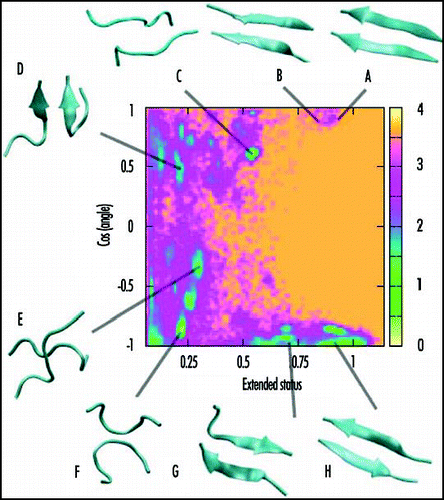
Figure 2 A generic aggregation picture derived from ART- and MD-OPEP simulations. Starting from a randomly chosen state, the peptides form amorphous aggregates. From there, the outcome changes with the oligomer size (OS) and chain length (L). For OS < 9 and L < 8, rapid aggregation proceeds directly to ordered β-sheets or indirectly through β-barrels. The double arrows indicate reversibility. For larger OS or L, aggregation into β-barrels and ordered β-sheets is very slow and rare.
Acknowledgements
G.W. thanks the National Natural Science Foundation of China under Grant No.10674029, the Young Foundation of Fudan University, the Senior Visiting Scholar Grant of Fudan University. N.M. acknowledges partial funding from the NSERC, FQRNT and the Canada Research Chair Program. P.D. acknowledges financial support from CNRS, University Paris 7 and the 6th European PCRD. N.M. and P.D. also thank financial support from the Alzheimer Society of Canada.
References
- Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol 1998; 7:447 - 453
- Gejyo F, Homma N, Suzuki Y, Arakawa M. Serum levels of beta 2-microglobulin as a new form of amyloid protein in patients undergoing long-term hemodialysis. New Engl J Med 1986; 314:585 - 586
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science 2004; 305:673 - 676
- Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJ, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. Solution NMR studies of the Abeta(1–40) and Abeta(1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc 2004; 126:1992 - 2005
- Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 2000; 97:145 - 150
- Sunde M, Serpell L, Bartlam M, Fraser P, Pepys M, Blake C. Common core structure of amyloid fibrils by synchrotron x-ray diffraction. J Mol Biol 1997; 273:729 - 739
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005; 435:773 - 778
- Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer's beta-amyloid fibrils. Biochemistry 2006; 45:498 - 512
- Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer's amyloid-beta(1–42) fibrils. Proc Natl Acad Sci USA 2005; 102:17342 - 17347
- Ferguson N, Becker J, Tidow H, Tremmel S, Sharpe TD, Krause G, Flinders J, Petrovich M, Berriman J, Oschkinat H, Fersht AR. Proc Natl Acad Sci USA 2006; 103:16248 - 16253
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416:483 - 484
- Lesne S, Teng Koh M, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallaher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006; 440:352 - 357
- Cohen F, Kelly J. Therapeutic approaches to protein-misfolding diseases. Nature 2003; 426:905 - 909
- Dobson C. Protein folding and misfolding. Nature 2003; 426:884 - 890
- Chiti F, Dobson C. Protein misfolding, functional amyloid, and human disease. Ann Rev Biochem 2006; 75:333 - 366
- Hardy J, Selkoe DJ. Science 2002; 297:353 - 356
- Bitan G, Vollers SS, Teplow DB. Elucidation of primary structure elements controlling early amyloid-b protein oligomerisation. J Biol Chem 2003; 278:34882 - 34889
- Mastrangelo I, Ahmed M, Sato T, Liu W, Wang C, Hough P, Smith SO. High-resolution atomic force microscopy of soluble abeta42 oligomers. J Mol Biol 2006; 358:106 - 109
- Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J Mol Biol 2001; 312:1103 - 1119
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis: Detection of a protofibrillar intermediate. J Biol Chem 1997; 272:22364 - 22372
- Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006; 66:S74 - S78
- Santini S, Wei GH, Mousseau N, Derreumaux P. Pathway complexity of Alzheimer's beta-amyloid Abeta16–22 peptide assembly. Structure 2004; 12:1245 - 1255
- Santini S, Mousseau N, Derreumaux P. In silico assembly of Alzheimer's Abeta16–22 peptide into beta-sheets. J Am Chem Soc 2004; 126:11509 - 11516
- Petty SA, Decatur SM. Experimental evidence for the reorganization of beta-strands within aggregates of the Abeta(16–22) peptide. J Am Chem Soc 2005; 127:13488 - 13489
- Ma B, Nussinov R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr Opin Chem Biol 2006; 10:445 - 452
- Hall CK, Wagoner VA. Computational approaches to fibril structure and formation. Methods Enzymol 2006; 412:338 - 365
- Caflisch A. Computational models for the prediction of polypeptide aggregation propensity. Curr Opin Chem Biol 2006; 10:437 - 444
- Ma B, Nussinov R. Molecular dynamics simulations of alanine rich β-sheet oligomers: Insight into amyloid formation. Protein Sci 2002; 11:2335 - 2350
- Zanuy D, Nussinov R. The sequence dependence of fiber organization: A comparative molecular dynamics study of the islet amyloid polypeptide segments 22–27 and 22–29. J Mol Biol 2003; 329:565 - 584
- Wu C, Lei H, Duan Y. Elongation of ordered peptide aggregate of an amyloidogenic hexapeptide NFGAIL observed in molecular dynamics simulations with explicit solvent. J Am Chem Soc 2005; 127:13530 - 13537
- Lopez de la Paz M, de Mori GM, Serrano L, Colombo G. Sequence dependence of amyloid fibril formation: Insights from molecular dynamics simulations. J Mol Biol 2005; 349:583 - 596
- Colombo G, Daidone I, Gazit E, Amadei A, Di Nola A. Molecular dynamics simulation of the aggregation of the core-recognition motif of the islet amyloid polypeptide in explicit water. Proteins 2005; 59:519 - 527
- Rohrig UF, Laio A, Tantalo N, Parrinello M, Petronzio R. Stability and structure of oligomers of the Alzheimer peptide Aβ16–22: From the dimer to the 32-mer. Biophys J 2006; 91:3217 - 3229
- Buchete NV, Tycko R, Hummer G. Molecular dynamics simulations of Alzheimer's beta-amyloid protofilaments. J Mol Biol 2005; 353:804 - 821
- Esposito L, Pedone C, Vitagliano L. Molecular dynamics analyses of cross-beta-spine steric zipper models: beta-sheet twisting and aggregation. Proc Natl Acad Sci USA 2006; 103:11533 - 11538
- Massi F, Straub JE. Probing the origins of increased activity of the E22Q “Dutch” mutant Alzheimer's beta-amyloid peptide. Biophys J 2001; 81:697 - 709
- Xu Y, Shen J, Luo X, Zhu W, Chen K, Ma J, Jiang H. Conformational transition of amyloid beta-peptide. Proc Natl Acad Sci USA 2005; 102:5403 - 5407
- Han W, Wu YD. A strand-loop-strand structure is a possible intermediate in fibril elongation: Long time simulations of amyloid-beta peptide (10–35). J Am Chem Soc 2005; 127:15408 - 15416
- Flock D, Colacino S, Colombo G, Di Nola A. Misfolding of the amyloid β-protein: A molecular dynamics study. Proteins 2006; 62:183 - 192
- Tarus B, Straub JE, Thirumalai D. Probing the initial stage of aggregation of the Abeta(10–35)-protein: Assessing the propensity for peptide dimerization. J Mol Biol 2005; 345:1141 - 1156
- Nguyen PH, Li MS, Stock G, Straub JE, Thirumalai D. Monomer adds to preformed structured oligomers of Abeta-peptides by a two-stage dock-lock mechanism. Proc Natl Acad Sci USA 2007; Jan 2 104:111 - 116
- Swendsen RH, Wang JS. Replica monte carlo simulation of spin-glasses. Phys Rev Letts 1986; 57:2607 - 2609
- Sugita Y, Okamoto Y. Replica-exchange molecular dynmaics method for protein folding. Chem Phys Lett 1999; 314:141 - 151
- Gnanakaran S, Nussinov R, Garcia AE. Atomic-level description of amyloid beta-dimer formation. J Am Chem Soc 2006; 128:2158 - 2159
- Baumketner A, Bernstein SL, Wyttenbach T, Bitan G, Teplow DB, Bowers MT, Shea JE. Amyloid beta-protein monomer structure: A computational and experimental study. Protein Sci 2006; 15:420 - 428
- Wei GH, Shea JE. Solvent effects on the structure of alzheimer's amyloid-β(25–35) peptide. Biophysical J 2006; 91:1638 - 1647
- Nishino M, Sugita Y, Yoda T, Okamoto Y. Structures of a peptide fragment of beta2-microglobulin studied by replica-exchange molecular dynamics simulations—Towards the understanding of the mechanism of amyloid formation. FEBS Lett 2005; 579:5425 - 5429
- Tsai HH, Reches M, Tsai CJ, Gunasekaran K, Gazit E, Nussinov R. Energy landscape of amyloidogenic peptide oligomerization by parallel-tempering molecular dynamics simulation: Significant role of Asn ladder. Proc Natl Acad Sci USA 2005; 102:8174 - 8179
- Gsponer J, Haberthur U, Caflisch A. The role of side-chain interactions in the early steps of aggregation: Molecular dynamics simulations of an amyloid-forming peptide from the yeast prion Sup35. Proc Natl Acad Sci USA 2003; 100:5154 - 5159
- Jang S, Shin S. Amyloid beta-peptide oligomerization in silico: Dimer and trimer. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys 2006; 110:1955 - 1958
- Cecchini M, Rao F, Seeber M, Caflisch A. Replica exchange molecular dynamics simulations of amyloid peptide aggregation. J Chem Phys 2004; 121:10748 - 10756
- Derreumaux P, Mousseau N. Coarse-grained protein molecular dynamics simulations. J Chem Phys 2007; 126:025101 - 025106
- Paci E, Gsponer J, Salvatella X, Vendruscolo M. Molecular dynamics studies of the process of amyloid aggregation of peptide fragments of transthyretin. J Mol Biol 2004; 340:555 - 569
- Zagrovic B, Pande V. Solvent viscosity dependence of the folding rate of a small protein: Distributed computing study. J Comput Chem 2003; 24:1432 - 1436
- Malek R, Mousseau N. Dynamics of Lennard-Jones clusters: A characterization of the activation-relaxation technique. Phys Rev E 2000; 62:7723 - 7728
- Wei GH, Mousseau N, Derreumaux P. Exploring the energy landscape of proteins: A characterization of the activation-relaxation technique. J Chem Phys 2002; 117:11379 - 11387
- Derreumaux P. From polypeptide sequences to structures using Monte Carlo simulations and an optimized potential. J Chem Phys 1999; 111:2301 - 2310
- Derreumaux P. Generating ensemble averages for small proteins from extended conformations by Monte Carlo simulations. Phys Rev Lett 2000; 85:206 - 209
- Chen W, Mousseau N, Derreumaux P. Families of structures for the Alzheimer's fragment Aβ(21–30) in solution by computer simulations. J Chem Phys 2006; 125:1 - 8
- Wei GH, Mousseau N, Derreumaux P. Sampling the self-assembly pathways of KFFE hexamers. Biophys J 2004; 87:3648 - 3656
- Wei G, Mousseau N, Derreumaux P. Assembly dynamics of KFFE octamers. J Phys Condensed Matter 2004; 16:5047 - 5054
- Boucher G, Mousseau N, Derreumaux P. Aggregating the amyloid Abeta(11–25) peptide into a four beta-sheet structure. Proteins 2006; 65:877 - 888
- Melquiond A, Boucher G, Mousseau N, Derreumaux P. Following the aggregation of amyloid-forming peptides by computer simulations. J Chem Phys 2005; 122:174904
- Melquiond A, Mousseau N, Derreumaux P. Structures of soluble amyloid oligomers from computer simulations. Proteins 2006; 65:180 - 191
- Mousseau N, Derreumaux P. Exploring the early steps of amyloid peptide aggregation. Acc Chem Res 2005; 38:885 - 891
- Melquiond A, Gelly JC, Mousseau N, Derreumaux P. Probing amyloid-fibril formation of the NFGAIL peptide by computer simulations. J Chem Phys 2007; 126:25101 - 25106
- Wei G, Derreumaux P, Mousseau N. Sampling the complex energy landscape of a simple beta-hairpin. J Chem Phys 2003; 119:6403 - 6406
- Favrin G, Irback A, Mohanty S. Oligomerization of amyloid Abeta16–22 peptides using hydrogen bonds and hydrophobicity forces. Biophys J 2004; 87:3657 - 3664
- Nguyen HD, Hall CK. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc Natl Acad Sci USA 2004; 101:16180 - 16185
- Ding F, LaRocque JJ, Dokholyan NV. Direct observation of protein folding, aggregation, and a prion-like conformational conversion. J Biol Chem 2005; 280:40235 - 40240
- Urbanc B, Cruz L, Yun S, Buldyrev SV, Bitan G, Teplow DB, Stanley HE. In silico study of amyloid beta-protein folding and oligomerization. Proc Natl Acad Sci USA 2004; 101:17345 - 17350
- Garzon-Rodriguez W, Sepulveda-Becerra M, Milton S, Glabe CG. Soluble amyloid Aβ-(1–40) exists as a stable dimer at low concentrations. J Biol Chem 1997; 272:21037 - 21044
- Huang T, Yang D, Plaskos N, Go S, Yip C, Fraser P, Chakrabartty A. Structural studies of soluble oligomers of the Alzheimer beta-amyloid peptide. J Mol Biol 2000; 297:73 - 87
- Harrison PM, Chan HS, Prusiner SB, Cohen FE. Conformational propagation with prion-like characteristics in a simple model of protein folding. Protein Sci 2001; 10:819 - 835
- Dima RI, Thirumalai D. Exploring protein aggregation and self-propagation using lattice models: Phase diagram and kinetics. Protein Sci 2002; 11:1036 - 1049
- Cellmer T, Bratko D, Prausnitz JM, Blanch H. Protein-folding landscapes in multichain systems. Proc Natl Acad Sci USA 2005; 102:11692 - 11697
- Hwang W, Zhang S, Kamm RD, Karplus M. Kinetic control of dimer structure formation in amyloid fibrillogenesis. Proc Natl Acad Sci USA 2004; 101:12916 - 12921
- Lei H, Wu C, Wang Z, Duan Y. Molecular dynamics simulations and free energy analyses on the dimer formation of an amyloidgenic heptapeptide from human beta2-microglobulin: Implication to the protofibril structure. J Mol Biol 2006; 356:1049 - 1063
- Dong X, Derreumaux P, Mousseau N. J Chem Phys In preparation
- Berendsen HJC, Postma JPM, Di Nola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984; 81:3684 - 3690
- Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. Solid state NMR reveals a pH-dependent antiparallel beta-sheet registry in fibrils formed by a beta-amyloid peptide. J Mol Biol 2004; 335:247 - 260
- Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 2000; 289:1317 - 1321
- Lashuel HA, Lansbury PT. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins?. Q Rev Biophys 2006; 39:167 - 201
- Song W, Wei GH, Mousseau N, Derreumaux P. Free Energy landscapes of four amyloid-forming peptides. J Am Chem Soc Submission
- Song W, Derreumaux P, Mousseau N, Wei GH. In preparation
- Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of prion propagation: Amyloid growth occurs by monomer addition. PLoS Biol 2004; 2:1582 - 1590
- Deng NJ, Yan L, Singh D, Cieplak P. Molecular basis for the Cu2+ binding-induced destabilization of beta2-microglobulin revealed by molecular dynamics simulation. Biophys J 2006; 90:3865 - 3879
- Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, Wisniewski T. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer's disease. Proc Natl Acad Sci USA 2006; 103:18787 - 18792
- Klimov DK, Thirumalai D. Dissecting the assembly of Abeta(16–22) amyloid peptides into antiparallel beta sheets. Structure 2003; 11:295 - 307
- Daggett V. Alpha-sheet: The toxic conformer in amyloid diseases?. Acc Chem Res 2006; 39:594 - 602
- Marchut AJ, Hall CK. Spontaneous formation of annular structures observed in molecular dynamics simulations of polyglutamine peptides. Comput Biol Chem 2006; 30:215 - 218
- Liu D, Xu Y, Feng Y, Liu H, Shen X, Chen K, Ma J, Jiang H. Inhibitor discovery targeting the intermediate structure of beta-amyloid peptide on the conformational transition pathway: Implications in the aggregation mechanism of beta-amyloid peptide. Biochemistry 2006; 45:10963 - 10972
- Wolfe MS. Therapeutic strategies for Alzheimer's disease. Nat Rev Drug Discov 2002; 1:859 - 866
- Kokkoni N, Stott K, Amijee H, Mason JM, Doig AJ. N-Methylated peptide inhibitors of beta-amyloid aggregation and toxicity: Optimization of the inhibitor structure. Biochemistry 2006; 45:9906 - 9918
- Gordon DJ, Sciarretta KL, Meredith SC. Inhibition of beta-amyloid(40) fibrillogenesis and disassembly of beta-amyloid(40) fibrils by short beta-amyloid congeners containing N-methyl amino acids at alternate residues. Biochemistry 2001; 40:8237 - 8245
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 2005; 280:5892 - 5901