Abstract
The serpins are the largest superfamily of protease inhibitors. They are found in almost all branches of life including viruses, prokaryotes and eukaryotes. They inhibit their target protease by a unique mechanism that involves a large conformational transition and the translocation of the enzyme from the upper to the lower pole of the protein. This complex mechanism, and the involvement of serpins in important biological regulatory processes, make them prone to mutation-related diseases. For example the polymerization of mutant α 1-antitrypsin leads to the accumulation of ordered polymers within the endoplasmic reticulum of hepatocytes in association with cirrhosis. An identical process in the neuron specific serpin, neuroserpin, results in the accumulation of polymers in neurons and the dementia FENIB. In both cases there is a clear correlation between the molecular instability, the rate of polymer formation and the severity of disease. A similar process underlies the hepatic retention and plasma deficiency of antithrombin, C1 inhibitor, α 1-antichymotrypsin and heparin co-factor II. The common mechanism of polymerization has allowed us to group these conditions together as a novel class of disease, the serpinopathies.
Serpins (or serine protease inhibitors) are the largest family of protease inhibitors. They have been found in all major branches of life including viruses, prokaryotes and eukaryotes.Citation1–Citation3 Despite their name there is increasing evidence that serpins can also inhibit other classes of proteases as demonstrated by the viral serpin CrmA and recently by a plant serpin, serpin1.Citation4,Citation5 They can even play a non-inhibitory role in events as diverse as blood pressure regulation (angiotensinogen), chromatin condensation (MENT), tumor progression (maspin), protein folding (hsp47) and hormone transport (cortisol and thyroxine binding globulin).Citation6
One of the most important roles of serpins is the regulation of enzymes involved in proteolytic cascades. Among these serpins are α1-antitrypsin, α1-antichymotrypsin, C1 inhibitor, antithrombin and plasminogen activator inhibitor-1, which play an important role in the control of proteases involved in the inflammatory, complement, coagulation and fibrinolytic pathways, respectively.Citation1,Citation3 The serpin superfamily is characterised by more than 30% homology with the archetypal serpin α1-antitrypsin and conservation of tertiary structure.Citation7,Citation8 Serpins adopt a metastable conformation composed in most cases of 9 α-helices, three β-sheet (A to C) and an exposed mobile reactive centre loop (RCL). This flexible RCL typically contains 20 residues that act as a pseudo substrate for the target protease ().Citation9–Citation15 After formation of a Michaelis complexCitation16,Citation17 the enzyme cleaves the P1-P1′ bond of the serpin, releasing the P1' residue and forming an ester bond between the protease and the serpin.Citation18,Citation19 This is then followed by a dramatic conformational transition from a stressed to relaxed conformation with the enzyme being pulled from the upper to the lower pole of the serpin and the insertion of the reactive loop as an extra strand in β-sheet A.Citation20–Citation25 As a consequence of this conformational change the thermal stability of the serpin is greatly enhanced. Whereas a typical serpin in its native state exhibits a midpoint of thermal denaturation of around 50–60°C, a cleaved serpin with its RCL fully incorporated into β-sheet A denatures at temperatures >120°C.Citation9,Citation26,Citation27 Another consequence is the inactivation of the enzyme, stabilised at the acyl-intermediate and unable to proceed further to deacylation of the complex.Citation24,Citation28 This serpin-protease complex then binds to members of the lipoprotein receptor family and is cleared from the circulation.Citation29–Citation31
Despite the evolutionary advantage conferred upon serpins by the remarkable mobility of the native state, their complexity is also their weak point.Citation19,Citation32 Mutations affecting the serpins can lead to a variety of diseases, resulting from either a gain or loss of function.Citation6,Citation19 For example mutations can cause aberrant conformational transitions that result in the retention of the serpin within the cell of synthesis. This will lead to either protein overload and death of the cell in which the serpin is synthesised, or disease as a consequence of the resulting plasma deficiency. Such a mechanism underlies diseases as diverse as cirrhosis, thrombosis, angio-oedema, emphysema and dementia. We review here the common mechanism underlying these diseases that we have grouped together as the serpinopathies.Citation33–Citation35 The aggregation and accumulation of conformationally destabilized proteins is an important feature of many neurodegenerative diseases, including Alzheimer's and Parkinson's disease and the spongiform encephalopathies. Indeed we have used the serpinopathies as a paradigm for these other ‘conformational diseases’.Citation36
Polymerization of α1-Antitrypsin in the Pathogenesis of Cirrhosis and Emphysema
α1-antitrypsin is an acute phase glycoprotein that is synthesised by, and secreted from, the liver. It inhibits neutrophil elastase and therefore plays an important role in the control of the inflammatory response. More than 100 allelic variants have been described with the most clinically relevant being the S (Glu264Val) and Z (Glu342Lys) alleles.Citation37–Citation40 The S allele is found in up to 28% of southern EuropeansCitation39 and reduces plasma levels of α1-antitrypsin to 60% of the normal M protein. The Z allele is present in 4% of northern EuropeansCitation39 and reduces the plasma level to 10–15% of normal. The decrease in plasma level is not associated with any clinical phenotype in the S homozygote but it has dramatic effects in those who are homozygous for the Z allele. The Z mutation causes the retention of α1-antitrypsin in hepatocytes as diastase resistant, periodic acid-Schiff (PAS) positive inclusions that cause both neonatal and adult liver disease ().Citation41–Citation44
Following synthesis, misfolded monomeric Z α1-antitrypsin is degraded by the proteosome but 10–15% folds normally and traffics through the secretory pathway to be released into the circulation. Disease results from the proportion of Z α1-antitrypsin that folds to form polymers. These accumulate within the endoplasmic reticulum (ER) of hepatocytes to form inclusions and hence cause disease.Citation44,Citation45 Biochemical, biophysical and crystallographic studies have elucidated the molecular basis of the polymerization of Z α1-antitrypsin. The mutation associated with the Z allele is located at the head of strand 5 of β-sheet A and the base of the mobile reactive loop (). This mutation causes a conformational transition and the formation of an unstable intermediate that we have called M*. M* is characterised by partial insertion of the RCL and opening of β-sheet A. The patent β-sheet A can then accept the loop of another molecule to form a loop-sheet dimer, which extends to form longer chains of loop-sheet polymers.Citation44–Citation49 Polymers activate the ER overload response but their ordered nature allows them to escape the surveillance of the unfolded protein response. Indeed this pathway is only activated in the presence of a secondary insult.Citation50,Citation51 More recently it has become apparent that polymers can be handled by autophagic pathways within hepatocytes.Citation52–Citation55
Further evidence of the importance of α1-antitrypsin polymers in the pathogenesis of liver disease is provided by two other mutants of α1-antitrypsin that are similarly associated with plasma deficiency and hepatic inclusions: α1-antitrypsin Siiyama (Ser53Phe)Citation56 and Mmalton (ΔPhe52).Citation57 The Siiyama variant is the commonest cause of α1-antitrypsin deficiency in Japan whilst the Mmalton variant is the commonest cause of α1-antitrypsin deficiency in the isolated island of Sardinia. Both of these mutants disrupt a hydrogen bond network based on His334 that bridges strands 3 and 5 of β-sheet A (the shutter domain; and ),Citation58 causing it to open and allow the formation of folding intermediatesCitation59 and loop-sheet polymers in vivo.Citation60,Citation61 The mild S (Glu264Val) and I (Arg39Cys) variants of α1-antitrypsin also lie in the shutter domain and can also form polymers in vivo. However they do so at a slower rate and this polymer formationCitation47 is associated with a mild plasma deficiency and no clinical phenotype.Citation62,Citation63 However, if a slowly polymerizing S (Glu264Val) or I (Arg39Cys) variant is inherited with a fast polymerizing Z variant then they will form heteropolymers that accumulate within hepatocytes and lead to cirrhosis.Citation63–Citation65 Thus there is a striking genotype-phenotype correlation between the rate of polymerization, the retention of α1-antitrypsin within the liver, and the severity of the plasma deficiency.
The reduction in the circulating level of α1-antitrypsin predisposes the Z homozygote to early onset, panlobular, basal emphysema.Citation66–Citation68 This predisposition is particularly apparent in Z α1-antitrypsin homozygotes who smoke as the combination of low levels of α1-antitrypsin within the lung and the inflammation caused by smoking have a dramatic effect on lung function.Citation69,Citation70 The intrapulmonary deficiency of α1-antitrypsin is exacerbated by the effect of the point mutation which reduces the association kinetics with neutrophil elastase by 5-fold and thus the ability of the protein to protect against proteolytic damage. Z α1-antitrypsin enters the lung by passive diffusion. It is also secreted by macrophages and bronchial epithelial cells. In both cases it contains the Z mutation and hence the propensity to form polymers. Indeed polymers of α1-antitrypsin have been detected in bronchial lavage and tissue sections from Z α1-antitrypsin homozygotes.Citation71,Citation72 These polymers are inactive as protease inhibitors and so further deplete the antiprotease screen within the lung. Pulmonary polymers of Z α1-antitrypsin are chemotactic for neutrophils in vitro and following instillation into the lungs of mice.Citation72–Citation74 Thus our understanding of the pathways of polymerization has provided new insights into the associated emphysema. However the importance of these polymers in driving the associated inflammation and emphysema remains to be clarified.Citation75,Citation76
Polymerization of Antithrombin, C1 Inhibitor, α1-Antichymotrypsin and Heparin Co-Factor II Causes the Retention of Protein within Hepatocytes and Plasma Deficiency
The phenomenon of loop-sheet polymerization is not restricted to α1-antitrypsin and has now been reported in mutants of other members of the serpin superfamily to cause disease. Naturally occurring mutations have been described in the shutter and other domains of the plasma proteins C1-inhibitor (Phe52Ser, Pro54Leu, Ala349Thr, Val366Met; Phe370Ser, Pro391Ser), antithrombin (Pro54Thr, Asn158Asp, Phe229Leu) and α1-antichymotrypsin (Leu55Pro, Pro229A1a). These mutations destabilise the serpin architecture to allow the formation of inactive reactive loop-β-sheet polymers that are also retained within hepatocytes. The associated plasma deficiency results in uncontrolled activation of proteolytic cascades and angio-oedema, thrombosis and chronic obstructive pulmonary disease respectively (reviewed in refs. 6 and 33–35). More recently a mutation in heparin co-factor II (Glu428Lys) has been associated with plasma deficiency but as yet this has not been shown to cause disease.Citation77 The mutation is of particular interest as it is the same as the Z allele that causes polymerization and deficiency of α1-antitrypsin. We have shown that this same mutation also causes temperature dependent polymerization and inactivation of mutants of the Drosophila serpin Necrotic.Citation78
Neuroserpin Polymers and the Dementia Familial Encephalopathy with Neuroserpin Inclusion Bodies (FENIB)
Perhaps the most striking disease associated with serpin polymerization is the dementia ‘familial encephalopathy with neuroserpin inclusion bodies’ or FENIB. This is characterised by the accumulation of mutant neuroserpin as PAS positive diastase-resistant inclusions or Collin's bodies within the deep layers of the cerebral cortex ().Citation79–Citation81 These inclusions, like those associated with Z α1-antitrypsin within hepatocytes, are formed of tangles of ordered polymers within the ER. Kindreds with FENIB present with presenile dementia and cognitive deficits unlike those of Alzheimer's or Huntington Diseases.Citation82
The first mutation of neuroserpin that causes FENIB was identified in a large Irish-American family and was termed Syracuse to recognise the origin of the pedigree. The Syracuse mutation (Ser49Pro) is in the shutter domain at an identical location to a previously described mutation in α1-antitrypsin that forms polymers in association with liver disease (the Siiyama mutation).Citation80,Citation83 An examination of the brain from affected individuals showed that the inclusions were composed solely of mutant neuroserpin that had formed chains of loop-β-sheet polymers.Citation80,Citation84 Three other mutations of neuroserpin have since been described, all of which are within the shutter domain: Portland (Ser52Arg), His338Arg and Gly392Glu.Citation81 There is a direct association between the severity of the mutation (as predicted by molecular modelling) and the number of inclusions and an inverse correlation with the age of onset of dementia. For example, the original family members with the Syracuse mutation showed small diffuse intraneuronal inclusions of neuroserpin and a late onset of dementia between 45 and 60 years old.Citation79,Citation80,Citation82 A family with the more severe Portland mutation showed larger inclusions and an onset of dementia in their mid-twenties. The family with the His338Arg mutation showed even more inclusions and an onset of dementia in their mid teens whilst those with the most severe mutation, Gly392Glu, have very large inclusions and an onset of dementia leading to death before the age of 20 years.Citation81
The direct correlation between the ‘polymerogenicity’ of the mutations and the onset of dementia strongly indicates that the intracellular accumulation of neuroserpin is by itself sufficient to cause neurodegeneration. The effect of polymerization on disease was corroborated by in vitro experiments showing a fast rate of polymerization for recombinant Ser49Pro neuroserpin, and an even faster rate for recombinant Ser52Arg neuroserpin which is associated with a more severe clinical phenotype.Citation85–Citation87 A cell model using transient transfection in COS-7 cells also showed the accumulation of mutant neuroserpin within the endoplasmic reticulum. The intracellular aggregates were composed of polymers similar to the loop-sheet polymers isolated from the brains of individuals affected by FENIB.Citation88 In keeping with the genotype-phenotype correlation observed in patients, the more severe Portland mutant accumulated more rapidly and its rate of secretion was lower than for the less severe Syracuse mutant.Citation88
Strategies to Prevent Polymerization and Ameliorate the Associated Disease
Understanding the pathway of polymerization has allowed the development of novel therapeutic strategies to block polymer formation and so ameliorate the associated disease. One strategy is to use peptides to block the aberrant linkage between the RCL of one molecule and β-sheet A of another. Polymerization of Z α1-antitrypsin can indeed be blocked in vitro by annealing 11–13 amino acid RCL peptides to β-sheet A.Citation45 The poor specificity of these peptides led to the design of smaller peptides and indeed 4–6-mer peptides can efficiently and specifically block the polymerization of Z β1-antitrypsin in vitro.Citation89,Citation90
Small sugar and alcohol molecules can reduce the rate of polymerization of both α1-antitrypsin and neuroserpin in vitro, most probably by stabilising β-sheet A.Citation91 Chemical chaperones stabilise intermediates on the folding pathwayCitation92,Citation93 but 4-phenylbutyric acid, which increased the secretion of Z α1-antitrypsin in a mouse model of disease,Citation92 has proved to be ineffective in clinical trials in patients with α1-antitrypsin deficiency.Citation94
Peptides and chaperones are poor therapeutic agents in vivo and so another approach is to use small molecules to block polymerization. A hydrophobic pocket has been identified on the lateral surface of α1-antitrypsin that is bounded by β-strand 2A and helices D and E but which is distinct from the polymerization interface. The introduction of bulky residues into this pocket retards the polymerization of M α1-antitrypsin and increases the secretion of Z α1-antitrypsin from a Xenopus oocyte expression system.Citation14,Citation95,Citation96 Consequently this pocket offers a novel target for rational drug design. The identification of chemical compounds that bind to this cavity is currently underway.
Abbreviations
| RCL | = | Reactive Centre Loop |
| PAS | = | periodic acid Schiff |
| FENIB | = | familial encephalopathy with neuroserpin inclusion bodies |
| ER | = | endoplasmic reticulum |
Figures and Tables
Figure 1 Inhibition of neutrophil elastase by α1-antitrypsin and the structural basis of polymerization. (A) After docking (left) the neutrophil elastase (grey) is inactivated by movement from the upper to the lower pole of the protein (right). This is associated with the insertion of the RCL (red) as an extra strand into β-sheet A (green). (B) The structure of α1-antitrypsin is centred on β-sheet A (green) and the mobile reactive centre loop (red). Polymer formation results from the Z variant of α1-antitrypsin (Glu342Lys at P17; indicated by arrow) or mutations in the shutter domain (blue circle) that open β-sheet A to favour partial loop insertion and the formation of an unstable intermediate (M*). The patent β-sheet A then accepts the loop of another molecule to form a dimer (D), which then extends into polymers (P). The individual molecules of α1-antitrypsin within the polymer, although identical, are coloured red, yellow and blue for clarity. Figure reproduced with permission from Lomas et al.Citation97
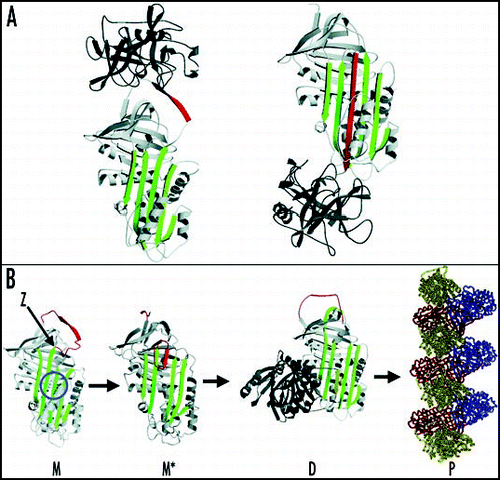
Figure 2 Z α1-antitrypsin is retained within hepatocytes as intracellular inclusions. These inclusions are PAS-positive and diastase resistant (A) and are associated with neonatal hepatitis and hepatocellular carcinoma. (B) Electron microscopy of a hepatocyte from the liver of a patient with Z α1-antitrypsin deficiency shows the accumulation of α1-antitrypsin within the rough ER (arrow). These inclusions are composed of chains of α1-antitrypsin polymers shown here from the plasma of a Siiyama α1-antitrypsin homozygote (C). More recently, polymers have been identified within PAS-positive inclusions with a monoclonal anti-polymer α1-antitrypsin antibody. (D and E) Immunochemistry of the liver from an individual with Z α1-antitrypsin deficiency, showing staining with an anti-α1-antitrypsin polyclonal antibody (D, arrow) and a monoclonal anti-polymer α1-antitrypsin antibody (E, arrow). It is these intracellular inclusions of polymers that are associated with neonatal hepatitis and hepatocellular carcinoma. Figure reproduced with permission from Lomas et al.Citation97
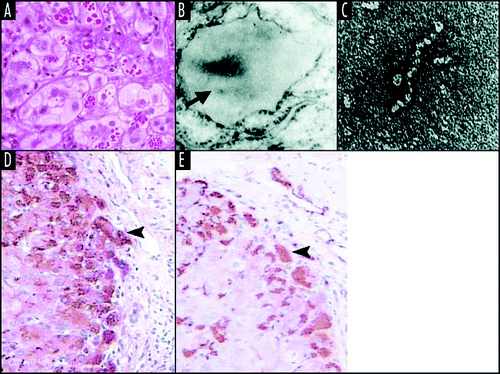
Figure 3 Mutant neuroserpin is retained within neurons as intracellular inclusions. These inclusions stain positive with PAS (A) and can be seen within the ER on electron microscopy (B). Electron microscopy of the isolated inclusions confirms that the mutant neuroserpin forms bead-like polymers identical to those of Z α1-antitrypsin (C). Figure reproduced with permission from Lomas et al.Citation97
Acknowledgements
This work was supported by the Medical Research Council (UK), the Wenner Gren Foundations, the Swedish Society for Medical Research and the Isaac Newton Trust Cambridge European Trust.
References
- Silverman GA, Bird PI, Carrell RW, Coughlin PB, Gettins PG, Irving JI, Lomas DA, Luke CJ, Moyer RW, Pemberton PA, Remold-O'Donnell E, Salvesen GS, Travis J, Whisstock JC. The serpins are an expanding superfamily of structurally similar but funtionally diverse proteins: Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem 2001; 276:33293 - 33296
- Irving JA, Steenbakkers PJ, Lesk AM, Op den Camp HJ, Pike RN, Whisstock JC. Serpins in prokaryotes. Mol Biol Evol 2002; 19:1881 - 1890
- Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, Rosado CJ, Langendorf CG, Pike RN, Bird PI, Whisstock JC. An overview of the serpin superfamily. Genome Biol 2006; 7:216
- Komiyama T, Ray CA, Pickup DJ, Howard AD, Thornberry NA, Peterson EP, Salvesen G. Inhibition of interleukin 1β converting enzyme by the cowpox virus serpin CrmA: An example of cross-class inhibition. J Biol Chem 1994; 269:19331 - 19337
- Vercammen D, Belenghi B, van de Cotte B, Beunens T, Gavigan JA, De Rycke R, Brackenier A, Inze D, Harris JL, Van Breusegem F. Serpin1 of Arabidopsis thaliana is a suicide inhibitor for metacaspase 9. J Mol Biol 2006; 364:625 - 636
- Kaiserman D, Whisstock JC, Bird PI. Mechanisms of serpin dysfunction in disease. Expert Rev Mol Med 2006; 8:1 - 19
- Hunt LT, Dayhoff MO. A surprising new protein superfamily containing ovalbumin, antithrombin-III, and α1-proteinase inhibitor. Biochem Biophys Res Commun 1980; 95:864 - 871
- Carrell R, Travis J. α1-antitrypsin and the serpins: Variation and countervariation. Trends Biochem Sci 1985; 10:20 - 24
- Elliott PR, Lomas DA, Carrell RW, Abrahams JP. Inhibitory conformation of the reactive loop of α1-antitrypsin. Nat Struct Biol 1996; 3:676 - 681
- Ryu SE, Choi HJ, Kwon KS, Lee KN, Yu MH. The native strains in the hydrophobic core and flexible reactive loop of a serine protease inhibitor: Crystal structure of an uncleaved α1-antitrypsin at 2.7 Å. Structure 1996; 4:1181 - 1192
- Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci USA 1997; 94:14683 - 14688
- Elliott PR, Abrahams JP, Lomas DA. Wild-type α1-antitrypsin is in the canonical inhibitory conformation. J Mol Biol 1998; 275:419 - 425
- Li J, Wang Z, Canagarajah B, Jiang H, Kanost M, Goldsmith EJ. The structure of active serpin 1K from Manduca sexta. Structure 1999; 7:103 - 109
- Elliott PR, Pei XY, Dafforn TR, Lomas DA. Topography of a 2.0Å structure of α1-antitrypsin reveals targets for rational drug design to prevent conformational disease. Protein Sci 2000; 9:1274 - 1281
- Kim SJ, Woo JR, Seo EJ, Yu MH, Ryu SE. A 2.1 Å resolution structure of an uncleaved α1-antitrypsin shows variability of the reactive center and other loops. J Mol Biol 2001; 306:109 - 119
- Ye S, Cech AL, Belmares R, Bergstrom RC, Tong Y, Corey DR, Kanost MR, Goldsmith EJ. The structure of a Michaelis serpin-protease complex. Nat Struct Biol 2001; 8:979 - 983
- Dementiev A, Simonovic M, Volz K, Gettins PG. Canonical inhibitor-like interactions explain reactivity of α1-proteinase inhibitor Pittsburgh and antithrombin with proteinases. J Biol Chem 2003; 278:37881 - 37887
- Wilczynska M, Fa M, Ohlsson PI, Ny T. The inhibition mechanism of serpins. Evidence that the mobile reactive center loop is cleaved in the native protease-inhibitor complex. J Biol Chem 1995; 270:29652 - 29655
- Huntington JA. Shape-shifting serpins - Advantages of a mobile mechanism. Trends Biochem Sci 2006; 31:427 - 435
- Wilczynska M, Fa M, Karolin J, Ohlsson PI, Johansson LBA, Ny T. Structural insights into serpin-protease complexes reveal the inhibitory mechanism of serpins. Nature Structural Biology 1997; 4:354 - 357
- Stratikos E, Gettins PGW. Major proteinase movement upon stable serpin-proteinase complex-formation. Proc Natl Acad Sci USA 1997; 94:453 - 458
- Stratikos E, Gettins PG. Mapping the serpin-proteinase complex using single cysteine variants of α1-proteinase inhibitor Pittsburgh. J Biol Chem 1998; 273:15582 - 15589
- Stratikos E, Gettins PG. Formation of the covalent serpin-proteinase complex involves translocation of the proteinase by more than 70Å and full insertion of the reactive center loop into β-sheet A. Proc Natl Acad Sci USA 1999; 96:4808 - 4813
- Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature 2000; 407:923 - 926
- Fa M, Bergstrom F, Hagglof P, Wilczynska M, Johansson LB, Ny T. The structure of a serpin-protease complex revealed by intramolecular distance measurements using donor-donor energy migration and mapping of interaction sites. Structure 2000; 8:397 - 405
- Carrell RW, Owen MC. Plakalbumin, α1-antitrypsin, antithrombin and the mechanism of inflammatory thrombosis. Nature 1985; 317:730 - 732
- Kaslik G, Kardos J, Szabo E, Szilagyi L, Zavodszky P, Westler WM, Markley JL, Graf L. Effects of serpin binding on the target proteinase: Global stabilization, localized increased structural flexibility, and conserved hydrogen bonding at the active site. Biochemistry 1997; 36:5455 - 5464
- Dementiev A, Dobo J, Gettins PG. Active site distortion is sufficient for proteinase inhibition by serpins: Structure of the covalent complex of α1-proteinase inhibitor with porcine pancreatic elastase. J Biol Chem 2006; 281:3452 - 3457
- Mast AE, Enghild JJ, Pizzo SV, Salvesen G. Analysis of the plasma elimination kinetics and conformational stabilities of native, proteinase-complexed, and reactive site cleaved serpins: Comparison of α1-proteinase inhibitor, α1-antichymotrypsin, antithrombin III, α2-antiplasmin, angiotensinogen, and ovalbumin. Biochemistry 1991; 30:1723 - 1730
- Nykjær A, Petersen CM, Moller B, Jensen PH, Moestrup SK, Holtet TL, Etzerodt M, Thøgersen HC, Munch M, Andreasen PA, Gliemann J. Purified α2-macroglobulin receptor/LDL receptor-related protein binds urokinase·plasminogen activator inhibitor type-1 complex: Evidence that the α2-macroglobulin receptor mediates cellular degradation of urokinase receptor-bound complexes. J Biol Chem 1992; 267:14543 - 14546
- Andreasen PA, Sottrup-Jensen L, Kjøller L, Nykjær A, Moestrup SK, Petersen CM, Gliemann J. Receptor-mediated endocytosis of plasminogen activators and activator/inhibitor complexes. FEBS Lett 1994; 338:239 - 245
- Whisstock JC, Bottomley SP. Molecular gymnastics: Serpin structure, folding and misfolding. Curr Opin Struct Biol 2006; 16:761 - 768
- Lomas DA, Mahadeva R. α1-antitrypsin polymerization and the serpinopathies: Pathobiology and prospects for therapy. J Clin Invest 2002; 110:1585 - 1590
- Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet 2002; 3:759 - 768
- Carrell RW, Lomas DA. α1-antitrypsin deficiency: A model for conformational diseases. N Engl J Med 2002; 346:45 - 53
- Carrell RW, Lomas DA. Conformational disease. Lancet 1997; 350:134 - 138
- Brantly M, Nukiwa T, Crystal RG. Molecular basis of α1-antitrypsin deficiency. Am J Med 1988; 84:13 - 31
- Billingsley GD, Walter MA, Hammond GL, Cox DW. Physical mapping of four serpin genes: α1-antitrypsin, α1-antichymotrypsin, corticosteroid-binding globulin, and protein C inhibitor, within a 280-kb region on chromosome 14q32.1. Am J Human Genet 1993; 52:343 - 353
- Blanco I, Fernández E, Bustillo EF. Alpha-1-antitrypsin PI phenotypes S and Z in Europe: An analysis of the published surveys. Clin Genet 2001; 60:31 - 41
- Blanco I, Bustillo EF, Rodriguez MC. Distribution of α1-antitrypsin PI S and PI Z frequencies in countries outside Europe: A meta-analysis. Clin Genet 2001; 60:431 - 441
- Sharp HL, Bridges RA, Krivit W, Freier EF. Cirrhosis associated with α1-antitrypsin deficiency: A previously unrecognized inherited disorder. J Lab Clin Med 1969; 73:934 - 939
- Eriksson S, Larsson C. Purification and partial characterization of PAS-positive inclusion bodies from the liver in α1-antitrypsin deficiency. N Engl J Med 1975; 292:176 - 180
- Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in α1-antitrypsin deficiency. N Engl J Med 1986; 314:736 - 739
- Janciauskiene S, Eriksson S, Callea F, Mallya M, Zhou A, Seyama K, Hata S, Lomas DA. Differential detection of PAS-positive inclusions formed by the Z, Siiyama, and Mmalton variants of α1-antitrypsin. Hepatology 2004; 40:1203 - 1210
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z α1-antitrypsin accumulation in the liver. Nature 1992; 357:605 - 607
- James EL, Bottomley SP. The mechanism of α1-antitrypsin polymerization probed by fluorescence spectroscopy. Arch Biochem Biophys 1998; 356:296 - 300
- Dafforn TR, Mahadeva R, Elliott PR, Sivasothy P, Lomas DA. A kinetic mechanism for the polymerization of α1-antitrypsin. J Biol Chem 1999; 274:9548 - 9555
- Sivasothy P, Dafforn TR, Gettins PG, Lomas DA. Pathogenic α1-antitrypsin polymers are formed by reactive loop-β-sheet A linkage. J Biol Chem 2000; 275:33663 - 33668
- Purkayastha P, Klemke JW, Lavender S, Oyola R, Cooperman BS, Gai F. α1-antitrypsin polymerization: A fluorescence correlation spectroscopic study. Biochemistry 2005; 44:2642 - 2649
- Lawless MW, Greene CM, Mulgrew A, Taggart CC, O'Neill SJ, McElvaney NG. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z α1-antitrypsin deficiency. J Immunol 2004; 172:5722 - 5726
- Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH. Accumulation of mutant α1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem 2005; 280:39002 - 39015
- Teckman JH, Perlmutter DH. Retention of mutant α1-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol 2000; 279:G961 - G974
- Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in α1-antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol 2004; 286:G851 - G862
- Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T. Intracellular inclusions containing mutant α1-antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem 2006; 281:4467 - 4476
- Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: One for soluble Z variant of human α1-proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell 2006; 17:203 - 212
- Seyama K, Nukiwa T, Takabe K, Takahashi H, Miyake K, Kira S. Siiyama (serine 53 (TCC) to phenylalanine 53 (TTC)): A new α1-antitrypsin-deficient variant with mutation on a predicted conserved residue of the serpin backbone. J Biol Chem 1991; 266:12627 - 12632
- Roberts EA, Cox DW, Medline A, Wanless IR. Occurrence of α1-antitrypsin deficiency in 155 patients with alcoholic liver disease. Am J Clin Pathol 1984; 82:424 - 427
- Zhou A, Stein PE, Huntington JA, Carrell RW. Serpin polymerisation is prevented by a hydrogen-bond network which is centred on His 334 and stabilised by glycerol. J Biol Chem 2003; 278:15116 - 15122
- Kang HA, Lee KN, Yu MH. Folding and stability of the Z and S(iiyama) genetic variants of human α1-antitrypsin. J Biol Chem 1997; 272:510 - 516
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. α1-antitrypsin Siiyama (Ser53➞Phe): Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333 - 15335
- Lomas DA, Elliott PR, Sidhar SK, Foreman RC, Finch JT, Cox DW, Whisstock JC, Carrell RW. α1-antitrypsin Mmalton (52Phe-deleted) forms loop-sheet polymers in vivo: Evidence for the C sheet mechanism of polymerization.. J Biol Chem 1995; 270:16864 - 16870
- Elliott PR, Stein PE, Bilton D, Carrell RW, Lomas DA. Structural explanation for the deficiency of S α1-antitrypsin. Nat Struct Biol 1996; 3:910 - 911
- Mahadeva R, Chang WS, Dafforn TR, Oakley DJ, Foreman RC, Calvin J, Wight DG, Lomas DA. Heteropolymerization of S, I, and Z α1-antitrypsin and liver cirrhosis. J Clin Invest 1999; 103:999 - 1006
- Campra JL, Craig JR, Peters RL, Reynolds TB. Cirrhosis associated with partial deficiency of α1-antitrypsin in an adult. Ann Intern Med 1973; 78:233 - 238
- Cruz M, Molina J, Pedrola D, Muñoz-López F. Cirrhosis and heterozygous α1-antitrypsin deficiency in a 4 year old girl. Helv Paediatr Acta 1975; 30:501 - 507
- Laurell CB, Eriksson S. The electrophoretic α1-globulin pattern of serum in α1-antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132 - 140
- Eriksson S. Studies in α1-antitrypsin deficiency. Acta Med Scand Suppl 1965; 432:1 - 85
- Stein PD, Leu JD, Welch MH, Guenter CA. Pathophysiology of the pulmonary circulation in emphysema associated with α1-antitrypsin deficiency. Circulation 1971; 43:227 - 239
- Larsson C. Natural history and life expectancy in severe α1-antitrypsin deficiency, PiZ. Acta Med Scand 1978; 204:345 - 351
- Janus ED, Phillips NT, Carrell RW. Smoking lung function, and α1-antitrypsin deficiency. Lancet 1985; 1:152 - 154
- Elliott PR, Bilton D, Lomas DA. Lung polymers in Z α1-antitrypsin deficiency-related emphysema. Am J Respir Cell Mol Biol 1998; 18:670 - 674
- Mulgrew AT, Taggart CC, Lawless MW, Greene CM, Brantly ML, O'Neill SJ, McElvaney NG. Z α1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest 2004; 125:1952 - 1957
- Parmar JS, Mahadeva R, Reed BJ, Farahi N, Cadwallader KA, Keogan MT, Bilton D, Chilvers ER, Lomas DA. Polymers of α1-antitrypsin are chemotactic for human neutrophils: A new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723 - 730
- Mahadeva R, Atkinson C, Li Z, Stewart S, Janciauskiene S, Kelley DG, Parmar J, Pitman R, Shapiro SD, Lomas DA. Polymers of Z α1-antitrypsin colocalize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol 2005; 166:377 - 386
- Janciauskiene S, Zelvyte I, Jansson L, Stevens T. Divergent effects of α1-antitrypsin on neutrophil activation, in vitro. Biochem Biophys Res Commun 2004; 315:288 - 296
- Persson C, Subramaniyam D, Stevens T, Janciauskiene S. Do native and polymeric α1-antitrypsin activate human neutrophils in vitro?. Chest 2006; 129:1683 - 1692
- Corral J, Aznar J, Gonzalez-Conejero R, Villa P, Minano A, Vaya A, Carrell RW, Huntington JA, Vicente V. Homozygous deficiency of heparin cofactor II: Relevance of P17 glutamate residue in serpins, relationship with conformational diseases, and role in thrombosis. Circulation 2004; 110:1303 - 1307
- Green C, Brown G, Dafforn TR, Reichhart JM, Morley T, Lomas DA, Gubb D. Drosophila necrotic mutations mirror disease-associated variants of human serpins. Development 2003; 130:1473 - 1478
- Davis RL, Holohan PD, Shrimpton AE, Tatum AH, Daucher J, Collins GH, Todd R, Bradshaw C, Kent P, Feiglin D, Rosenbaum A, Yerby MS, Shaw CM, Lacbawan F, Lawrence DA. Familial encephalopathy with neuroserpin inclusion bodies. Am J Pathol 1999; 155:1901 - 1913
- Davis RL, Shrimpton AE, Holohan PD, Bradshaw C, Feiglin D, Collins GH, Sonderegger P, Kinter J, Becker LM, Lacbawan F, Krasnewich D, Muenke M, Lawrence DA, Yerby MS, Shaw CM, Gooptu B, Elliott PR, Finch JT, Carrell RW, Lomas DA. Familial dementia caused by polymerization of mutant neuroserpin. Nature 1999; 401:376 - 379
- Davis RL, Shrimpton AE, Carrell RW, Lomas DA, Gerhard L, Baumann B, Lawrence DA, Yepes M, Kim TS, Ghetti B, Piccardo P, Takao M, Lacbawan F, Muenke M, Sifers RN, Bradshaw CB, Kent PF, Collins GH, Larocca D, Holohan PD. Association between conformational mutations in neuroserpin and onset and severity of dementia (erratum 2002, 360, 1102). Lancet 2002; 359:2242 - 2247
- Bradshaw CB, Davis RL, Shrimpton AE, Holohan PD, Rea CB, Fieglin D, Kent P, Collins GH. Cognitive deficits associated with a recently reported familial neurodegenerative disease: Familial encephalopathy with neuroserpin inclusion bodies. Arch Neurol 2001; 58:1429 - 1434
- Briand C, Kozlov SV, Sonderegger P, Grutter MG. Crystal structure of neuroserpin: A neuronal serpin involved in a conformational disease. FEBS Lett 2001; 505:18 - 22
- Takao M, Benson MD, Murrell JR, Yazaki M, Piccardo P, Unverzagt FW, Davis RL, Holohan PD, Lawrence DA, Richardson R, Farlow MR, Ghetti B. Neuroserpin mutation S52R causes neuroserpin accumulation in neurons and is associated with progressive myoclonus epilepsy. J Neuropathol Exp Neurol 2000; 59:1070 - 1086
- Belorgey D, Crowther DC, Mahadeva R, Lomas DA. Mutant neuroserpin (S49P) that causes familial encephalopathy with neuroserpin inclusion bodies is a poor proteinase inhibitor and readily forms polymers in vitro. J Biol Chem 2002; 277:17367 - 17373
- Belorgey D, Sharp LK, Crowther DC, Onda M, Johansson J, Lomas DA. Neuroserpin Portland (Ser52Arg) is trapped as an inactive intermediate that rapidly forms polymers: Implications for the epilepsy seen in the dementia FENIB. Eur J Biochem 2004; 271:3360 - 3367
- Onda M, Belorgey D, Sharp LK, Lomas DA. Latent S49P neuroserpin forms polymers in the dementia familial encephalopathy with neuroserpin inclusion bodies. J Biol Chem 2005; 280:13735 - 13741
- Miranda E, Romisch K, Lomas DA. Mutants of neuroserpin that cause dementia accumulate as polymers within the endoplasmic reticulum. J Biol Chem 2004; 279:28283 - 28291
- Mahadeva R, Dafforn TR, Carrell RW, Lomas DA. Six-mer peptide selectively anneals to a pathogenic serpin conformation and blocks polymerisation: Implications for the prevention of Z α1-antitrypsin related cirrhosis. J Biol Chem 2002; 277:6771 - 6774
- Chang YP, Mahadeva R, Chang WS, Shukla A, Dafforn TR, Chu YH. Identification of a 4-mer peptide inhibitor that effectively blocks the polymerization of pathogenic Z α1-antitrypsin. Am J Respir Cell Mol Biol 2006; 35:540 - 548
- Sharp LK, Mallya M, Kinghorn KJ, Wang Z, Crowther DC, Huntington JA, Belorgey D, Lomas DA. Sugar and alcohol molecules provide a therapeutic strategy for the serpinopathies that cause dementia and cirrhosis. FEBS J 2006; 273:2540 - 2552
- Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant α1-antitrypsin (α1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in α1-AT deficiency. Proc Natl Acad Sci USA 2000; 97:1796 - 1801
- Devlin GL, Parfrey H, Tew DJ, Lomas DA, Bottomley SP. Prevention of polymerization of M and Z α1-Antitrypsin (α1-AT) with trimethylamine N-oxide: Implications for the treatment of α1-AT deficiency. Am J Respir Cell Mol Biol 2001; 24:727 - 732
- Teckman JH. Lack of effect of oral 4-phenylbutyrate on serum α1-antitrypsin in patients with α1-antitrypsin deficiency: A preliminary study. J Pediatr Gastroenterol Nutr 2004; 39:34 - 37
- Lee C, Maeng JS, Kocher JP, Lee B, Yu MH. Cavities of α1-antitrypsin that play structural and functional roles. Protein Sci 2001; 10:1446 - 1453
- Parfrey H, Mahadeva R, Ravenhill NA, Zhou A, Dafforn TR, Foreman RC, Lomas DA. Targeting a surface cavity of α1-antitrypsin to prevent conformational disease. J Biol Chem 2003; 278:33060 - 33066
- Lomas DA, Belorgey D, Mallya M, Miranda E, Kinghorn KJ, Sharp LK, Phillips RL, Page R, Robertson AS, Crowther DC. Molecular Mousetraps and the Serpinopathies. Biochem Soc Trans 2005; 33:321 - 330