Abstract
A number of drugs active against prions either in vitro, in cellular systems or in vivo in animal models have been isolated in various screening assays. In this minireview, we would like to suggest, that in addition to their direct interest as potential therapeutic agents, these molecules could be used as original research tools to understand prion propagation. The use of antiprion compounds as tool to understand fundamentals of prion propagation relies on reverse screening approaches. These global genetic and/or biochemical approaches aim to identify the intracellular target(s) and mechanism of action of the drugs. Once those are known, the biological activity of the compounds can be optimized on a rational basis, their potential side effects understood and minimized. In vitro enzyme-based screening assays can then be designed to allow discovery of new, more potent and selective molecules. Here we describe the main comprehensive biochemical and genetical approaches to realize reverse screening approaches based on antiprion drugs. We will finish by discussing the interest of using drug inactivation of specific targets as a substitute to genetic inactivation.
A number of drugs have been isolated as active against mammalian prion (reviewed in ref. Citation1). For most of these molecules, the mode of action and targets remain largely unknown. In principle, two main modes of action for antiprion drugs can be envisioned: either in cis directly on PrPC/PrPSc, or in trans by interfering with the activity of cellular factors required for prion propagation. Some compounds are thought to bind directly to PrPC or PrPSc (action in cis). Among these compounds are Congo Red (CR), Pentosan Polysulfate (PPS) or Glycosaminoglycans (GAGs). Other compounds are thought to act in trans by affecting PrPC or PrPSc indirectly. Among these molecules are various lysosomotropic factors including the antimalarial drugs Quinacrine (QC) and Chloroquine. Indeed, the lysosome is a potential site of conversion of PrPC to PrPSc.Citation2 In addition, a recent report,Citation3 proposes that QC's antiprion activity is related to its ability to redistribute cholesterol from the plasma membrane to intracellular compartments, thereby destabilizing membrane domains. This conclusion was drawn from correlation experiments indicating that drugs known to display cholesterol-redistributing activity (but structurally unrelated to QC) also have antiprion potency. Finally, QC was also shown to interact directly with PrP.Citation4,Citation5 The example of QC, with these conflicting results, thus illustrates the need for unbiaised and global approaches not driven by a preconcieved hypothesis on the drug mode of action. To our knowledge, no such approaches were applied for any of the known antiprion drugs, with the noticeable exception of chlorpromazine for which a haploinsufficiency profiling screen (HIP, see in the later section) has been published.Citation6 In this minireview, we discuss the various advantages of defining extensively intracellular targets of antiprion drugs. We then would like to present some global approaches that can be applied to uncover, in an unbiaised manner, cellular mechanism(s) of action of compounds active against prions. To finish we propose that, once intracellular targets have been clearly identified, the drugs can be used to do “chemical genetics” to inactivate cellular target(s) which could be especially useful in situations where classical inactivation by mutagenesis is unpractical (for instance if redundant multigenic families are involved).
The Benefits of the Use of Global Approaches to Determine the Mode of Action of Antiprion Drugs
Similar to classical genetic screens, global approaches allow to address the question of the drug mode of action in an unbiaised manner and without any preconceived idea. As such, these approaches can lead to discovery of new prion controlling mechanisms, especially mechanisms in trans which remain largely unknown. Once the target and drug mechanism are known, drug efficiency can be improved on a rational basis. In addition, these global approaches allow identifying and understanding potential side effects of the molecules. This point is of special importance for compounds evaluated in preclinical and clinical trials. Together, these approaches can also help to define new screening targets for the development of highly selective mechanism-based, cell-free screening assays.
Antiprion Drugs as Baits in Affinity Chromatography on Immobilized Compounds
Protein purification using affinity chromatography based on immobilized substrates or cofactors has been used for several decades.Citation7–Citation10 In contrast, the use of affinity chromatography with immobilized drugs to comprehensively purify and identify cellular factors that interact with these compounds is relatively recent and undoubtedly linked to the improvements of proteomics and mass spectrometry (MS). Affinity chromatography experiments on immobilized drugs has been already successfully applied to protein kinase inhibitors like flavopiridol,Citation11 purvalanol,Citation12 (R)-roscovitine (CYC202, Seliciclib, at present in phase IIb of clinical trials)Citation13 or Gefitinib (ZD1839, Iressa, already in clinic for the treatment of non small cells lung cancers)Citation14 or to Hsp90 chaperone inhibitor geldanamycin.Citation15 It should be noted that the affinity chromatography approach on immobilized drugs to investigate the actual antiprion mechanism of a compound of interest should be considered only as a first step of the analysis, as it only shows a specific interaction between the drug and a cellular component. Whether or not this interaction affects the biological activity of this component (inhibition or activation) needs to be determined in a second step. In a third step, the link between this biological effect and the antiprion activity of the drug also needs to be established. Finally, it should be kept in mind that affinity chromatography allows purification of protein (or nucleic acid) targets but potentially misses other types of targets (lipids for example).
The first important technical point when starting the affinity chromatography approach is the preparation of the drug-linked affinity matrix. This point is of the uppermost importance. Indeed, to avoid possible steric hindrance between target(s) and the matrix (generally Sepharose beads), it is necessary to introduce a spacer between the antiprion drug of interest and the Sepharose beads. A good linker should be long enough to avoid steric hindrance and should not be too hydrophobic to avoid auto-aggregation or unspecific hydrophobic interactions with macromolecules from the cell lysate (). Different linkers have been described. Among them () are Polyethylene glycol (PEG) linkers,Citation13,Citation16–Citation18 divalent epoxide linkers (ethyleneglycol diglycidylether, EGDE)19 and amino-caproylaminopentyloxy (ACAP) linkers (Gug F and Galons H, in preparation). We believe the ACAP linker is an interesting choice because, in addition to its optimal length, its internal peptidic bond lowers its hydrophobicity. Furthermore, the amide function increases the rigidity of the linker. An important point is also to check that introduction of the linker does not interfere with the antiprion activity of the considered drug (or at least that some of its activity is retained). Several positions should be tested in order to determine the most appropriate.
These matrices can then be used for affinity chromatography experiments to purify biological targets of the considered antiprion drug (). Crude extracts of various tissues or cell types, prion-infected or not, should then be incubated with the immobilized drug, the matrix extensively washed and the bound proteins eluted with electrophoresis sample buffer, and analyzed by SDS-PAGE.Citation20 Several specificity controls should be included (, right panel). In a first control, the Sepharose beads without the drug are used. In second control, competitions with an excess of free antiprion drug are performed. Proteins interacting specifically with the antiprion compound should be displaced by the free drug. A last important control is to realize the same type of competition with an inactive, close chemical derivative of the antiprion drug, if available. The macromolecules specifically bound to the antiprion drug are thus those which bind the drug matrix, but not the matrix alone and which are competed away by the active antiprion drug but not by its inactive derivative. However, it should be kept in mind, as a limit of this biochemical method, that some other potential biological targets (in particular those not related to the antiprion activity but rather to undesired side effects) might be missed due to the position chosen for the linker. For this reason global genetic approaches can be considered as complement to the biochemical approach with immobilized antiprion compounds.
Antiprion Drugs as Baits in Yeast-based Genetic Approaches
A number of global approaches based on the use of the budding yeast Saccharomyces cerevisiae have been developed to uncover drug targets.Citation21 The main advantage of these methods compared to the biochemical approach described above is the absence of chemical linker added onto the tested compound. Among these cell-based, high-throughput screening methods are: (i) haploinsufficiency profiling (HIP), (ii) synthetic lethality screens (SLS) and (iii) genome wide overexpression screens (OES).
In the HIP approach, a library of diploid yeast strains with heterozygous deletion of each single gene is screened for drug sensitivity either in a single culture with a competitive growth assay or by screening the 6000+ strains separately in parallel. This assay is based on the observation that reducing the copy number of a gene encoding a drug target from two copies (in diploid strain) to one copy (in a diploid strain heterozygote for deletion of this gene) often results in a strain sensitized to the drug of interest. In one of these HIP assays, Chlorpromazine, a tricyclic antidepressant shown to promote PrPSc clearance in N2a cell systemCitation22 exhibited genetic interaction with integral membrane ATPase activities.Citation6 It should be noted, however, that in this screen only the effect of chlorpromazine on toxicity was analyzed and not its effect on prion propagation.
In the SLS approach, the drug is screened at a concentration that is normally subefficient against a library of haploid yeast strains with individual gene deletions. Genes whose deletion results in increased drug sensitivity might be one of several direct drug targets or genes that are involved in the same cellular pathways as the drug target.Citation23
In the OES method (), the rationale is to identify genes or cDNA whose overexpression confers resistance to a drug in yeast. This screen is based on the idea that cells overexpressing a target or a component related to the target, should tolerate higher levels of the active compound. The main interest of this approach is that overexpressed genes or cDNAs can be of any origin (from yeast but also from mouse or human). In addition, because this method can be based on yeast prions, convenient red/white colony color assays can be used in conjunction with GAL-inducible promoters (highly active when yeast cells are grown in galactose medium and inactive when cells are grown in glucose medium). The utility of this approach in the analysis of small molecules targets has been demonstrated.Citation24 Naturally, typical false positive in such approaches might correspond to proteins involved in drug import/export and stability. In addition, genes whose overexpression is toxic in yeast cells are counterslected.
Finally, another yeast-based method can be used: the yeast three-hybrid system (Y3H, ). This method is derived from the yeast two-hybrid system (Y2H) which has proved to be a powerful tool for detecting protein-protein interactions.Citation25 In the Y2H system, protein-protein interaction lead to reconstitution of a transcriptional activator by close positioning in space of its DNA binding (DBD) and activation domain (AD) expressed separately as fusion proteins with the two potentially interacting proteins. Activity of the reconstituted transcriptional activator is evaluated using convenient reporter genes like LacZ or HIS3. In 1996, Licitra and Liu extended this method to small molecule target identification by dimerization of two receptor proteins via heterodimeric ligand.Citation26 As described in , one ligand/receptor pair (for example methotrexate/DHFR in fusion with DBD) serves as bait. The ligand (e.g., methotrexate) is covalently attached to the antiprion molecule of interest (6AP in the example given) via a linker. DHFR-DBD fusion protein is expressed in cells treated with the dimer methotrexate-6AP. In the same cells, a library of protein (here again of any origin) in fusion with the AD is expressed. If some of these fusion proteins have affinity with 6AP, an active transcriptional activator can be reconstituted, as in the case of Y2H experiments. Y3H can thus be considered as another way of scanning the proteome (of any species) to identify the targets of antiprion drugs. However, one of the limit of this method is the same than the one highlighted for affinity chromatography on immobilized drug. Because a linker is attached to the antiprion drug it is necessary to verify that the modified drug retains activity. Also, the linker can potentially prevent binding of cellular factors—not related to the antiprion mechanism—that can be important for the understanding of undesired side effects.
Screening for Drugs Instead of Mutants, Antiprion Drugs as Tools in Genetic Experiments
In this last section, we would like to present how antiprion drugs could be used as fundamental tools in model systems like yeast as substitutes to genetic inactivation.
A classical random mutagenesis screen aimed at the isolation of mutations destabilizing or exacerbating a prion phenotype can theoretically only lead to identification of non-redundant genes. In contrast, a multigenic family involved in prion propagation and/or stability would not be identified in such classical genetic screen. Conversely, all members of a family could be targeted by a single drug. Therefore, a drug screen could potentially uncover involvement of a redundant gene family in prion propagation while a classical genetic screen cannot.
In addition, drugs can be used as alternative approaches to genetic inactivation in fundamental studies. There are several advantages of using a drug rather than mutations to observe an effect on a prion phenotype. First, it is much more rapid and simple; second, it is reversible; and third, the effect can be adjusted (dose-dependent effect), which is especially important in the case of essential genes. Thus, drug induced inactivation can be considered as analogous to conditional mutations with the additional and important advantage to be able to affect multigenic families. This aspect is perfectly illustrated by the widespread use of GuHCl, as inhibitor of Hsp104 in yeast prion studies. In budding yeast, Guanidine hydrochoride (GuHCl) is known to efficiently cure all known prions. The GuHCl is a chemical inhibitor of Hsp104p,Citation27–Citation31 a protein chaperone essential for propagation of prions in budding yeast but not conserved in animals. GuHCl is probably the only known antiprion molecule for which the critical biological target is clearly identified and numerous studies made use of GuHCl as a convenient tool for rapid and transient Hsp104-inactivation. It should be noted, however, that some published data tend to indicate that, in addition to Hsp104p, GuHCl might affect additional targetsCitation32–Citation34 involved in prion propagation.
To finish, drugs can be used in more sophisticated genetic screens like those leading to synthetic phenotype. In such screens, classically, geneticists look for a second site mutation able to either exacerbate or suppress the effect of a first mutation. If the target of an antiprion drug is known, this compound can be used to mimic the effect of inactivation of the corresponding gene. By looking among libraries of mutants (for example Euroscarf collection of dele- tion of every non essential budding yeast single gene) for those hypersensitive or resistant to the effect of the drug, it is possible to easily identify second site mutation able to exacerbate or suppress effect of inactivation of the drug target. For instance, by looking for mutations that suppress or exacerbate, the curing effect of GuHCl, when cells are treated by subefficient dose of this chemical compound, new cellular factors involved in yeast prions propagation might be identified.
Closing Remarks
Recently, we have developed a rapid two-step yeast-based assay to screen for antiprion drugs.Citation35,Citation36 The first step of the screening is based on the [PSI+] phenotype and then, active molecules are tested against [URE3] phenotype. Given the structural and functional divergences between the proteins responsible for these prion phenotypes (respectively Sup35p for the [PSI+] phenotype and Ure2p for the [URE3] phenotype), compounds active against both yeast prions can be considered as active against all yeast prions. Our initial assumption was that mechanisms controlling prion appearance and/or propagation could be conserved from yeast to human, as it is the case for most of the major cell biology regu- latory mechanisms. Indeed, the vast majority of drugs we isolated as active against both [PSI+] and [URE3] prions turned out to be also active against mammalian prion in three different mammalian cell-based assaysCitation35,Citation37 but also in vivo in animal models (Tribouillard et al in preparation). These results strongly argue in favor of common prion controlling mechanisms conserved in eukaryotes, from yeast to human, thus validating the yeast-based assay to isolate antiprion compounds but also the yeast model to study prions in general.
In this way, in the prion field, in addition to be an appropriate model system, yeast is also a nice tool not only for drug screening but also for reverse screening approaches aiming at understanding the mode of action of active compounds and thus potential new prion controlling mechanisms.
Figures and Tables
Figure 1 Antiprion drugs as baits in affinity chromatography on immobilized compounds. (A) The structure of the linker connecting the drug to the matrix (Sepharose bead) is crucial. A short spacer can prevent interaction with cellular targets because of steric hindrance (upper scheme). If the spacer is too hydrophobic (depicted in red in the middle panel), it can lead to its auto aggregation thus also preventing target binding. In addition, a hydrophobic linker can generate unspecific hydrophobic interactions with macromolecules from cell lysates. An optimal spacer (lower panel) should be only mildly hydrophobic (hydrophilic part depicted in blue) and long enough to avoid steric hindrance. In this case, specific interactions can occur, as long as the covalent link of the linker to the drug does not affect its antiprion activity too strongly. (B) Possible linker chemical structures are depicted. Polyethylene glycol (PEG) (upper panel) and amino caproylaminopentyloxy linker (lower panel). (C) Affinity chromatography purification on immobilized 6AP. In the example given, the antiprion drug 6AP was covalently liked to Sepharose beads via an amino caproylaminopentyloxy linker. Cell extracts were then incubated with this matrix. After extensive washing, the affinity matrix bound cellular components were analyzed by SDS PAGE followed by silver staining. The interacting proteins were identified by mass spectrometry.
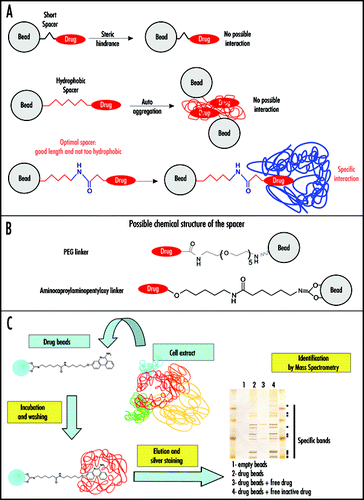
Figure 2 Antiprion drugs as baits in genetic approaches. (A) Genome wide overexpression screen (OES). A cDNA library (of any origin) expressed from the GAL-inducible promoter in a yeast plasmid was transformed in a [PRION+] yeast strain (in the example given [PSI+] forming white colonies) and cells were streaked on a Galactose medium (in condition where strong expression of cDNA was induced) and containing an antiprion drug. The curing effect produces red [psi-] colonies. Colonies which remain white despite presence of the antiprion compound might express a cDNA whose overexpression prevent the curing effect of the drug. These colonies are restreaked on the same Galactose medium containing the antiprion drug to check for resistance to the antiprion drug and also on Glucose medium containing antiprion drug to check that the isolated clone is sensitive to the drug in this condition where expression of the cDNA is largely repressed. In the example given, one clone appears to be a false positive for which resistance is not caused by expression of the cDNA (it remains white on Glucose medium). (B) Three-hybrid assay (Y3H). This method is based on the use of a heterodimeric ligand constituted of a ligand of the receptor used as bait covalently linked to the antiprion drug (6AP in the example given). The heterodimeric ligand can be used to screen against a library of cDNAs (from any origin) in fusion with the activation domain (AD) of a transcriptional activator (here Gal4p). The HIS3 reporter gene will only be activated in cells expressing a cDNA encoding a protein to which the antiprion drug (6AP here) binds, and thus only these cells will be able to grow in a medium lacking histidine.
![Figure 2 Antiprion drugs as baits in genetic approaches. (A) Genome wide overexpression screen (OES). A cDNA library (of any origin) expressed from the GAL-inducible promoter in a yeast plasmid was transformed in a [PRION+] yeast strain (in the example given [PSI+] forming white colonies) and cells were streaked on a Galactose medium (in condition where strong expression of cDNA was induced) and containing an antiprion drug. The curing effect produces red [psi-] colonies. Colonies which remain white despite presence of the antiprion compound might express a cDNA whose overexpression prevent the curing effect of the drug. These colonies are restreaked on the same Galactose medium containing the antiprion drug to check for resistance to the antiprion drug and also on Glucose medium containing antiprion drug to check that the isolated clone is sensitive to the drug in this condition where expression of the cDNA is largely repressed. In the example given, one clone appears to be a false positive for which resistance is not caused by expression of the cDNA (it remains white on Glucose medium). (B) Three-hybrid assay (Y3H). This method is based on the use of a heterodimeric ligand constituted of a ligand of the receptor used as bait covalently linked to the antiprion drug (6AP in the example given). The heterodimeric ligand can be used to screen against a library of cDNAs (from any origin) in fusion with the activation domain (AD) of a transcriptional activator (here Gal4p). The HIS3 reporter gene will only be activated in cells expressing a cDNA encoding a protein to which the antiprion drug (6AP here) binds, and thus only these cells will be able to grow in a medium lacking histidine.](/cms/asset/ab364c31-fac4-4cd9-955f-30206ff2c438/kprn_a_10904053_f0002.gif)
Acknowledgements
Most of our work in the prion field was carried out in the laboratory of Laurent Meijer (CNRS Roscoff ) who is warmly acknowledged for his continuing support, friendship and helpful discussions. Thanks are also due to Elodie Couplan for critical proof-reading of the manuscript and for helpful suggestions. This work was supported by the following grants: GIS “Infections à prion”, ACI “Jeune Chercheur” from the french government, “CRITT Santé Bretagne”, “Ministère de la Recherche” and FRM grants (to Déborah Tribouillard), “Ingénieur de valorisation” fellowship from the CNRS and ANR “blanche” from the french government.
References
- Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain 2006; 129:2241 - 2265
- Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol 2000; 74:4894 - 4897
- Klingenstein R, Lober S, Kujala P, Godsave S, Leliveld SR, Gmeiner P, Peters PJ, Korth C. Tricyclic antidepressants, quinacrine and a novel, synthetic chimera thereof clear prions by destabilizing detergent-resistant membrane compartments. J Neurochem 2006;
- Vogtherr M, Grimme S, Elshorst B, Jacobs DM, Fiebig K, Griesinger C, Zahn R. Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J Med Chem 2003; 46:3563 - 3564
- Georgieva D, Schwark D, von Bergen M, Redecke L, Genov N, Betzel C. Interactions of recombinant prions with compounds of therapeutical significance. Biochem Biophys Res Commun 2006; 344:463 - 470
- Lum PY, Armour CD, Stepaniants SB, Cavet G, Wolf MK, Butler JS, Hinshaw JC, Garnier P, Prestwich GD, Leonardson A, Garrett-Engele P, Rush CM, Bard M, Schimmack G, Phillips JW, Roberts CJ, Shoemaker DD. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell 2004; 116:121 - 137
- Cuatrecasas P, Wilchek M, Anfinsen CB. Selective enzyme purification by affinity chromatography. Proc Natl Acad Sci USA 1968; 61:636 - 643
- Lerman LS. A biochemically specific method for enzyme isolation. Proc Natl Acad Sci USA 1953; 39:232 - 236
- Arsenis C, McCormick DB. Purification of liver flavokinase by column chromatography on flavin-cellulose compounds. J Biol Chem 1964; 239:3093 - 3097
- McCormick DB. Specific purification of avidin by column chromatography on biotin-cellulose. Anal Biochem 1965; 13:194 - 198
- Schnier JB, Kaur G, Kaiser A, Stinson SF, Sausville EA, Gardner J, Nishi K, Bradbury EM, Senderowicz AM. Identification of cytosolic aldehyde dehydrogenase 1 from non-small cell lung carcinomas as a flavopiridol-binding protein. FEBS Lett 1999; 454:100 - 104
- Knockaert M, Gray N, Damiens E, Chang YT, Grellier P, Grant K, Fergusson D, Mottram J, Soete M, Dubremetz JF, LeRoch K, Doerig C, Schultz P, Meijer L. Intracellular targets of cyclin-dependent kinase inhibitors: Identification by affinity chromatography using immobilised inhibitors. Chem Biol 2000; 7:411 - 422
- Bach S, Knockaert M, Reinhardt J, Lozach O, Schmitt S, Baratte B, Koken M, Coburn SP, Tang L, Jiang T, Liang DC, Galons H, Dierick JF, Pinna LA, Meggio F, Totzke F, Schachtele C, Lerman AS, Carnero A, Wan Y, Gray N, Meijer L. Roscovitine targets, protein kinases and pyridoxal kinase. J Biol Chem 2005; 280:31208 - 31219
- Brehmer D, Greff Z, Godl K, Blencke S, Kurtenbach A, Weber M, Muller S, Klebl B, Cotten M, Keri G, Wissing J, Daub H. Cellular targets of gefitinib. Cancer Res 2005; 65:379 - 382
- Whitesell L, Mimnaugh EG, DeCosta B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA 1994; 91:8324 - 8328
- Wan Y, Hur W, Cho CY, Liu Y, Adrian FJ, Lozach O, Bach S, Mayer T, Fabbro D, Meijer L, Gray NS. Synthesis and target identification of hymenialdisine analogs. Chem Biol 2004; 11:247 - 259
- Knockaert M, Lenormand P, Gray N, Schultz P, Pouyssegur J, Meijer L. p42/p44 MAPKs are intracellular targets of the CDK inhibitor purvalanol. Oncogene 2002; 21:6413 - 6424
- Godl K, Wissing J, Kurtenbach A, Habenberger P, Blencke S, Gutbrod H, Salassidis K, Stein-Gerlach M, Missio A, Cotten M, Daub H. An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc Natl Acad Sci USA 2003; 100:15434 - 15449
- Shimizu N, Sugimoto K, Tang J, Nishi T, Sato I, Hiramoto M, Aizawa S, Hatakeyama M, Ohba R, Hatori H, Yoshikawa T, Suzuki F, Oomori A, Tanaka H, Kawaguchi H, Watanabe H, Handa H. High-performance affinity beads for identifying drug receptors. Nat Biotechnol 2000; 18:877 - 881
- Guiffant D, Tribouillard D, Gug F, Galons H, Meijer L, Blondel M, Bach S. Identification of intracellular targets of small molecular weight chemical compounds using affinity chromatography. Biotechnol J 2007; 2:68 - 75
- Sturgeon CM, Kemmer D, Anderson HJ, Roberge M. Yeast as a tool to uncover the cellular targets of drugs. Biotechnol J 2006; 1:289 - 298
- Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA 2001; 98:9836 - 9841
- Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol 2004; 22:62 - 69
- Luesch H, Wu TY, Ren P, Gray NS, Schultz PG, Supek F. A genome-wide overexpression screen in yeast for small-molecule target identification. Chem Biol 2005; 12:55 - 63
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature 1989; 340:245 - 246
- Licitra EJ, Liu JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc Natl Acad Sci USA 1996; 93:12817 - 12821
- Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI(+)] of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 2000; 97:240 - 244
- Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol 2001; 40:1357 - 1369
- Jung G, Masison DC. Guanidine hydrochloride inhibits Hsp104 activity in vivo: A possible explanation for its effect in curing yeast prions. Curr Microbiol 2001; 43:7 - 10
- Jung G, Jones G, Masison DC. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion- curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA 2002; 99:9936 - 9941
- Grimminger V, Richter K, Imhof A, Buchner J, Walter S. The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J Biol Chem 2004; 279:7378 - 7383
- Wegrzyn RD, Bapat K, Newnam GP, Zink AD, Chernoff YO. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol Cell Biol 2001; 21:4656 - 4669
- Zhou P, Derkatch IL, Liebman SW. The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI(+)] and [PIN(+)]. Mol Microbiol 2001; 39:37 - 46
- Bradley ME, Bagriantsev S, Vishveshwara N, Liebman SW. Guanidine reduces stop codon read-through caused by missense mutations in SUP35 or SUP45. Yeast 2003; 20:625 - 632
- Bach S, Talarek N, Andrieu T, Vierfond JM, Mettey Y, Galons H, Dormont D, Meijer L, Cullin C, Blondel M. Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nat Biotechnol 2003; 21:1075 - 1081
- Bach S, Tribouillard D, Talarek N, Desban N, Gug F, Galons H, Blondel M. A yeast-based assay to isolate drugs active against mammalian prions. Methods 2006; 39:72 - 77
- Tribouillard D, Bach S, Gug F, Desban N, Beringue V, Andrieu T, Dormont D, Galons H, Laude H, Vilette D, Blondel M. Using budding yeast to screen for anti-prion drugs. Biotechnol J 2006; 1:58 - 67