Abstract
The crucial role of the neuronal Tau protein in microtubule stabilization and axonal transport suggests that too little or too much Tau might lead to neuronal dysfunction. The presence of a hyper-phosphorylated but non-aggregated molecule as a toxic species that might sequester normal Tau is discussed. We present recent in vitro results that might allow to dissect the role of individual phosphorylation sites on its structure and function. We also discuss in this review the role of phosphorylation for the aggregation of the neuronal Tau protein, and compare it to the aggregation induced by external poly-anions.
Alzheimer's disease and its concomitant cognitive decline form a grim perspective for society, especially as the average life span of our population is expected to increase. This is even more the case as part of the basic knowledge concerning the disease is still under discussion. Indeed, contrary to other major disease classes such as cancer where a number of biological players have been well defined and have turned into potential targets for drug development, the molecular events leading to neuronal degeneration and ensuing cognitive decline are not completely understood. Even more, in a remarkable paradigm shift, both the extracellular β amyloid plaques and intracellular neuronal filaments and tangles, previously thought of as the main molecular markers and also the main culprits for the disease, are now considered as an ultimate rescue mechanism of the diseased brain.Citation1 This change in perception, equally found in other neuronal diseases such as Huntington's disease,Citation2 deprives us from an obvious pharmacological target, even though it is not clear what we get in exchange. Alzheimer derived diffusible ligands (ADDLs) are β amyloid peptide oligomers,Citation3 without a clear definition though of their precise molecular content and conformation. As for Tau, the major component of the neuronal filaments and tangles,Citation4 a hyperphosphorylated but soluble form rather than the aggregated protein might be the toxic species. Finally, even the two opposing viewpoints of “gain of toxic function” versus “loss of physiological function” have not yet been sorted out for neither molecular marker, be it for β amyloidCitation5 or Tau.Citation6 The functional overlap between Tau and other microtubule associated proteins (MAPs), leading to the absence of a clear phenotype for Tau knockout mice, does not even lead to a clear cut answer to this question. However, both an overproduction of the longer β amyloid [1–42] peptide and an abnormal (hyper)phosphorylation of Tau seem related to the disease, and it remains a major challenge to tease out the precise role of these components in the disease progression.
From a clinical perspective, Alzheimer's diseased neurofibrillary pathology has been post mortem scored according to the Braak rules.Citation7 This latter staging is based upon quantifying the neurofibrillary lesions in distinct regions from the brain using a silver iodate technique originally proposed by Gallyas.Citation8 Technical constraints, however, have limited this staging to research centers, and have prompted the same group to develop a more routinely accessible immunochemical method.Citation9 The latter uses the AT8 antibody, that immunostains a hyperphosphorylated Tau form, be it in its soluble or aggregated form.Citation10 Importantly, the comparative imaging of brain slices with silver or with the antibody allow a nearly identical staging, establishing an unambiguous link between hyperphosphorylation and the presence of tangles. In this review, we want to focus on the link between (hyper)phosphorylation and disease related aspects of Tau, and want to discuss how in vitro studies might shed further light on the link between Tau post translational modifications, its aggregation and the general modulation of its functional aspects.
A first question concerns a clear definition of “hyper ” and “abnormal” phosphorylation. Normal Tau contains 2–3 phosphate groups, assuring the dynamic character of the microtubule network (see below). Tau in its aggregated form (as Paired Helical Filaments (PHFs) or Straight Filaments (SF)) contains 5–9 moles of phosphate/ mole of the protein, defining it as hyper phosphorylated.Citation11 Overlap exists between the AD and normal adult patterns of phosphorylation, making the quantitative differences in the level of phosphate incorporation one of the decisive parameters.Citation12 Specific phosphorylation patterns also seem generated in the disease, and these form the basis for a large class of AD specific antibodies, including the above mentioned AT-8 antibodyCitation10 with specificity for both phospho-Ser199/phospho-Ser202 and phosphoThr205. All these data indicate that some phospho “bar code” might exist, whereby some sites are important for its physiological role of microtubule dynamics regulator, whereas another set (overlapping or not with the previous one) leads to aggregation into PHFs, degradation and/or toxic function.
Untangling this code will be a major enterprise, largely due to the large number of phosphorylation sites on Tau together with the complex interplay of the different kinases involved. Underlying many of the difficulties is the analytical problem of characterizing samples at a qualitative (what sites?) and quantitative (what degree of phosphate incorporation?) level. Both mass spectrometry and immunochemistry have well recognized advantages of sensitivity for characterizing a phosphorylation pattern of Tau samples derived from in vivo material, and the recent demonstration of top down mass spectrometry for the characterization of complex protein molecules without previous digestion has the potential of opening up a novel observation window for such complex patterns.Citation13 We recently have demonstrated that NMR spectroscopy equally might play a role in characterizing a complex phosphorylation pattern.Citation14 Although plagued by an extremely low sensitivity compared to the above mentioned methods and requiring a stable isotope labelled substrate protein, it has the potential to answer both questions of what site(s) are modified and to what extent, and its non destructive character leads to well-characterized samples that then can be used for structural and functional assays. As an example, after a full characterization of the cAMP dependent kinase (PKA) generated phospho-pattern (), we acrylodan labelled the same sample and used it to quantify the binding parameters to taxol-stabilized microtubules. This allowed us to demonstrate that phosphorylation of the Ser214 position causes an affinity drop by two orders of magnitude (), without detaching this part of the protein from the microtubule surface. Despite this, the protein is not completely devoid of polymerization activity (), underscoring the complexity of relationship between phosphorylation status and activity.
The axon of the mature neuron is characterized by a polarized microtubule orientation, while dendrites contain microtubules of mixed polarity.Citation16 At the core of the complex neuronal transport machinery that assures correct subcellular localization of organelles, mRNAs and proteins, the dynamic stability of the microtubular network is of uttermost importance for the correct functioning of the neuron.Citation17 Tau localizes mostly to axons, whereas MAP2 localization is largely restriced to the somatodendritic compartment. As long-distance trafficking uses mainly the axonal microtubule railway and Tau does (de)stabilize this network, it is of no surprise that a deregulation of its expression and/or phosphorylation level can lead to defects in axonal transport such as found in the early stages of ADCitation18 or even at the later stages of the disease.Citation19 Overexpression of the longest human tau isoform in wild-type mice leads to motor defects similar to those observed in progressive supranuclear palsy, another tauopathy,Citation20 but crossing these mice with constitutively active Gsk3β transgenic mice reduces importantly the number of axonal dilatations in brain and spinal cord, the axonal degeneration and muscular atrophy, and alleviates practically all motor problems.Citation21 The amount of Tau associated with microtubules was reduced by 50% in preparations from brain and spinal cord of these mice that overexpress both human Tau and Gsk3β compared to the hTau transgenic mice. In vitro, Tau accumulation at the surface of taxol stabilized microtubules has been observed, albeit with a lower affinity than the direct interaction,Citation22 and these aggregates might correspond to the traffick deregulating patches of Tau causing the axonopathy. In the hTau/Gsk3β mouse model, however, the authors did not detect true Tau filaments or neurofibrillary tangles, suggesting that the phosphorylation by this sole enzyme is enough to create a phosphorylation pattern of Tau that avoids its accumulation on the microtubule surface, but that does not lead to its aggregation into PHFs. Using a different mouse model overexpressing the naturally aggregating P301L hTau mutant, Le Corre et al. tested a small molecule inhibitor of the Erk2 kinase (with, however, a similar inhibition for other kinases such as cdc2, Gsk3β, PKA and PKC), and found that this compound does reduce motor impairments in a P301L Tau transgenic mouse model.Citation23 Without affecting tangle counts, the inhibitor causes a reduction of soluble aggregated hyperphosphorylated tau, although the exact nature of this species remains to be defined. The conclusions of this study with the kinase inhibitor build upon previous work by the group of Iqbal, who showed that (i) a hyperphosphorylated but soluble form of Tau (AD P-Tau) existsCitation11 (ii) it interacts with normal Tau,Citation24 and (iii) aggregation of this AD P-Tau into filaments neutralizes this interaction.Citation25 This species would not interact with tubulin, but even when present in a minor concentration, it would form a sink for the normal Tau, thereby leading to disruption of the microtubular network. When the disease progresses, the concentration of AD P-Tau increases, leading eventually to its aggregated form. Using an inducible model of the same hTau (P301L) transgenic mouse, Santa Cruz et al. showed that the soluble hyperphosphorylated species leads to neuronal degeneration, and this irrespective of ongoing tangle formation after the shutting off of the transgene.Citation26
Whereas these studies tend to indicate that the filaments of aggregated Tau are not the main culprit, questions remain as for the identity of the species that would be responsible for the sequestration of normal Tau, and hence lead to microtubule impairment. In vitro studies hereby can play an important role, as they can hopefully reproduce and subsequently allow the identification of the molecular features that define these species. We have thus set out to identify the kinase(s) that might lead to a species that (i) interacts with normal Tau with such an affinity that it might disrupt the Tau:microtubule interaction, and (ii) that might lead to the formation of amyloid aggregates without the addition of any anionic cofactors. As for the first requirement, recent data by FRET spectroscopy using an acrylodan labelled Tau and taxol stabilized microtubules have shown that the affinity of Tau for the microtubular surface is high, with a dissociation constant KD of the order of 20 nM.Citation15,Citation27 When Tau is the polymerizing agent, the binding is even characterized by a quasi irreversible component.Citation27 Finally, the tubulin concentration in the neuronal axon is high, so for P-Tau to compete successfully for this MT associated Tau, one would need a very high affinity constant. Alternatively, two aspects of the same phosphorylation event(s) might reinforce this scenario, whereby a given subset of kinases generates the AD P-Tau species, and another one leads to destabilization of the Tau:MT interaction. In this aspect, it is interesting to note that the sole phosphorylation of Ser214 by PKA can lead to a hundred fold decrease in affinity for the MT surface ().
As for the molecular aspects of Tau aggregation, most if not all of the present work has followed up on the initial observation that the addition of poly anions such as heparin can promote the aggregation of Tau into PHFs that under the electron microscope have the same aspect as those fibers isolated form the brains of AD patients.Citation28 Because later on, other poly anions such as the surface of arachidonic acid micelles or RNA equally were found to promote the fibrilization process,Citation29,Citation30 charge compensation rather than the precise anion seems to play an important role in the process. Following up on earlier mapping studies of the core region with proteases,Citation31 we have recently NMR spectroscopy to (i) define the immobile core region with a per-residue precisionCitation32 and (ii) map the interaction between heparin fragments and Tau.Citation33 We confirmed the pronounced interaction of heparin with both the regions up and downstream of the microtubule binding repeats (MTBRs), but observed equally a strong interaction with the second and third repeat.Citation33 Of special interest was the observation that all heparin seems integrated in the fiber, as all visible signals of mobility retaining regions resonated at exactly the same frequency as the free Tau protein. EPR data on full length Tau PHF suggested a model for the fibers with a parallel in-register stacking of β strand,Citation34 similar to the recently obtained high resolution data on crystalsCitation35 or fibersCitation36 derived from a prion peptide, or of fibers from β amyloid peptide.Citation37 Those peptide arrangements show a “dry” interface where the shape complementarity of facing side chains leaves little room even for water molecules,Citation35 further excluding the possibility that heparin intercalates between the facing β sheets. Considering that the NMR invisible solid like core region in the heparin induced PHFs spanning over hundred amino acids is not necessarily the true amyloid region, we can imagine that this region consists both of immobilized but disordered regions, and of genuine amyloid regions defined by some regular stacking. In other systems, this amyloid region consists of rather small peptides, and if we consider the R2 and R3 repeats, we can imagine that they would be no longer than the V275QIINK280 (in PHF6*) or V306QIVYK311 (in PHF6) hexapeptides, previously identified by peptide mapping studies as the aggregation nuclei.Citation38,Citation39 Stacking of these peptides into parallel in register β sheets would however lead to the formation of ladder-like intermolecular stretches of the same residues. The K280–K281 and K311P312 motifs would create a continuous stretch of positive charges with accompanying strong electrostatic repulsion, unless a mechanism of charge neutralization is provided. One of such mechanisms is the deletion of at least one lysine, and ΔK280 indeed is a mutant which aggregates more rapidly.Citation40 The in vitro study suggests a model where the heparin polymer wraps tightly around the outer surface of the (double) pleated sheets, and thereby neutralizes the inhibitory charge repulsions that would occur in a continuous but intermolecularly formed polylysine stretch. The heterogeneous nature of most heparin preparations thereby might lead to fibers of lesser regularity than those formed by isolated synthetic peptides. Solid state NMR and/or crystallography, as techniques that might resolve the recent controversy concerning the structure of the core region—only cross β strandsCitation41,Citation42 or equally some α helical structureCitation43,Citation44—will require homogeneous preparations of filaments, and will have to distinguish between the amorphous and truly amyloid phases in this core. Our finding that subtle variations in size and/or charge distribution between two batches of the same commercial heparin can lead to fiber formation of all Tau moleculesCitation32,Citation45 or only less than 30% (Sillen A, Lippens G, unpublished data) suggests that other poly anions might be better suited to prepare fibers for structural biology. Careful dosing and characterization of the fibers through a combination of biochemical and low resolution spectroscopic methods will thereby be one of the corner stones for the structural elucidation of the PHF core region.
Can phosphorylation lead to an equivalent charge compensation mechanisms, or are other structural factors in play? The AD P-Tau was shown to form filaments upon incubation at physiologically relevant conditions.Citation46 Upon dephosphorylation, however, all isoforms loose this capacity, suggesting that time wise, phosphorylation comes before aggregation. However, the same study revealed that the three or four microtubule binding domains cannot be phosphorylated for aggregation to occur, limiting the role of phosphorylation to charge compensation of the inhibitory regions up- and downstream of the MTBRs. Although in agreement with the finding that the isolated MTBRs aggregate more readily than the full length protein, this seems in contradiction with the aggregation model induced by exogeneous poly anions (), where charge compensation is not limited to the MTBR flanking regions, but also within the repeats itself. We are presently working with recombinant Tau and different kinases to reproduce the aggregation without additional poly anions. This should allow us to apply our structural biology tools to the fiber formation induced by the sole event of phosphorylation, and hence get better insight in the physiological aggregation mechanism. At the same time, we hope to get insight in the important but unanswered question to what certain AD specific antibodies really detect. Indeed, some of these have been classified as “conformational antibodies.” Structural elucidation of the antibody in complex with its phosphorylated Tau antigen could be a major step forward in the understanding of the distinguishing features of PHF-Tau, and hence in the aggregation mechanism.
Phosphorylation is evidently not only a signal for aggregation, but is present in very many physiological processes. Just to mention a few aspects concerning Tau, it promotes equally the interaction with Hsc70, which acts as a linker of the CHIP E3 ligase, thereby establishing a link with its degradation.Citation47 Other protein components such as the prolyl cis/trans isomerase Pin1 equally interact with the phosphorylated Tau, and might even restore its incapacity of microtubule formation.Citation48 Very recently, phosphorylated Tau was shown to interact with actin filaments, thereby influencing their bundling and association into so-called Hirano bodies.Citation49 A detailed biochemical/biophysical study of these different pathways might lead to a better understanding of Tau's role in Alzheimer's disease, and hopefully open a novel therapeutic window on this disease.
Figures and Tables
Figure 1 (Left) NMR assignment of the phosphorylation pattern of Tau after incubation with PKACitation14. (Right) Effect of a single phosphorylation event at Ser214 on the microtubule binding propertiesCitation15 (right, top) or on its tubulin polymerizing capacity (right, bottom). In both panes, the upper curve is Tau, and the lower one pSer214 Tau. The affinity of acrylodan labeled Tau towards taxol stabilzed microtubules was measured by FRET for Tau (solid curve) or pSer214 Tau (dotted line), whereas turbidity was used to evaluate the polymerization of tubulin into microtubules.
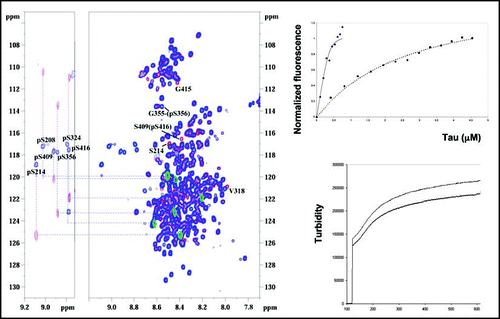
Figure 2 (Left) Electron microscopy picture of a Tau PHF promoted by incubation with heparin. (Right) Model for the amyloid core of the fiber, with heparin providing for the negative charges essential to compensate for the positive stretch formed by parallel in register stacking of lysine containing peptides.Citation33

Acknowledgments
Alain Sillen was funded by a European Training and Mobility Grant (HPRN CT 2002 00241). Part of this work was funded by the Agence National de la Recherche Grant ANR 05 BLANC 0320 01 to Guy Lippens. Nathalie Sibille was funded by a fellowship of the AIRMA (Association internationale de recherche contre la maladie d'Alzheimer). The 600 MHz facility used in this study was funded by the Région Nord Pas de Calais (France), the CNRS, and the Institut Pasteur de Lille. The 800MHz spectrometer was funded within the French National project “NMR et Radiocristallographie structurale - Grand Bassin Parisien” by the Région Nord Pas de Calais (FEDER), CNRS, USTL and Research Ministry.
References
- Ross CA, Poirier MA. What is the role of protein aggregation in neurodegeneration?. Nat Rev Mol Cell Biol 2005; 6:891 - 898
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004; 431:805 - 810
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1 42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 1998; 95:6448 - 6453
- Buee L, Bussiere T, Buee Scherrer V, Delacourte A, Hof P R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 2000; 33:95 - 130
- Shen J, Kelleher RJ 3rd. The presenilin hypothesis of Alzheimer's disease: evidence for a loss of function pathogenic mechanism. Proc Natl Acad Sci USA 2007; 104:403 - 409
- Trojanowski JQ, Lee VM. Pathological tau: a loss of normal function or a gain in toxicity?. Nat Neurosci 2005; 8:1136 - 1137
- Braak H, Braak E. Neuropathological stageing of Alzheimer related changes. Acta Neuropathol 1991; 82:239 - 259
- Gallyas F. Silver staining of Alzheimer's neuro Wbrillary changes by means of physical development. Acta Morph Acad Sci Hung 1971; 19:1 - 8
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol (Berl) 2006; 112:389 - 404
- Biernat J, Mandelkow EM, Schroter C, Lichtenberg Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M. The switch of tau protein to an Alzheimer like state includes the phosphorylation of two serine proline motifs upstream of the microtubule binding region. EMBO J 1992; 11:1593 - 1597
- Köpke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke Iqbal I. J Biol Chem 1993; 268:24374 - 24380
- Wischik CM, Edwards PC, Lai RY, Gertz HN, Xuereb JH, Paykel ES, Brayne C, Huppert FA, Mukaetova Ladinska EB, Mena R, et al. Quantitative analysis of tau protein in paired helical filament preparations: implications for the role of tau protein phosphorylation in PHF assembly in Alzheimer's disease. Neurobiol Aging 1995; 16:409 - 417
- Han X, Jin M, Breuker K, McLafferty FW. Extending top down mass spectrometry to proteins with masses greater than 200 kilodaltons. Science 2006; 314:109 - 112
- Landrieu I, Lacosse L, Leroy A, Wieruszeski JM, Trivelli X, Sillen A, Sibille N, Schwalbe H, Saxena K, Langer T, Lippens G. NMR analysis of a Tau phosphorylation pattern. J Am Chem Soc 2006; 128:3575 - 3583
- Sillen A, Barbier P, Landrieu I, Lefevbre S, Wieruszeski JM, Leroy A, Peyrot V, Lippens G. NMR investigation of the interaction between the neuronal protein Tau and the microtubules. Biochemistry 2007; In press.
- Baas PW, Black MM, Banker GA. Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J Cell Biol 1989; 109:3085 - 3094
- Gotz J, Ittner LM, Kins S. Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer's disease?. J Neurochem 2006; 98:993 - 1006
- Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, Raina AK, Vinters HV, Tabaton M, Johnson AB, Paula Barbosa M, Avila J, Jones PK, Castellani RJ, Smith MA, Perry G. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. Am J Pathol 2003; 162:1623 - 1627
- Khatoon S, Grundke Iqbal I, Iqbal K. Brain levels of microtubule associated protein tau are elevated in Alzheimer's disease: a radioimmuno slot blot assay for nanograms of the protein. J Neurochem 1992; 59:750 - 753
- Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VM, Trojanowski JQ, Jakes R, Crowther RA, Spillantini MG, Burki K, Goedert M. Axonopathy and amyotrophy in mice transgenic for human four repeat tau protein. Acta Neuropathol (Berl) 2000; 99:469 - 481
- Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four repeat human tau protein. Am J Pathol 1999; 155:2153 - 2165
- Ackmann M, Wiech H, Mandelkow E. Nonsaturable binding indicates clustering of tau on the microtubule surface in a paired helical filament like conformation. J Biol Chem 2000; 275:30335 - 30343
- Le Corre S, Klafki HW, Plesnila N, Hubinger G, Obermeier A, Sahagun H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, Dickson DW, Hutton M, Roder HM. An inhibitor of tau hyper phosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci USA 2006; 103:9673 - 9678
- Alonso AC, Zaidi T, Grundke Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA 1994; 91:5562 - 5566
- Alonso AC, Li B, Grundke Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci USA 2006; 103:8864 - 8869
- SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005; 309:476 - 481
- Makrides V, Massie MR, Feinstein SC, Lew J. Evidence for two distinct binding sites for tau on microtubules. Proc Natl Acad Sci USA 2004; 101:6746 - 6751
- Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule associated protein tau into Alzheimer like filaments induced by sulphated glycosaminoglycans. Nature 1996; 383:550 - 553
- Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer's disease. Am J Pathol 1997; 150:2181 - 2195
- Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule associated protein tau into Alzheimer like paired helical filaments. FEBS Lett 1996; 399:344 - 349
- Novak M, Kabat J, Wischik CM. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer's disease paired helical filament. EMBO J 1993; 12:365 - 370
- Sillen A, Leroy A, Wieruszeski JM, Loyens A, Beauvillain JC, Buee L, Landrieu I, Lippens G. Regions of tau implicated in the paired helical fragment core as defined by NMR. Chembiochem 2005; 6:1849 - 1856
- Sibille N, Sillen A, Leroy A, Wieruszeski JM, Mulloy B, Landrieu I, Lippens G. Structural impact of heparin binding to full length Tau as studied by NMR spectroscopy. Biochemistry 2006; 45:12560 - 12572
- Margittai M, Langen R. Template assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci USA 2004; 101:10278 - 10283
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross beta spine of amyloid like fibrils. Nature 2005; 435:773 - 778
- Ritter C, Maddelein ML, Siemer AB, Luhrs T, Ernst M, Meier BH, Saupe SJ, Riek R. Correlation of structural elements and infectivity of the HET s prion. Nature 2005; 435:844 - 848
- Tycko R. Progress towards a molecular level structural understanding of amyloid fibrils. Curr Opin Struct Biol 2004; 14:96 - 103
- von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, Mandelkow E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta structure. J Biol Chem 2001; 276:48165 - 48174
- von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci USA 2000; 97:5129 - 5134
- Goedert M, Jakes R, Crowther RA. Effects of frontotemporal dementia FTDP 17 mutations on heparin induced assembly of tau filaments. FEBS Lett 1999; 450:306 - 311
- Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross beta structure. Proc Natl Acad Sci USA 2003; 100:9034 - 9038
- Barghorn S, Davies P, Mandelkow E. Tau paired helical filaments from Alzheimer's disease brain and assembled in vitro are based on beta structure in the core domain. Biochemistry 2004; 43:1694 - 1703
- Sadqi M, Hernandez F, Pa U, Perez M, Schaeberle MD, Avila J, Munoz V. Alpha helix structure in Alzheimer's disease aggregates of tau protein. Biochemistry 2002; 41:7150 - 7155
- Kunjithapatham R, Oliva FY, Doshi U, Perez M, Avila J, Munoz V. Role for the alpha helix in aberrant protein aggregation. Biochemistry 2005; 44:149 - 156
- Sillen A, Wieruszeski JM, Leroy A, Younes AB, Landrieu I, Lippens G. High resolution magic angle spinning NMR of the neuronal tau protein integrated in Alzheimer's like paired helical fragments. J Am Chem Soc 2005; 127:10138 - 10139
- Alonso AC, Zaidi T, Novak M, Grundke Iqbal I, Iqbal K. Hyperphosphorylation induces self assembly of tau into tangles of paired helical filaments /straight filaments. Proc Natl Acad Sci USA 2001; 98:6923 - 6928
- Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 2004; 279:4869 - 4876
- Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer associated phosphorylated tau protein. Nature 1999; 399:784 - 788
- Fulga TA, Elson Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F actin mediates tau induced neuronal degeneration in vivo. Nat Cell Biol 2007; 9:139 - 148