Abstract
Huntingtin containing an expanded polyglutamine causes neuronal death and Huntington disease. Although expanded huntingtin is found in virtually every cell type, its toxicity is limited to neurons of certain areas of the brain, such as cortex and caudate/putamen. In affected areas of the brain, expanded huntingtin is not found in its intact monomeric form. It is found instead in the form of N-terminal fragments, oligomers and polymers, all of which accumulate in the cortex. Whereas the oligomer is mostly soluble, the polymers and the fragments associate with each other and with other proteins to form the insoluble inclusions characteristic of the disease. It is likely that the aggregates containing expanded huntingtin are toxic to neurons, but it remains to be determined whether the oligomer or the inclusion is the toxic species.
Introduction
The cause of Huntington disease is the presence of an abnormally long polyglutamine (polyQ) sequence close to the N-terminus of huntingtin, a protein of unknown function.Citation1,Citation2 Huntington disease is one of nine diseases of the central nervous system caused by polyglutamine expansion in nine unrelated proteins. Diseases of polyglutamine expansion are characterized by the presence of insoluble inclusions in neurons of the central nervous system.Citation3,Citation4 These inclusions, whose average size is about 5–7 µm, contain a variety of proteins in addition to the mutant protein bearing the expanded polyglutamine. Although the causative role of inclusions in Huntington disease has been challenged, considerable evidence supports the notion that some form of huntingtin aggregation contributes to neuronal death.Citation5–Citation7
Mechanism of Aggregation of Proteins Bearing Expanded Polyglutamine
In 1993, Howard Green hypothesized that, as a result of excessive polyglutamine length, huntingtin could become aggregated by transglutaminase.Citation8 Transglutaminases catalyze the formation of isopeptide cross-links between the side chains of glutaminyl and lysyl residues of adjacent proteins. In 1994, Max Perutz proposed that polyglutamine sequences could form multimers stabilized by hydrogen-bonded polar zippers between the main chain and side chain amides.Citation9
Polar zippers.
The first evidence for aggregate formation by polyQ sequences came from the discovery that a small protein containing polyQ of 10 residues (Q10) formed oligomers in vitro.Citation10 It was later shown that transgenic mice expressing the first exon of the human huntingtin gene, which encoded about 130 glutamine residues flanked by 17 N-terminal and 52 C-terminal residues developed neuronal inclusions and neurological disease.Citation11 The bacterially-produced peptide encoded by exon 1 formed aggregates in vitro when its polyQ exceeded 51 residues, but not when the polyQ was 20 or 30 residues.Citation12 Since purified peptide was used, aggregation must have resulted from multimerization by polar zipper formation. The authors concluded that the inclusions found in exon 1 transgenic mice were similarly stabilized by hydrogen-bonded polar zippers, but no direct analysis of the intermolecular bonds was carried out.
Hazeki et al.Citation13 introduced the N-terminal fragment of huntingtin bearing Q77 into cultured cells by transfection, and observed the formation of inclusions. Reduction of the inclusions to monomeric fragments after incubation in concentrated formic acid showed that the aggregates were stabilized by hydrogen bonds, possibly polar zippers.
Most diseases of polyQ expansion occur when the polyQ sequence exceeds 40 residues. This threshold has been explained by the fact that the polyQ sequence forms a helical fiber with 20 residues per turn and that two turns (40 residues) are required for stabilization of the helix and promotion of multimerization.Citation14
Transglutaminase-catalyzed cross-linking.
There is ample evidence that polyQ is a substrate of transglutaminase in vitro. Model peptides containing Q5–Q18 have been shown to be much better substrates than those containing Q1–Q3.Citation15 Experiments with full-length huntingtin containing polyQ sequences of various lengths have shown that the ability of huntingtin to act as a substrate of transglutaminase improves sharply with length of polyQ.Citation16 Numerous other reports have provided evidence supporting a causative role of transglutaminase in diseases of polyQ expansion, but this role has also been challenged.Citation17–Citation22
A possible function of transglutaminase in the stabilization of neuronal aggregates is corroborated by the fact that the enzyme is widely distributed in the brain, where it can be activated, since ε-(γ-glutamyl) lysine cross-links are found in normal brain.Citation23–Citation25
Forms of Aggregated Huntingtin Detected by Immunofluorescence Staining
Immunofluorescence staining shows that inclusions are found in affected areas of the brain such as cortex and caudate/putamen, but not in unaffected areas such as cerebellum. Within affected areas, inclusions are limited to neurons: they are not detected in glial cells.Citation21 Inclusions tend to be nuclear in patients with juvenile Huntington disease, which is characterized by large polyQ expansions, whereas they are mostly cytoplasmic in patients with adult Huntington disease and shorter polyQ expansions.Citation4 Using an antibody directed against the 17-most N-terminal amino acids of huntingtin, we were able to show that the frequency of nuclear inclusions, but not that of cytoplasmic inclusions correlates with the severity of the disease.Citation26 This suggests that nuclear aggregation but not cytoplasmic aggregation might participate in neuronal death.
By using the EM48 antibody raised against the first 256 amino acids of huntingtin and which specifically stains aggregated huntingtin, Gutekunst et al.Citation27 identified many more aggregates than when antibodies raised against more N-terminal parts of the molecule were used. Most of these aggregates were located in neuropils, which are likely to correspond to dendrites. Dendritic aggregates could contribute to neuronal death by disturbing dendritic transport.Citation27 EM48 also detected small puncta ranging in diameter from 0.3 to 1.4 µm, but it was not clear whether these puncta corresponded to microaggregates or to immunostained organelles.Citation27
Forms of Aggregates Containing Expanded PolyQ Detected by Western Blotting and Filter Assays
Two main kinds of antibodies have been used: those directed against determinants outside of the polyQ sequence and which therefore are expected to stain both normal and expanded huntingtin, and those specific to the expanded polyQ sequence.Citation28
Analysis of the forms of huntingtin present in aggregates was made possible by the use of concentrated formic acid. Concentrated formic acid can dissolve proteins insoluble in solutions containing SDS.Citation29 Hazeki et al.Citation13 were the first to demonstrate that aggregates containing an expanded polyQ were dissolved by concentrated formic acid. Peptides containing 20 consecutive Q residues are insoluble when incubated at 100°C in a solution of SDS. The addition of concentrated formic acid instead of SDS dissolves the peptides completely.Citation30 This means that if the insolubility of peptides containing polyQ is the consequence of polar zipper formation, these hydrogen bonds are dissociated by concentrated formic acid.Citation9 The resistance of any polyQ-bearing aggregate to dissociation by formic acid suggests its stabilization by bonds stronger than hydrogen bonds, possibly covalent bonds.Citation8,Citation30
Monomer and formic acid-resistant oligomers of expanded polyQ are present in the inclusions of cultured cells.
PC12 cells stably transfected with an inducible plasmid expressing Q205 flanked by androgen receptor sequences form inclusions within 48 h of induction with ponasterone A (). These inclusions can be purified on the basis of their insolubility in SDS, 2-mercaptoethanol (2-ME) and concentrated guanidinium chloride. Purified inclusions submitted to electrophoresis yield no detectable product after staining with the 1C2 antibody. This is because the inclusions are insoluble in SDS/2-ME and are too large to enter the gel. In contrast when incubated in concentrated formic acid, the inclusions are completely dissolved and release a large amount of monomeric Q205 in addition to a smaller amount of an oligomer with a considerably higher molecular mass. The effect of formic acid treatment may be explained by its ability to dissociate noncovalently stabilized aggregates otherwise unable to enter the gel. It may be concluded that the inclusions formed in PC12 cells are composed of monomers and oligomers of the Q205-bearing peptide. Monomer and oligomer are associated through hydrogen bonds. Both monomer and oligomer are readily detected in the cytoplasm without formic acid pre-treatment.
Very large polymers can be identified by a filter retention assay that takes advantage of the fact that they are unable to pass through a 0.2 µm cellulose acetate filter.Citation12 After filtration of formic acid-treated extract of PC12 cells expressing Q205 through cellulose acetate filters, no retained aggregates are detected.Citation30
A soluble cytoplasmic oligomer containing expanded huntingtin in the cortex of patients with Huntington disease.
In most cell types from patients with Huntington disease, the normal and expanded huntingtin are found in virtually equal amounts. But when extracts are prepared from affected areas of the brain and analyzed by Western blotting using an anti-N-terminal huntingtin antibody, the expanded protein is found to be much less abundant and there appears a faint smear extending above the position of the mutant protein.Citation16,Citation31,Citation32 This smear is strongly stained by the 1C2 antibody, which is specific for expanded polyQ.Citation31 No such smear is observed in either cerebellum or lymphoblastoid cells of patients with Huntington disease.Citation30 This suggests specific oligomerization of mutant huntingtin in the more affected regions of the brain.
The oligomer of expanded huntingtin appears to accumulate in the cortex where it is 3–6 times more abundant than normal monomeric hutingtin. Pre-treatment of the cortical extract with concentrated formic acid prior to the electrophoresis does not appreciably affect the appearance of the oligomer.Citation30 The oligomer is likely to be stabilized by covalent bonds, since it is not reduced to monomers by formic acid (). Subcellular fractionation of cortical extracts prior to formic acid treatment shows that the soluble oligomer is located in the cytoplasm and not in the nuclei. Incubation of cortical nuclei in formic acid releases a significant amount of oligomer (). This water insoluble oligomer is presumably a component of the nuclear inclusions together with polymerized and fragmented huntingtin (see below).
An insoluble polymer in nuclei of cerebral cortex.
Electrophoresis of cortical nuclear extracts in 4% polyacrylamide gels followed by staining with the 1C2 antibody shows the presence of a component at the top of the gel. This component was deemed to be a polymer too large to enter the gel (). Such polymers can be detected based on their retention by cellulose acetate filters. In some patients with Huntington disease, particularly juvenile cases, inclusions are mostly nuclear. In order to determine whether the inclusions of these patients contained insoluble polymers, nuclei were isolated from the cortex of such patients, purified by density gradient centrifugation, incubated in concentrated formic acid and submitted to the filter retention assay. The presence of insoluble polymer was revealed by a dark spot stained by the 1C2 antibody. No stained polymer was detected in cerebellar nuclei (). Therefore the nuclear inclusions of patients with juvenile Huntington disease contain a formic acid-insoluble polymer.
N-terminal fragments of expanded huntingtin in brain inclusions.
A great deal of indirect evidence has supported the notion that the inclusions contain N-terminal fragments of expanded huntingtin and no intact protein. N-terminal fragments of huntingtin bearing an expanded polyglutamine are more lethal to cells and more readily form inclusions than the intact protein when they are expressed in cells or in transgenic mice.Citation33,Citation34 Inclusions present in the brain of patients with Huntington disease are stained by antibodies directed against the residues of huntingtin located in the vicinity of the expanded polyQ, but not by those raised against more C-terminal parts of the protein.Citation4,Citation35
From evidence largely derived from cultured cells containing an expanded huntingtin, it has been concluded that inclusions originated from a discrete fragment generated by a specific endoprotease such as caspase 3, calpain or an aspartic endopeptidase.Citation36–Citation38 However it remained unclear whether such fragments were present in the brain inclusions of patients with Huntington disease.
Brain inclusions can be purified based on their insolubility in solutions containing concentrated urea and SDS.Citation26 As the inclusions isolated from PC12 cells, cortical inclusions can be dissolved in concentrated formic acid. The forms of expanded huntingtin released by formic acid are then identified by electrophoresis and staining with the 1C2 antibody.
Polyacrylamide gel electrophoresis of the purified inclusions treated with formic acid reveals oligomer and a broad smear of fragmented huntingtin, with a molecular weight range of 50–150 kDa. No such material is released from cerebellum (). A smear of soluble fragmented huntingtin extending from ∼70–200 kDa is also present in cortical extract untreated with formic acid, whereas no smear of fragments or a very weak smear is detected in cerebellum. Pre-incubation of the cortical extract with formic acid leads to an increase in the intensity of the smear and to its extension towards lower molecular weights (). This effect must result from the release by formic acid of fragments present in the inclusions. Incubation of cerebellar extracts with formic acid does not release fragments.
The expanded polyQ sequence of huntingtin is located very close to the N-terminus of the protein (beginning at residue 18). Therefore the smear stained by the 1C2 antibody must be composed of N-terminal fragments ranging between about 350 and 1300 residues. This is confirmed by the fact that the 4C8 monoclonal antibody, which recognizes an epitope located between residues 490 and 580 fails to stain the lower part of the smear stained by 1C2 ().Citation38 Such fragments are likely to result from the action of multiple proteolytic activities with little specificity. It remains possible that a small specific fragment may nucleate the inclusions and that larger fragments may then be recruited to the inclusions.
In order to determine whether the cortical inclusions contain polymeric huntingtin in addition to fragmented huntingtin, we submitted purified inclusions pre-treated with formic acid to electrophoresis through 0.75% agarose gels; such gels have been designed for resolution of extremely large proteins such as titin.Citation39 After completion of the electrophoresis and transfer to nitrocellulose, the cortical samples show intense 1C2 staining at the well; no such staining is observed in cerebellum. This staining is likely to correspond to a polymer of expanded huntingtin, which is unable to enter the agarose gel because of its large size (). Such a polymer may be identical to the polymer detected by the filter retention assay in cortical nuclei.Citation30
Concluding Remarks
In unaffected areas of the brain of patients with Huntington disease, such as cerebellum, normal and expanded huntingtin are equally abundant and are both found entirely in a soluble monomeric form.
In affected areas of the brain, such as cortex, the normal huntingtin also exists entirely as a soluble monomer, but the expanded huntingtin does not. Instead, the expanded huntingtin is found in fragmented, oligomerized and polymerized forms, which can be either soluble or insoluble in water.
The water-soluble forms of the expanded huntingtin of cortex consist of oligomerized and fragmented huntingtin. The soluble oligomers of huntingtin appear to accumulate in cortex, where their abundance largely exceeds that of normal huntingtin.
The water-insoluble expanded huntingtin, which is mostly associated with the inclusions, consists of oligomeric, polymeric and fragmented protein. These three forms of expanded huntingtin are associated with each other by non-covalent bonds since they are released by formic acid. In contrast, the oligomer and the polymer themselves are likely to be stabilized by covalent bonds since they are resistant to formic acid.
Expansion of the polyQ sequence, found in the androgen receptor, causes spinobulbar muscular atrophy. Li et alCitation40 have recently shown that soluble oligomers of the androgen receptor with expanded polyQ are present in the brain of mice bearing an androgen receptor with expanded polyQ. Therefore the presence of oligomers containing the expanded polyQ may be a general property of diseases of polyQ expansion.
Abbreviations
| PolyQ | = | polyglutamine |
| SDS | = | sodium dodecyl sulfate |
| 2-ME | = | 2-mercaptoethanol |
Figures and Tables
Figure 1 Aggregation of Q205 in PC12 cells. (A) A 24 h induction by ponasterone A was suffisant to induce the formation of large aggregates containing EGFP-Q205 in the cell line PC12-EGFP-Q205. Each cell contained a small number (one to three usually) of large aggregates and showed a very low level of diffuse cytoplasmic fluorescence. This suggested that most of the soluble recombinant protein had been recruited to large aggregates. However, a number of cells displayed either a multitude of small aggregates, or a high level of diffuse cytoplasmic fluorescence with no evidence of aggregation. These differences are probably related to differences in the time-course of aggregation in different cells. (B) Purified inclusions from the cell line PC12-Q205 were either treated or not with formic acid (FA), boiled in SDS/2-ME and submitted to electrophoresis through 7.5% polyacrylamide. After transfer to nitrocellulose, the proteins were stained with the 1C2 antibody. In the absence of formic acid treatment, no signal was detected. After treatment with 90% formic acid for one hour at 37°C, electrophoresis revealed a strong band corresponding to a monomer (mnQ205) with an apparent molecular mass of ∼100 kDa; the deduced molecular mass is 36,340, but expanded polyQ reduces mobility in gel electrophoresis. A second band with a considerably higher molecular mass was clearly stained and corresponded to an oligomer (oligQ205). Reproduced from Iuchi et al.Citation30
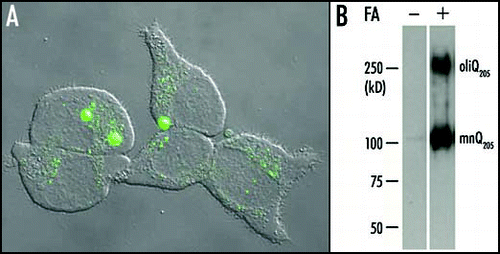
Figure 2 State of expanded huntingtin in Huntington disease. (A) Crude extracts of cortex (Cx) and cerebellum (Cb) were pre-incubated in formic acid for 1 h at 37°C. Western blots prepared after electrophoresis on a 4% polyacrylamide gel were stained consecutively with the anti-N-terminal and the 1C2 antibody. All samples of cortex produced a broad oligomeric band of expanded huntingtin. No such band was found in the cerebellum. In contrast, all of the cerebellar samples, but none of the cortical ones, yielded a clear band of monomeric expanded huntingtin. (B) Iodixanol-purified nuclei, either treated or not with formic acid, were analyzed by electrophoresis through an 8% polyacrylamide gel. After transfer to nitrocellulose, proteins were stained with the 1C2 antibody. In the absence of formic acid pre-treatment, no resolved band is present. The only detectable signal consist of a weakly stained band at the top of the 4% stacking gel. After treatment with formic acid, electrophoresis reveals a strongly stained polymer (pol.) at the well, a compressed oligomer (olig.) band whose mean mobility corresponds to that of the free cytosolic oligomer in A, and a broad band of fragments (frag.) with molecular masses ranging from ∼50 to 150 kDa. We may conclude that the nuclei contain insoluble polymer, oligomer, and fragments, all of which possess expanded polyQ. Reproduced from Iuchi et al.Citation30
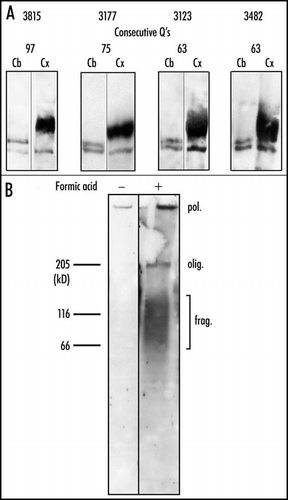
Figure 3 Polymeric state of expanded huntingtin in Huntington disease. Purified nuclei of cortex and cerebellum of Huntington disease patient 3815 and of controls were extracted with concentrated formic acid. The suspension was diluted 10-fold in Tris/SDS/2-ME, boiled for five minutes, and immediately passed through cellulose acetate filters. The retained material was visualized with antibody 1C2. Control SALDAV 080010 was a lymphoblast line possessing a huntingtin with 79 glutamine residues. Rat cortex, whose huntingtin possesses Q8, should not be stainable by 1C2. Cortex 3815: 20,000 nuclei gave a strong signal. The corresponding cerebellum, SALDAV 080010, and rat cortex gave weak signals. Cortex of patient 3123: 120,000 nuclei produced a weaker signal than 20,000 nuclei of cortex 3815, in keeping with a number of inclusions smaller in case 3123 than in case 3815. Arrow indicates control without nuclei. Resistance to formic acid of the cortical polymers of the two patients strongly suggests stabilization of the polymers by covalent bonds. Reproduced from Iuchi et al.Citation30
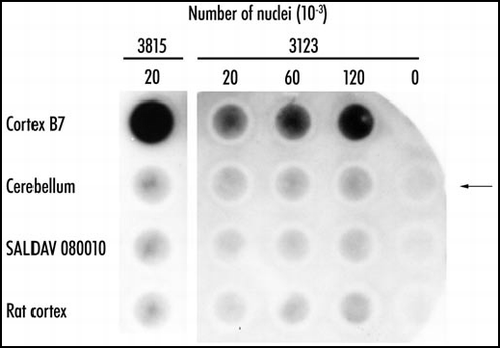
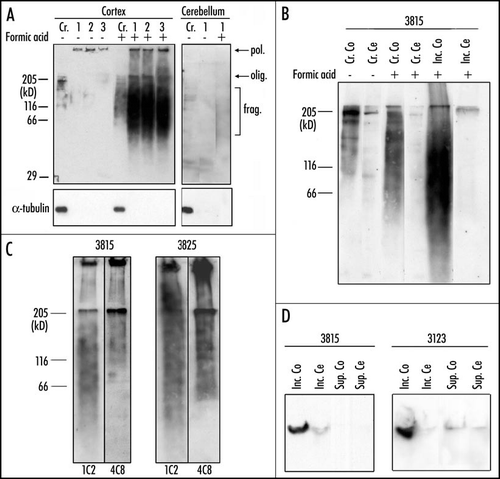
Figure 4 State of expanded huntingtin in purified aggregates of patients with Huntington disease. (A) Inclusions were purified from cortex of patient 3815. Samples at the various stages of purification were preincubated or not in formic acid and analyzed by Western blotting with the 1C2 antibody: (Cr) Crude extract, (1) after dialysis, (2) after low speed centrifugation, (3) final pellet of purified inclusions. In the absence of formic acid treatment, only a faint signal was detected in the well. After treatment with 90% formic acid for one hour at 37°C, electrophoresis of the cortical extract revealed an oligomer band (olig.), a polymer (pol.) and a broad range of fragments (frag.) with molecular masses ranging from ∼50 to 150 kDa. No such material was released from the cerebellum. α-tubulin, an extremely abundant neuronal protein, was not detectable after purification. (B) Crude extracts (Cr.) and purified inclusions (Inc.) from cortex (Co) of patient 3815 were either treated or not with formic acid. Similarly treated cerebellar extracts (Cr. Ce and Inc. Ce) were used as control. Crude extracts loaded on the gel contained 100 µg of protein; inclusions were purified from 1 mg of crude extract. Proteins were resolved through an 8% polyacrylamide gel and analyzed by Western blotting, using the 1C2 antibody. A smear of water-soluble fragmented huntingtin was detected in the absence of formic acid treatment in cortex, but not in cerebellum. Treatment with formic acid increased the intensity of the smear, presumably because further fragments were released from the purified inclusions. The smear of fragments released from the purified inclusions of patient 3815 overlapped with that present in the untreated extract, but extended to lower molecular weights. The absence of fragmented huntingtin in cerebellum is likely to explain the absence of inclusions in this region of the brain. (C) Inclusions purified from the brain of two patients (3815 and 3825) were incubated in the presence of formic acid and stained sequentially with the 4C8 and the 1C2 antibody. The 4C8 antibody failed to detect the lower part of the smear of fragmented huntingtin detected by the 1C2 antibody; the staining was abruptly interrupted at ∼60–70 kDa presumably because fragments smaller than 70 kDa did not contain the more C-terminal epitope recognized by 4C8. The signal was stronger for case 3825, because four times more material was loaded on the gel. (D) Inclusions were purified as described in (26) except that centrifugation was at 300 000 g rather than 100 000 g. After the high-speed centrifugation, the pellet containing the inclusions and the supernatant were submitted to electrophoresis through a 0.75% agarose gel and stained with the 1C2 antibody. A polymer was present in the cortical (Co) inclusions (pellet) (Inc.) of patient 3815 and 3123, but not in the supernatant (Sup.). The polymer was therefore entirely associated with the inclusions. No such polymer was found in the cerebellum (Ce). Reproduced from Hoffner et al.Citation26
Acknowledgements
Guylaine Hoffner was supported by the Bettencourt-Schueller Foundation and the Region Ile de France, and research was financed by CNRS and the Institut des Maladies Rares. We are grateful to the Proceedings of the National Academy of Science, the Journal of Neurochemistry, The International Society for Neurochemistry and Blackwell Publishing for permission to reproduce the figures.
References
- The Huntington's disease collaborative research group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993; 72:971 - 983
- Gusella JF, MacDonald ME. Huntington's disease: Seeing the pathogenic process through a genetic lens. Trends Genet 2006; 31:533 - 540
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997; 90:537 - 548
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997; 277:1990 - 1993
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004; 431:805 - 810
- Bates G. Huntingtin aggregation and toxicity in Huntington's disease. Lancet 2003; 361:1642 - 1644
- Hoffner G, Djian P. Protein aggregation in Huntington's disease. Biochimie 2002; 84:273 - 278
- Green H. Human genetic diseases due to codon reiteration: Relationship to an evolutionary mechanism. Cell 1993; 74:955 - 956
- Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci USA 1994; 91:5355 - 5358
- Stott K, Blackburn JM, Butler PJ, Perutz M. Incorporation of glutamine repeats makes protein oligomerize: Implication for neurodegenerative diseases. Proc Natl Acad Sci USA 1995; 92:6509 - 6513
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996; 87:493 - 506
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997; 90:549 - 558
- Hazeki N, Tukamoto T, Goto J, Kanazawa I. Formic acid dissolves aggregates of an N-terminal huntingtin fragment containing an expanded polyglutamine tract: Applying to quantification of protein components of the aggregates. Biochem Biophys Res Commun 2000; 277:386 - 393
- Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci USA 2002; 99:5591 - 5595
- Kahlem P, Terré C, Green H, Djian P. Peptides containing glutamine repeats as substrates for transglutaminase-catalyzed cross-linking: Relevance to diseases of the nervous system. Proc Natl Acad Sci USA 1996; 93:14580 - 14585
- Kahlem P, Green H, Djian P. Transglutaminase action imitates Huntington's disease: Selective polymerization of huntingtin containing expanded polyglutamine. Mol Cell 1998; 1:595 - 601
- Igarashi S, Koide R, Shimohata T, Yamada M, Hayashi Y, Takano H, Date H, Oyake M, Sato T, Sato A, Egawa S, Ikeuchi T, Tanaka H, Nakano R, Tanaka K, Hozumi I, Inuzuka T, Takahashi H, Tsuji S. Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA protein with an expanded polyglutamine stretch. Nat Genet 1998; 18:111 - 117
- Lesort M, Chun W, Johnson GV, Ferrante RJ. Tissue transglutaminase is increased in Huntington's disease brain. J Neurochem 1999; 73:2018 - 2027
- Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, Mitchell D, Steinman L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med 2002; 8:143 - 149
- Bailey CD, Johnson GV. Tissue transglutaminase contributes to disease progression in the R6/2 Huntington's disease mouse model via aggregate-independent mechanisms. J Neurochem 2005; 92:83 - 92
- Hoffner G, Djian P. Transglutaminase and diseases of the central nervous system. Frontiers Biosc 2005; 10:3078 - 3092
- Chun W, Lesort M, Tucholski J, Ross CA, Johnson GV. Tissue transglutaminase does not contribute to the formation of mutant huntingtin aggregates. J Cell Biol 2001; 153:25 - 34
- Gilad GM, Varon LE. Transglutaminase activity in rat brain: Characterization, distribution, and changes with age. J Neurochem 1985; 45:1522 - 1526
- Kim SY, Grant P, Lee JH, Pant HC, Steinert PM. Differential expression of multiple transglutaminases in human brain. J Biol Chem 1999; 274:30715 - 30721
- Lorand L. Neurodegenerative diseases and transglutaminase. Proc Natl Acad Sci USA 1996; 93:14310 - 14313
- Hoffner G, Island ML, Djian P. Purification of neuronal inclusions of patients with Huntington disease reveals a broad range of N-terminal fragments of expanded huntingtin and insoluble polymers. J Neurochem 2005; 95:125 - 136
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: Relationship to neuropathology. J Neurosci 1999; 19:2522 - 2534
- Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, et al. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature 1995; 378:403 - 406
- Mokrasch LC. Spontaneous incorporation of biogenic amines into purified proteolipid. Biochem Biophys Acta 1965; 107:608 - 610
- Iuchi S, Hoffner G, Verbeke P, Djian P, Green H. Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. Proc Natl Acad Sci USA 2003; 100:2409 - 2414
- Schilling G, Sharp AH, Love SJ, Wagster MV, Li S H, Stine OC, Ross CA. Expression of the Huntington's disease (IT15) protein product in HD patients. Hum Mol Genet 1995; 4:1365 - 1371
- Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y, Weber C, Agid Y, Hirsch EC, Mandel JL. Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat Genet 1995; 10:104 - 110
- Lunkes A, Mandel JL. A cellular model that recapitulates major pathogenic steps of Huntington's disease. Hum Mol Genet 1998; 7:1355 - 1361
- Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCucheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet 1998; 18:150 - 154
- Sieradzan KA, Mechan AO, Jones L, Wanker EE, Nukina N, Mann DM. Huntington's disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp Neurol 1999; 156:92 - 99
- Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet 1996; 13:442 - 449
- Gafni J, Ellerby LM. Calpain activation in Huntington's disease. J Neurosci 2002; 22:4842 - 4849
- Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 2002; 10:259 - 269
- Warren CM, Krzesinski PR, Greaser ML. Vertical agarose gel electrophoresis and electroblotting of high molecular weight proteins. Electrophoresis 2003; 24:1695 - 1702
- Li M, Chevalier Larsen ES, Merry DE, Diamond MI. Soluble androgen receptor oligomers underlie pathology in a mouse model of spinobulbar muscular atrophy. J Biol Chem 2007; 282:3157 - 3164