Abstract
Many research efforts in the last years have been directed towards understanding the factors determining protein misfolding and amyloid formation. Protein stability and amino acid composition have been identified as the two major factors in vitro. The research of our group has been focused on understanding the relationship between amino acid sequence and amyloid formation. Our approach has been the design of simple model systems that reproduce the biophysical properties of natural amyloids. An amyloid sequence pattern was extracted that can be used to detect amyloidogenic hexapeptide stretches in proteins. We have added evidence supporting that these amyloidogenic stretches can trigger amyloid formation by non-amyloidogenic proteins. Some experimental results in other amyloid proteins will be analyzed under the conclusions obtained in these studies. Our studies together with evidences from other groups suggest that amyloid formation is the result of the interplay between a decrease of protein stability, and the presence of highly amyloidogenic regions in proteins. As many of these results have been obtained in vitro, the challenge for the next years will be to demonstrate their validity in in vivo systems.
Introduction
The phenomenon of protein misfolding and subsequent formation of amyloid fibrils is attracting considerably attention from the biological and chemical research community for different reasons. The most obvious one is its relationship with the pathology of the range of human diseases known as amyloidoses.Citation1 Many efforts are motivated for the need of finding points of intervention against these, at the moment, incurable diseases. There is also a strong motivation in understanding how normally soluble monomeric proteins can self-assemble to form highly-organized quaternary structures as amyloid fibrils. This question constitutes not only a fascinating supramolecular assembly problem but also could hold the key to understand how to interfere with this process and design new therapeutic strategies. In addition the lessons learned from amyloid self-assembly could have also applications for the design of protein and peptide based nanomaterials.Citation2,Citation3
In the early 70's, the demonstration that lysosomal extracts were sufficient to convert amyloidogenic proteins into amyloid fibrils introduced the assumption that proteolytic processing was the major amyloidogenic determinant.Citation4,Citation5 Currently, based on many evidences from independent investigations (for a review see refs. Citation6–Citation8), it has become widely accepted in the past years that protein misfolding is the molecular basis of all the amyloid related disorders. Conformational changes of the natively folded or unfolded protein have been demonstrated to precede the formation of amyloid structures. Under certain circumstances, all the amyloidogenic proteins can adopt an amyloid prone state at least partially unfolded that favors the intermolecular interactions that lead to the formation of oligomeric species. In these amyloid prone states, the protein can nucleate initial oligomeric assemblies where the content of secondary β structure is generally increased. These “seeds” or “nuclei” provide a sort of template where other misfolded or partially folded molecules are recruited increasing the size of the assemblies to final give rise to the final fibrils.Citation9 Extensive biophysical research has been conducted aimed at understanding the different steps of this process and also to characterize the different intermediates formed in the processCitation10,Citation11 and mature amyloid fibrils.Citation12 One of the key questions in the field has been, and still is, to understand why only certain proteins enter this misfolding and amyloid formation pathway. Approaches borrowed from protein folding and engineering have led to the identification of two major players in vitro: protein stability and amino acid sequence.
Here, we will focus on the study of the relationship between amino acid sequence and amyloid formation carried out in our group during the last years. Several biological relevant examples will be examined under the light of these studies. Finally, we will describe the ongoing applications of the amyloid stretch hypothesis.
Amyloid Protein Family: Diversity and Common Denominators
Up to now, there are more than 20 different proteins or peptides associated with human amyloidosis. In their nonfibrillar form natural occurring amyloidogenic proteins display a wide range of native folds. For example, transthyretin is a homotetramer with high β sheet structure, and only one helix per monomer.Citation13 In contrast, the native form of lysozyme is predominantly α-helix with a small amount of β structure.Citation14 Many amyloidogenic polypeptides consist primarily—of random-coil structures in their native, soluble states. These include the Aβ peptide,Citation15 IAPP,Citation16 α synucleinCitation17 and tau.Citation18 Additionally, in the last years the amyloid field has been widened by nonpathogenic proteins,Citation19–Citation22 and designed peptidesCitation23,Citation24 that can form fibrils in vitro with similar byophisical properties from those formed in vivo. Therefore, the group of peptides and proteins able to form amyloid fibrils in vivo and/or in vitro is very heterogeneous and they do not share any apparent fold or sequence homology.
X-ray fiber diffraction data indicate, however, that all amyloid fibrils from pathogenic and non pathogenic proteins share a common cross β structure.Citation25 Moreover, it has been demonstrated that some nonpathogenic proteins can exert a similar toxic effect in cell culture as natural occurring amyloids.Citation26–Citation28 These results provide strong support for the increasingly adopted view in the field that the ability to form amyloid fibrils is a general property of the polypeptide backboneCitation19,Citation29 modulated by the side chains,Citation24 and that there might be certain general principles governing protein fibrillization. According with this hypothesis, the propensity to form amyloid structures is related to some physico-chemical properties of the polypeptide chain such as charge, hydrophobicity, and secondary structure propensity, together with a consideration of the distribution of hydrophobic and polar residues.Citation30,Citation31
Amino Acid Sequence and Amyloid Formation
One of the widely accepted principles in the amyloid field is that proteins have to be partially or fully unfolded to form amyloid fibrils.Citation6–Citation8 However, most natively unfolded proteins in vivo do not undergo fibril formation,Citation32 indicating that unfolding is necessary, but not sufficient to promote protein polymerization. Hence, there must be some sequence motifs that are more prone to self-assemble into amyloid material than others.
Different independent investigations point in the direction that the amyloidogenic capability of a protein seems to be concentrated in particular protein regions and, more specifically, in small sequence fragments therein.Citation33–Citation38 It has been shown, for example, that a hexamer of human IAPP (residues 22–27, NFGAIL) and even a pentamer (residues 23–27, FGAIL) are already sufficient for amyloid formation and cytotoxicity.Citation35 Recent studies on protein self-assembly indicate that only 6 residues of the molecule need to be ordered to give rise to an ordered filament and point mutations at the hexapeptide region prevent protein aggregation.Citation38 In the case of the Alzheimer peptide (Aβ 1–42), the sequence KLVFFA (residues 16–21) has been identified as the shortest sequence able to form fibrils in vitro.Citation36 Furthermore, the comparison of two homologous proteins that differ in amyloidogenic properties has served to identify short divergent sequence fragments, that once swapped into the nonamyloidogenic protein can trigger amyloid formation.Citation33,Citation37
Extraction of an amyloid sequence pattern.
The research carried out in our group during the past years has mainly focused on establishing the sequence and structural bases of amyloid formation. Since amyloid deposits formed by natural proteins are difficult to study using standard biophysical techniques, our strategy has consisted in devising peptide and protein model systems that possess suitable physical properties such as reversibility, good solubility, etc. to allow biophysical characterization, and that are small enough to permit a detailed analysis of the aggregation process.Citation22,Citation24,Citation39 If amyloid formation is actually driven by short fragments of a misfolded protein, small model peptides should be more suitable than proteins to investigate those elements in sequences that favor amyloid formation. Whereas a mutation would alter just the self-assembly properties of a small peptide, it might lead to protein destabilization, complicating the extraction of pure sequence propensities to form amyloid fibrils.
Initially, we reported the computer-aided design of an hexapeptide-based model system for amyloidogenesis.Citation24 Its simplicity served to highlight that fibril formation is due to a very delicate balance between specific side-chain and electrostatic interactions within a sequence and to propose a structural model of the fibrils, which is consistent with the organization of the protofilament core of naturally occurring proteins. This result validates its use to study amyloid fibril formation and structure.
The small size of this model peptide system has been exploited to determine how exact sequence details modulate, or completely disrupt, the apparent generality of amyloid fibril formation in proteins.Citation39 This question has been addressed by systematically replacing the residues of a de novo designed amyloid peptide with all natural amino acids. Previously, only alanineCitation40 and prolineCitation38 scanning had been carried out in amyloid fragments found experimentally to determine the role of each residue.
From this saturation mutagenesis experiment, a sequence pattern that can be used to identify amyloidogenic stretches in proteins was extracted (). The positional scanning mutagenesis revealed as well that there is a position dependence upon mutation of amyloid fibril formation, and also that mutationally very tolerant (edges) and restrictive (core) positions can be found within an amyloid sequence.Citation39 This amyloidogenic pattern was validated experimentally and by the in silico sequence scanning of amyloid proteins and protein databases. Analysis of protein databases showed that highly amyloidogenic sequences matching the pattern are less frequent in proteins than innocuous amino acid combinations; and that if present they are surrounded by amino acids that disrupt their aggregating capability called amyloid breakers. Usually these positions are occupied by proline, which is a β sheet breaker, or charged residues.
As a final test, a set of amyloid peptides and proteins that form amyloid fibrils in vitro and/or in vivo and whose amyloid regions have been investigated experimentally were scanned using the amyloid pattern. The results suggest that the pattern is a successful tool that can be used to detect hexapeptide stretches that overlap with the amyloid regions found experimentally. For example, for the Aβ(1–42) peptide, the region that agrees with the pattern consists of residues 16–21 (16KLVFFA21). Interestingly, this region overlaps with the minimal sequence shown to be essential for Aβ polymerization, residues 16–20.Citation36
Another tool that can be used for the same purpose has been developed by combining a 3D profile generated from the crystal structure of an amyloidogenic peptideCitation41 and an energy prediction method.Citation42 Algorithms to predict β-aggregation propensity have been also developedCitation43,Citation44 although it is important to keep in mind that β-aggregation not always leads to amyloid formation (for a review see ref. Citation45).
The Amyloid Stretch Hypothesis
The previous evidences added by different groupsCitation33–Citation38 together with the amyloid patternCitation39 suggest that even a short amino acid stretch bearing a highly amyloidogenic motif could provide most of the driving force needed to trigger the self-assembly process of a protein. We will refer to this as the amyloid stretch hypothesis. Previously, it has been shown that insertion of long amyloid domains (30–80 amino acids) from naturally occurring amyloid proteins can trigger amyloid formation of some nonamyloidogenic proteins both in vitroCitation46,Citation47 and in vivo.Citation48 Although shorter amyloidogenic fragments (10-mer and 7-mer) from the N-terminus region of the yeast prion Sup35 have been already used with the same aim, these fragments have been inserted either into an already amyloidogenic proteinCitation49 or no compelling evidenceCitation50 has been provided in favor of the amyloid stretch hypothesis.
Our strategy to demonstrate the amyloid stretch hypothesis was the conversion of a nonamyloidogenic protein into an amyloid-prone molecule by inserting just a six-residue amyloidogenic stretch.Citation22 We chose the α-spectrin SH3 domain (α-SH3) as a target nonamyloidogenic protein. This protein has been shown not to form amyloid fibrils under any conditions tested in our laboratory.Citation51 As amyloidogenic sequences, we selected a de novo designed peptide, STVIIE, highly amyloidogenic in vitro and in a set of mutants.Citation24,Citation39 To validate the amyloid pattern within the context of a protein, we designed point mutants of this sequence at different position categories (core and edges). We also introduced an amyloidogenic 6-residue fragment of the Aβ amyloid peptide identified with the amyloid pattern, 16KLVFFA21. Sequences were inserted at different locations within the structure of the protein.
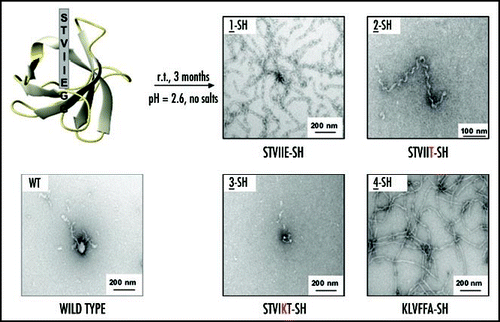
A destabilization or dramatic change in the structure of the domain due to the exogenous sequences would confound the demonstration of the amyloid stretch hypothesis. Hence, first we verified that the insertions had not affected the folding of the domain, and also investigated the stability of the different chimeric proteins. Next, we carried out fibrillation assays to test the amyloidogenic properties of the amyloid SH3 variants. Modified versions of the α-SH3 carrying these short amyloidogenic sequences in the N-terminus are as stable as the WT protein, but they fibrillate under conditions where the original domain still remains soluble (). Thus, the amyloidogenic behavior shown by these proteins is not due to an extra destabilization of the protein, but rather to the amyloidogenic properties of the inserted sequence. The amyloidogenicity shown by the proteins with the insertion at the N-terminus correlates with the prediction of the amyloid pattern. Mutation of the amyloidogenic insertion at the edges keeps the amyloidogenic feature of the chimeric protein while the introduction of a positive charge at the core of the amyloidogenic insertion completely abolishes amyloid fibril formation. Our results also show that the hexapeptide sequence derived from the Aβ peptide, 16KLVFFA21, is also able to trigger amyloid formation by a completely unrelated domain. Proteolysis experiments carried out with one of the amyloidogenic SH3 variants proved that the hexapeptide insertion is not only able to trigger amyloid formation, but also is forming part of the core of the fibril resistant to proteolysis.
Despite being slightly destabilized the C-terminus variants do not form any kind of amyloid material under the same conditions the N-terminus do. The N-terminus of α SH3 is disordered, while the C-terminus is structured into a β strand.Citation52 Therefore, the amyloid tail should be more exposed for mutants with the insertion at the N-terminus, and thus more accessible for intermolecular processes such as amyloid fibril formation. This result highlights that the structural environment of the amyloidogenic stretch plays a fundamental role on whether or not an amyloid-prone sequence can productively trigger the amyloid self-assembly process. Hence, in order to be amyloidogenic a protein must carry an appropriate amyloid stretch (sequence determinant) that must be or become locally unfolded to initiate the process of amyloid formation (structural determinant).
Some Experimental Results Revisited with the Amyloid Stretch Hypothesis
However, somebody might raise concerns against the generality of these conclusions due to the use of a model protein system that is not associated to any known human pathology. To provide further support we have analyzed some experimental results obtained by other groups in different amyloidogenic proteins under the light of the amyloid stretch hypothesis.
As an example, β2 microglobulin (β2m) forms fibrils at neutral pH only under destabilizing conditions that allow for the exposure of the β strand shown experimentally to be important for amyloid formationCitation53 and identified as amyloidogenic by the amyloid pattern.Citation39 Therefore, as in the case of the C-terminus proteins, β2m contains a sequence encoding for amyloid formation but trapped into a stable conformation.
Mutational analysis have shown that the aggregation rate of human muscle acylphosphatase is very sensitive to mutation at the region 87–97.Citation54 In fact, this region contains a six-residues fragment matching the amyloid pattern (90EYSNFS95).Citation39 Limited proteolysis experiments indicate that already under native conditions, the region immediately adjacent to the amyloid stretch (residues 74–90) is accessible to proteolytic digestion and therefore solvent exposed.Citation55
In the case of gelsolin only proteolysis fragments of this protein (residues 173–243 and 173–225) are found in amyloid deposits associated with familial amyloidoses of Finnish type (FAF).Citation56 Mutation of the aspartic residue at position 187 to asparagine or tyrosine results in variants unable to bind Ca+2. Studies in vitro has shown that this causes a reduction of the stability of the protein and an increase to its susceptibility to general proteolysis.Citation57 The initial cleavage by furin protease of the mutant gelsolin seems to occurs during secretion in the trans-Golgi network (TNG).Citation58 The further proteolytic processing and the conditions that promote fibril formation in vivo still remain unclear.
Scanning of the secreted isoform of human gelsolin with the amyloid pattern revealed six different amino acid stretches matching the pattern. This is a good example to show that carrying amyloid stretches is a necessary but not sufficient requirement to determine amyloid formation by a protein. Only one of the six amyloid stretches is located in the fragment forming amyloid fibrils in vivo, 186GDCFIL191. Interestingly, mutations associated to disease (D187N and D187Y) occur at position two of this sequence. In both cases the amyloid pattern predicts that the mutations should keep the amyloidogenicity of the amyloid stretch. Hence, they are causing the aberrant proteolytic processing of the mutant protein while keeping the amyloidogenicity of the sequence. We can hypothesize that the shorter size of the fragment, the mildly acidic pH of the TNG and the concentration of these fragments in secretory vesicles might be favouring the formation of amyloid species.
The zipper spine model recently proposed by the group of Eisenberg would be also in agreement with the results of the amyloid stretch hypothesis. In this model a specific region of a protein binds to the same region in another molecule, forming the β-sheet spine of the amyloid, while the rest of the molecule decorates the periphery of the spine.Citation33,Citation59 The design of the N-terminus variants clearly suggests an organization of the fibrils similar to this model. One would expect that amyloid formation results as a gain of interaction acquired by the insertion of the short amino acid stretch,Citation60 and that the insertion is forming part of the proteolysis resistant amyloid core as proved by the results of the proteolysis experiments.
Amyloid Formation Signals in Proteins
One of the dogmas in biology is that all the information required for a protein to fold is encoded in its primary sequence. In the past years, however, it has been demonstrated that consensus short sequences within this primary sequence also determine important events in the life of a protein, such as localization, post-traslational modifications or degradation.Citation61,Citation62 The verification of the amyloid stretch hypothesis adds a new function to this list: amyloid formation.Citation22
In the last years, numerous examples of functional roles of amyloid-like structures have been found. The egg-shell of the silkmoth is almost exclusively composed of chorion, an amyloid type structure that appears to protect the developing embryo.Citation63 Several studies demonstrate that fibril formation by some yeast prion proteins gives the host yeast a selective growth advantage over prion-free yeast under some conditions.Citation64 However, this beneficial effect might not be general as recent studies indicate that some yeast prion proteins are indeed deleterious in their host.Citation65 Melanosomes are specialized lysosome-related organelles that generate and store the pigment in melanocytes and retinal pigment epithelial cells. Pmel17 processing and amyloid formation is essential for melanosome biogenesis.Citation66 Hence, amyloid stretches would be not only coding for pathological process, but in some cases also for function. From an evolutionary perspective, it would be interesting to investigate if originally amyloid formation was a “mistake” that it was later on recycled to perform physiological roles, or if it happened the other way round.
Conclusions and Outlook
The amyloid pattern and the demonstration of the amyloid stretch hypothesis have significantly contributed to the understanding of the sequence and structural factors responsible for amyloid self-assembly. Currently, we are using them to tackle some other problems within the amyloid field. Compounds able to interfere with the process of amyloid formation are expected to prevent or reverse the pathology of amyloid diseases.Citation67,Citation68 As we have demonstrated that amyloid stretches can trigger amyloid self-assembly we propose them as new therapeutic targets. Molecules can be developed that specifically bind to amyloid stretches in proteins, hence preventing and/or reversing not only the process of amyloid formation but also amyloid induced toxicity in cell culture (Esteras-Chopo et al., submitted).
Different studies have proved that the polymerization course of protein amyloidogenic fragments reproduces many of the physicochemical properties of the analogous reaction undertaken by the full-length protein.Citation23,Citation24 It is thus plausible that the diverse species formed upon self-assembly of small peptides may also reproduce the cytotoxic mechanism of natural amyloids. We have carried out an extensive study with different amyloid stretches identified using the amyloid pattern in several human amyloid proteins. Our results confirm that they reproduce many aspect of amyloid-induced toxicity in cell culture and we have also identified new factors involved in this process (Pastor et al Submitted).
Fortunately, only a limited number of proteins have access to an amyloid prone state in vivo. According with the amyloid stretch hypothesis, a necessary condition for amyloid formation is the presence of an appropriate amyloid sequence. Therefore, sequence requirement restricts the amount of proteins able to form amyloid fibrils. But also the sequence must be accessible for the establishment of intermolecular interactions, which is the protein must become at least locally destabilized. This view integrates the two main proposals up to date to explain the phenomenon of amyloid formation. In addition to the amyloid patternCitation39 and the 3D profile methodCitation42 to detect amyloid sequences, several tools have been developed to detect β-aggregating sequences and to evaluate the propensity of a protein for β-aggregationCitation43,Citation44 which in some cases lead to amyloid formation.Citation45 In the near future, elegant and clean experiments should be conducted to validate these prediction tools in in vivo systems.
Abbreviations
| IAPP | = | islet amyloid polypeptide |
| α-SH3 | = | α spectrin SH3 domain |
| β2m | = | β2 microglobulin |
| FAF | = | familial amyloidoses of Finnish type |
| TNG | = | trans Golgi network |
Figures and Tables
Figure 1 The amyloid sequence pattern used to detect amyloid stretches in proteins. PROSITE syntax (www.expasy.org/prosite/). ‘[ ]’residues allowed at the position; ‘{ }’ residues forbidden at the position; ‘-’ separates each pattern element. Taken from Lòpez de la Paz and Serrano, 2004 © by The National Academy of Sciences of the USA.
![Figure 1 The amyloid sequence pattern used to detect amyloid stretches in proteins. PROSITE syntax (www.expasy.org/prosite/). ‘[ ]’residues allowed at the position; ‘{ }’ residues forbidden at the position; ‘-’ separates each pattern element. Taken from Lòpez de la Paz and Serrano, 2004 © by The National Academy of Sciences of the USA.](/cms/asset/df319365-5091-4a1f-8515-e6a7cf41ae29/kprn_a_10904100_f0001.gif)
Figure 2 Amyloid formation by the different N-terminus α-SH3 variants. All the proteins were incubated under similar conditions (pH 2.6, c ∼ 300 µM, t = 3 months and room temperature). 2-SH was assayed only at the lowest concentration (c ∼ 100 µM) because of solubility problems. r.t.: room temperature. Adapted from Esteras-Chopo et al, 2005 © by The National Academy of Sciences of the USA.
Acknowledgments
We acknowledge Dr. Manuela Lopez de la Paz for her past and current input into these investigations. We acknowledge the support of a Spanish Ministry of Education and Science fellowship (to M. Teresa Pastor) and by a European Union grant (APOPIS, Abnormal Proteins in the Pathogenesis of Neurodegenerative Disorders) (to Alexandra Esteras-Chopo and M. Teresa Pastor).
References
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science 2006; 314:777 - 781
- Pastor MT, Esteras-Chopo A, Lopez de la Paz M. Design of model systems for amyloid formation: Lessons for prediction and inhibition. Curr Opin Struct Biol 2005; 15:57 - 63
- Baldwin AJ, Bader R, Christodoulou J, MacPhee CE, Dobson CM, Barker PD. Cytochrome display on amyloid fibrils. J Am Chem Soc 2006; 128:2162 - 2163
- Glenner GG, Ein D, Eanes ED, Bladen HA, Terry W, Page DL. Creation of “amyloid” fibrils from Bence Jones proteins in vitro. Science 1971; 174:712 - 714
- Shirahama T, Cohen AS. Intralysosomal formation of amyloid fibrils. Am J Pathol 1975; 81:101 - 116
- Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci 1999; 24:329 - 332
- Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol 1998; 8:101 - 106
- Rochet JC, Lansbury PT Jr. Amyloid fibrillogenesis: Themes and variations. Curr Opin Struct Biol 2000; 10:60 - 68
- Harper JD, Lansbury PT Jr. Models of amyloid seeding in Alzheimer's disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 1997; 66:385 - 407
- Smith AM, Jahn TR, Ashcroft AE, Radford SE. Direct observation of oligomeric species formed in the early stages of amyloid fibril formation using electrospray ionisation mass spectrometry. J Mol Biol 2006; 364:9 - 19
- Mastrangelo IA, Ahmed M, Sato T, Liu W, Wang C, Hough P, Smith SO. High-resolution atomic force microscopy of soluble Abeta42 oligomers. J Mol Biol 2006; 358:106 - 119
- Makin OS, Serpell LC. Structures for amyloid fibrils. Febs J 2005; 272:5950 - 5961
- Blake CC, Geisow MJ, Oatley SJ, Rerat B, Rerat C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol 1978; 121:339 - 356
- Artymiuk PJ, Blake CC. Refinement of human lysozyme at 1.5 A resolution analysis of non-bonded and hydrogen-bond interactions. J Mol Biol 1981; 152:737 - 762
- Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis: Structure and biological activity of protofibrillar intermediates. J Biol Chem 1999; 274:25945 - 25952
- Kayed R, Bernhagen J, Greenfield N, Sweimeh K, Brunner H, Voelter W, Kapurniotu A. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J Mol Biol 1999; 287:781 - 796
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT Jr. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry 1996; 35:13709 - 13715
- Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem 1994; 269:24290 - 24297
- Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson CM. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci USA 1999; 96:3590 - 3594
- Fandrich M, Fletcher MA, Dobson CM. Amyloid fibrils from muscle myoglobin. Nature 2001; 410:165 - 166
- Guijarro JI, Sunde M, Jones JA, Campbell ID, Dobson CM. Amyloid fibril formation by an SH3 domain. Proc Natl Acad Sci USA 1998; 95:4224 - 4228
- Esteras-Chopo A, Serrano L, Lopez de la Paz M. The amyloid stretch hypothesis: Recruiting proteins toward the dark side. Proc Natl Acad Sci USA 2005; 102:16672 - 16677
- Fezoui Y, Hartley DM, Walsh DM, Selkoe DJ, Osterhout JJ, Teplow DB. A de novo designed helix-turn-helix peptide forms nontoxic amyloid fibrils. Nat Struct Biol 2000; 7:1095 - 1099
- López de la Paz M, Goldie K, Zurdo J, Lacroix E, Dobson CM, Hoenger A, Serrano L. De novo designed peptide-based amyloid fibrils. Proc Natl Acad Sci USA 2002; 99:16052 - 16057
- Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol 1997; 273:729 - 739
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002; 416:507 - 511
- Baglioni S, Casamenti F, Bucciantini M, Luheshi LM, Taddei N, Chiti F, Dobson CM, Stefani M. Prefibrillar amyloid aggregates could be generic toxins in higher organisms. J Neurosci 2006; 26:8160 - 8167
- Kranenburg O, Kroon-Batenburg LM, Reijerkerk A, Wu YP, Voest EE, Gebbink MF. Recombinant endostatin forms amyloid fibrils that bind and are cytotoxic to murine neuroblastoma cells in vitro. FEBS Lett 2003; 539:149 - 155
- Dobson CM. Protein folding and its links with human disease. Biochem Soc Symp 2001; 00:1 - 26
- Chiti F, Calamai M, Taddei N, Stefani M, Ramponi G, Dobson CM. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc Natl Acad Sci USA 2002; 99:Suppl 4 16419 - 16426
- Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003; 424:805 - 808
- Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions?. Proteins 2000; 41:415 - 427
- Ivanova MI, Sawaya MR, Gingery M, Attinger A, Eisenberg D. An amyloid-forming segment of beta2-microglobulin suggests a molecular model for the fibril. Proc Natl Acad Sci USA 2004; 101:10584 - 10589
- Jones S, Manning J, Kad NM, Radford SE. Amyloid-forming peptides from beta2-microglobulin-Insights into the mechanism of fibril formation in vitro. J Mol Biol 2003; 325:249 - 257
- Tenidis K, Waldner M, Bernhagen J, Fischle W, Bergmann M, Weber M, Merkle ML, Voelter W, Brunner H, Kapurniotu A. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J Mol Biol 2000; 295:1055 - 1071
- Tjernberg LO, Naslund J, Lindqvist F, Johansson J, Karlstrom AR, Thyberg J, Terenius L, Nordstedt C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem 1996; 271:8545 - 8548
- Ventura S, Zurdo J, Narayanan S, Parreno M, Mangues R, Reif B, Chiti F, Giannoni E, Dobson CM, Aviles FX, Serrano L. Short amino acid stretches can mediate amyloid formation in globular proteins: The Src homology 3 (SH3) case. Proc Natl Acad Sci USA 2004; 101:7258 - 7263
- von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci USA 2000; 97:5129 - 5134
- López de la Paz M, Serrano L. Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci USA 2004; 101:87 - 92
- Azriel R, Gazit E. Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide: An experimental support for the key role of the phenylalanine residue in amyloid formation. J Biol Chem 2001; 276:34156 - 34161
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005; 435:773 - 778
- Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D. The 3D profile method for identifying fibril-forming segments of proteins. Proc Natl Acad Sci USA 2006; 103:4074 - 4078
- Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J Mol Biol 2005; 350:379 - 392
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 2004; 22:1302 - 1306
- Rousseau F, Schymkowitz J, Serrano L. Protein aggregation and amyloidosis: Confusion of the kinds?. Curr Opin Struct Biol 2006; 16:118 - 126
- Tanaka M, Machida Y, Nishikawa Y, Akagi T, Morishima I, Hashikawa T, Fujisawa T, Nukina N. The effects of aggregation-inducing motifs on amyloid formation of model proteins related to neurodegenerative diseases. Biochemistry 2002; 41:10277 - 10286
- Baxa U, Taylor KL, Wall JS, Simon MN, Cheng N, Wickner RB, Steven AC. Architecture of Ure2p prion filaments: The N-terminal domains form a central core fiber. J Biol Chem 2003; 278:43717 - 43727
- Wigley WC, Stidham RD, Smith NM, Hunt JF, Thomas PJ. Protein solubility and folding monitored in vivo by structural complementation of a genetic marker protein. Nat Biotechnol 2001; 19:131 - 136
- He Y, Tang H, Yi Z, Zhou H, Luo Y. Fibrillogenesis of apomyoglobin facilitated by aggregation sequence of yeast Sup35 in various regions. FEBS Lett 2005; 579:1503 - 1508
- Chae YK, Cho KS, Chun W. A prionogenic peptide derived from Sup35 can force the whole GST fusion protein to show amyloid characteristics. Protein Pept Lett 2002; 9:315 - 321
- Ventura S, Lacroix E, Serrano L. Insights into the origin of the tendency of the PI3-SH3 domain to form amyloid fibrils. J Mol Biol 2002; 322:1147 - 1158
- Musacchio A, Noble M, Pauptit R, Wierenga R, Saraste M. Crystal structure of a Src-homology 3 (SH3) domain. Natur 1992; 359:851 - 855
- McParland VJ, Kalverda AP, Homans SW, Radford SE. Structural properties of an amyloid precursor of beta(2)-microglobulin. Nat Struct Biol 2002; 9:326 - 331
- Chiti F, Taddei N, Baroni F, Capanni C, Stefani M, Ramponi G, Dobson CM. Kinetic partitioning of protein folding and aggregation. Nat Struct Biol 2002; 9:137 - 143
- Monti M, Garolla di Bard BL, Calloni G, Chiti F, Amoresano A, Ramponi G, Pucci P. The regions of the sequence most exposed to the solvent within the amyloidogenic state of a protein initiate the aggregation process. J Mol Biol 2004; 336:253 - 262
- Kiuru S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid 1998; 5:55 - 66
- Huff ME, Page LJ, Balch WE, Kelly JW. Gelsolin domain 2 Ca2+ affinity determines susceptibility to furin proteolysis and familial amyloidosis of finnish type. J Mol Biol 2003; 334:119 - 127
- Chen CD, Huff ME, Matteson J, Page L, Phillips R, Kelly JW, Balch WE. Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca(2+) stabilization. Embo J 2001; 20:6277 - 6287
- Sambashivan S, Liu Y, Sawaya MR, Gingery M, Eisenberg D. Amyloid-like fibrils of ribonuclease A with three-dimensional domain-swapped and native-like structure. Nature 2005; 437:266 - 269
- Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol 2003; 10:461 - 467
- Aitken A. Protein consensus sequence motifs. Mol Biotechnol 1999; 12:241 - 253
- House CM, Frew IJ, Huang HL, Wiche G, Traficante N, Nice E, Catimel B, Bowtell DD. A binding motif for Siah ubiquitin ligase. Proc Natl Acad Sci USA 2003; 100:3101 - 3106
- Hamodrakas SJ, Hoenger A, Iconomidou VA. Amyloid fibrillogenesis of silkmoth chorion protein peptide-analogues via a liquid-crystalline intermediate phase. J Struct Biol 2004; 145:226 - 235
- Osherovich LZ, Weissman JS. The utility of prions. Dev Cell 2002; 2:143 - 151
- Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci USA 2005; 102:10575 - 10580
- Huff ME, Balch WE, Kelly JW. Pathological and functional amyloid formation orchestrated by the secretory pathway. Curr Opin Struct Biol 2003; 13:674 - 682
- Citron M. Strategies for disease modification in Alzheimer's disease. Nat Rev Neurosci 2004; 5:677 - 685
- Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 2003; 4:49 - 60