Abstract
The common feature of many neurodegenerative diseases is emergence of protein aggregates. Identifying their composition can provide valuable insights into the cellular mechanisms of protein aggregation and neuronal death. No reliable method for identification of the aggregate-associated proteins has been available. Here we describe a method for characterization of protein aggregates based on sedimentation of immunocomplexes without involvement of a solid support. As a model, we used the aggregates formed in yeast by a polyglutamine-containing segment of mutant huntingtin. Sixteen proteins associated with the isolated aggregates were identified with 2D gel electrophoresis followed by mass spectrometry. We found that the aggregates in cells lacking Rnq1 prion recruited lesser amounts of chaperones than those in the wild type cells. The method can be utilized for characterization of various types of aggregates, prions and very large protein complexes under mild conditions that preserve associated proteins.
Introduction
In many neurodegenerative diseases, such as amyotrophic lateral sclerosis, Alzheimer's disease, Parkinson's disease, and Huntington's disease, the pathology and the eventual death of specific neuronal populations occur as a result of accumulation of specific abnormal polypeptides. These polypeptides usually aggregate and form insoluble intracellular inclusions. The formation of the inclusion bodies generally precedes neurodegeneration and cell death. Thus, the mechanisms of intracellular protein aggregation as well as the composition of the aggregates associated with various diseases attract much attention because of their relevance to a number of known pathological conditions.
In this paper we addressed these questions with the model of Huntington's disease (HD). Certain neurodegenerative disorders, including HD, are caused by an extension of polyglutamine domains in mutant proteins.Citation1 A unifying feature of these disorders is that proteins with extended polyglutamine domains (polyQ) tend to aggregate in the nuclei and/or cytoplasm of affected neurons.Citation2,Citation3 A broad body of evidence suggests that the extension of the polyglutamine tract can cause neurotoxicity through abnormal protein-protein interactions. Accordingly, the “sequestration” hypothesisCitation4–Citation6 was introduced, proposing that polyQ aggregates recruit critical cellular factors, thereby compromising their function and leading to toxicity. Therefore, development and application of biochemical methods for isolation of the aggregates and identification of the associated proteins can be essential for understanding the cellular processes underlying the polyQ toxicity. Moreover, defining proteins involved in polyQ aggregates will help to uncover basic cellular mechanisms controlling the aggregate formation.Citation7,Citation8 Obviously, the approaches for isolation of the disease-associated inclusions and identification of their compositions will be beneficial for therapies related not only to HD, but also to many other protein conformation disorders.Citation9
Multiple attempts have been undertaken to identify proteins associated with polyQ; most have employed two-hybrid screens for polypeptides that interact with expanded polyglutamine domains.Citation10,Citation11 These reports indicated that polyQ binds to a plethora of cellular proteins. The two-hybrid approach, however, does not allow a comprehensive search for proteins sequestered in the polyQ aggregates, since some of these proteins may either interact only with the soluble forms of polyQ, or be recruited via indirect interactions, or be involved only in the process of polyQ aggregation.
Broad analysis of the components associated with aggregates is hampered by the difficulties in isolation of aggregates. In fact, extreme heterogeneity of the aggregates and their enormous sizes preclude application of routine biochemical methods to the purification of aggregates. Isolation of aggregates was performed previously by utilizing the ionic detergent insolubility of amyloidsCitation12 or density gradient fractionation.Citation13 While these methods may be useful to address certain questions about the structure of protein aggregates, they are inadequate for identification of the aggregate-associated proteins. In fact, SDS treatment causes dissociation of most of associated proteins, while density gradient isolation yields a high fraction of non-specifically-associated polypeptides. Another method using a fluorescence-activated cell sorter to separate GFP-tagged aggregates overcame some of these problems. However, the cell sorter approach has several obvious limitations: it requires large and highly homogeneous aggregates, gives low yields of purified aggregates, and allows identification of only abundant proteins, such as Hsps (Hsp70, HDJ-1, HDI-2 and Hsp84) and EF-1a.Citation14 Furthermore, this method is not applicable to the isolation of polyQ aggregates from clinical samples.
In the present study, we have developed a novel method for isolation of polyQ aggregates. As a model, we isolated aggregates from yeast cells expressing a construct that was FLAG-tagged at the N-terminus and EGFP-tagged at the C-terminus, encoding the first 17 amino acids of huntingtin exon1 followed by a tract of 103 glutamines (103Q).Citation15 Under these conditions, polyQ aggregation directly depends on the length of the polyglutamine tract. 103Q forms heterogeneous multiple aggregates in cytoplasm causing severe toxicity.Citation16
Our newly developed method is based on affinity purification without involvement of a solid phase. It allowed isolation of these aggregates under mild conditions that preserve associated proteins. Using this method, we initiated a proteomics studyCitation17,Citation18 to identify associated proteins, and the results we present here demonstrate that diverse molecular chaperones and components of glycolysis were over-represented among these proteins. We further confirmed the specificity of this method by showing that dramatically reduced amounts of these proteins are associated with unusual aggregates isolated from the strains that lack the Rnq1 prion.
Materials and Methods
Strains.
Wild-type strain W303 (MATa ade2-1 trp1-1 leu2-3,112 his3-11,15 ura3-52 can1-100 ssd1-d) and strains with GFP-tagged endogenous proteins in a parental strain (MATa his3Δ leu2Δ met15Δ ura3Δ) were obtained from the Yeast GFP Clone Collection (Invitrogen). Deletion mutants rnq1, hsp104 (BY4742 (MATα his3Δ leu2Δ lys2Δ ura3Δ)) were obtained from the deletion library (Invitrogen) of yeast nonessential genes.
Yeast constructs.
The pYES2-based vector for expression of polyQ constructs under control of the Gal1 promoter was described previously.Citation15 Briefly, the N-terminal huntingtin sequence containing the first 17 amino acids and 103 glutamines was FLAG-tagged at the N-terminus and tagged with EGFP at the C-terminus. In addition, for colocalization experiments, we prepared constructs where GFP was replaced with mRFP.
Yeast growth and induction.
Cells were routinely grown at 30°C on selective minimal medium with 2% raffinose and, for induction of 103Q, were transferred into the selective media with 2% galactose for six hours.
Antibodies.
Mouse anti-FLAG monoclonal antibody was purchased from Sigma (St. Louis, MO). AffiniPure Rabbit anti-mouse and goat anti-rabbit IgGs (H+L) were from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Rabbit Anti-GFP polyclonal antibody was from Invitrogen (Carlsbad, CA). Rabbit true blot HRP-conjugated anti-rabbit IgG was purchased from Biosciences (San Diego, CA).
Isolation of aggregate.
Yeast cells grown at the logarithmic phase were collected by centrifugation and then disrupted with glass beads in lysis buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 1% TritonX-100 with protease inhibitors). After centrifugation at 1000 x g for 2.5 min to remove the debris, the lysates were adjusted to have equal protein concentrations before they were loaded onto the 2.5 cm (dia) x 10 cm gel filtration column (50 mL Sephacryl S-400 HR). Fractions (1.7 mL) were collected at 0.85 mL/min flow rate and analyzed by microscope for the presence of the GFP-labeled aggregates. Four fractions with the highest content of aggregates were combined. The corresponding fractions from the cell lysates with vector only were used as a control. The primary antibody, mouse anti-FLAG IgG, was added to the pooled fractions to achieve a final concentration of 70mg/mL and the solution was incubated for 1.5 h. The secondary antibody, rabbit anti-mouse IgG, was added to a final concentration of 140 µg/mL and the solution was incubated for 1 h. The tertiary antibody, goat anti-rabbit IgG, was added at 300 µg/mL and the solution was incubated for 1.5 h. Finally, the samples were loaded onto the 50% sucrose cushion and spun down at 600 x g for 2.5 min. The supernatant was aspirated and the purified aggregate pellet was stored at -20°C.
Two-dimensional polyacrylamide (2D) gel electrophoresis.
Purified aggregate complexes (300–500 µg) were resuspended in 200 µL rehydration buffer (Bio-Rad) and separated with 2D gel electrophoresis. IEF was performed by using immobilized pH gradient strips with a pH range from 3 to 10 on a Bio-Rad IEF cell with a programmed voltage gradient. SDS/PAGE was performed on Bio-Rad precast 12.5% polyacrylamide gel. Gels were stained overnight with Coomassie blue.
Mass spectrometry and protein identification.
Two- dimensional gel spots of interest were excised, destained, DTT-reduced, iodoacetamide-modified and digested with trypsin. After extracting peptides from gel pieces using 1% TFA and 50% acetonitrile, the samples were Zip-tipTM cleaned, resuspended with 0.1% TFA and 50% acetonitrile and spotted onto target with 2,5-dihydroxybenzoic acid (DHB) as matrix. The resulting tryptic peptides of each spot were analyzed with a Bruker Reflex IV matrix-assisted laser-desorption/ionization time-of-flight (MALDI-TOF) mass spectrometer equipped with a Laser Science nitrogen laser (Franklin, MA) having a 3-ns pulse width at 337 nm. Spectra were acquired by summing the signal recorded after 150–200 laser shots. Singly charged monoisotopic peptide masses were generated and used as inputs for database searching using MoverZ software (ProteoMetrics, LCC, New York, NY), after external and internal calibration of spectra.Citation19 Database searching was performed against the NCBlnr database by using the online PROFOUND search engines at http://prowl.rockefeller.edu/prowl-cgi/profound.exe.Citation20 Search parameters were as follows: S. cerevisiae for the taxonomic category; protein mass range of 0–200 kDa; iodoacetamide modified cysteines; maximum of two missed cleavage sites; mass tolerance of 0.1 Da.
Tandem MS/MS analyses were performed on an electrospray ionization (ESI) QSTAR Pulsar i quadrupole-orthogonal TOF mass spectrometer (MS and MS/MS) (Applied Biosystems, Foster City, CA). Peptides cleaned by Zip-TipTM were eluted with acetonitrile: water: formic acid (50/50/0.5, v/v/v) and analyzed. Collision-induced decomposition MS/MS spectra were acquired at 30–60 V collision cell voltage and the resulting spectra were examined manually. The data were analyzed using QAnalyst (Applied Biosystems) software.
For protein identification, a minimum of four matched peptides and the probability greater than 0.99 are search criteria. Theoretical masses of tryptic digestion of the predicted proteins were also manually checked with the mass spectra to be certain of the identification.
Western blot analysis.
Samples were analyzed by 12% SDS/PAGE gel followed by immunoblotting with antibodies indicated in the text.
Fluorescence microscopy.
Fluorescence microscopy was performed with an Axiovert 200 (Carl Zeiss) microscope with a 100x objective. For colocalization study the cells were fixed for 10 min in 4% formaldehyde, washed with PBS and analyzed on the glass slides.
Results
Isolation of 103Q aggregate.
To gain insights into the mechanisms of polyQ aggregation and toxicity we used previously described yeast model of HD.Citation16 In wild-type yeast strains transformed with 103Q, all cells contained multiple aggregates of various sizes () after 6 h of galactose induction. These cells were physically disrupted in the buffer containing 1% Triton X-1a, and the lysates were clarified from unbroken cells and debris by a low-speed centrifugation. To separate 103Q aggregates from any soluble mono- and oligomeric forms, we subjected cell lysates to gel filtration through Sephacryl S-400 HR and collected fractions highly enriched with aggregates detectable under a microscope. For the most part, these fractions corresponded to the void volume of the column, with a cut-out range of about 8 MDa (not shown), thus being effectively separated from the soluble 103Q (see for the efficiency of the separation). Accordingly this step gave about 7-fold purification of 103Q. We recovered about 95% of this polypeptide, since, as we demonstrated previously, in the wild-type yeast cells only a small fraction of 103Q remains soluble when assessed by differential centrifugation.Citation16 Furthermore, it allowed us to get rid of soluble 103Q molecules, which was especially important for isolation aggregates from the strains with suppressed aggregation (see below).
To isolate polyQ aggregates from the enriched fraction, we first employed conventional biochemical approaches including ion exchange chromatography and gel filtration, but did not succeed because of enormous heterogeneity of the aggregates (, top panel). Affinity purification did not succeed due to the large size of the aggregates, since the shear force applied during washing of the beads was so strong that the majority of the aggregates were lost during the washing procedure (not shown). Therefore, we employed a novel approach eliminating the need for a solid support in affinity purification (). We built a 3-D antibody mesh that incorporated polyQ aggregates by sequential addition to the lysates of primary anti-FLAG antibody (since our model polyQ were FLAG-tagged, we have chosen to employ anti-FLAG antibody which can be substituted with any other antibody recognizing polyQ epitopes), then of 2-fold excess of the secondary antibody, and finally of 4-fold excess of the tertiary antibody. As a result of the consecutive incubations, the aggregates became impregnated into the formed IgG mesh (, lower panel). These macroscopic structures were collected by a slow centrifugation, which recovered about half of 103Q present in the sample before centrifugation (not shown). For centrifugation, the samples were loaded on a 50% sucrose cushion to effectively separate the immunocomplexes from the soluble unbound proteins. Indeed, as a result of this centrifugation, the immunocomplexes were almost completely separated from BSA, which was used as a stabilizer for the antibodies (). Since the centrifugation through the sucrose cushion was run at a speed lower than the one used to clarify the yeast lysates before the gel filtration, even the largest particles were not precipitated under these conditions unless they were associated with the IgG mesh. Similarly, the IgG mesh-containing fraction was isolated from the lysate containing the vector plasmid, and was used as a control.
Identification of aggregate-associated proteins.
The immunoisolates were reconstituted in a buffer containing 8M urea and analyzed with 2D gel electrophoresis. After Coomassie blue staining many protein spots were detected in precipitates from the 103Q lysates (, upper panel). Precipitates from control lysates without 103Q yielded about three times less IgG (not shown), probably because formation of an extensive IgG mesh in the solution depends on the presence of polyQ aggregates. Accordingly, to normalize samples on the basis of the amount of IgG present, we loaded on 2D gels the entire precipitate from control cells and only 1/3 of the precipitate from 103Q-expressing cells. Increased loading volume resulted in poorer quality of 2D gels of the control samples (, lower panel). Higher loading was expected to increase the amounts of nonspecifically associated proteins, nevertheless, with the control samples, only heavy and light chains of IgG and traces of BSA were detected in 2D gels (, lower panel). This finding indicates that polypeptides found in 103Q precipitates specifically associate with aggregates.
To identify aggregate-interacting proteins, protein spots specifically detected on 2D gels of isolated 103Q aggregates were excised and subjected to in-gel digestion with trypsin. MALDI-TOF MS was applied and the resulting mass spectra were analyzed. Protein assignments were made after searching Profound. shows the representative mass spectrum of Sgt2 with sequence coverage more than 50%. ESI MS/MS was subsequently performed to achieve further sequence information and confirmed the protein assignments made by MALDI-TOF MS (). We identified various chaperones, including three members of the Hsp70 family, Hsp40, small heat shock protein, a cochaperone TPR-containing protein Sgt2; five glycolytic enzymes; a signaling 14-3-3 protein; two proteins with prion domains; and two unknown proteins. Hsp70, Hsp40, 14-3-3 and GAPDH have already been reported to associate with PolyQ aggregates,Citation21,Citation22 while the ten other proteins are newly identified aggregate-associated polypeptides ().
To verify that the identified proteins indeed associate with 103Q aggregates in vivo, we assessed colocalization of some of these proteins with aggregates in fixed cells. For this study we used the Yeast GFP Collection, which contains clones with GFP-tagged endogenous proteins.Citation23 We have chosen several GFP-tagged clones representing all groups of the identified aggregate-associated proteins, including molecular chaperones (Ssa1, Hsp42 and Sgt2), glycolytic enzymes (Fba1 and GAPDH), signaling protein (Bmh1) and a prion domain-containing polypeptide Pin3. We expressed in these clones 103Q tagged with mRFP in place of originally used EGFP (103Q-mRFP) to view the 103Q colocalization with tested proteins. In fixed cells no leakage of either EGFP fluorescence to Texas Red channel or 103Q-mRFP fluorescence to FITC channel occurs (not shown). As shown in , Ssa1 and Pin3 proteins almost completely colocalized with 103Q aggregate. Sgt2, GAPDH, Fba1, and Bmh1 proteins (the last two not shown) were only partially recruited to 103Q aggregates, which may be explained by the high cellular levels of these proteins. Hsp42-GFP formed a single aggregate per cell in noninduced cells and upon 103Q induction partially redistributed into, the polyQ aggregates. These data clearly confirm the interactions detected through Mass spectrometry analysis.
Different types of 103Q aggregates display different chaperone content.
Previous studies indicated that yeast prions, especially Rnq1, are essential for polyQ aggregation.Citation16 In fact, conversion of yeast prions to their soluble forms, caused by deletion of HSP104 geneCitation24,Citation25 led to a dramatic suppression of the 103Q aggregation and relief of the 103Q toxicity.Citation16 Similar effects were seen with deletion of the RNQ1 gene alone.Citation26 While in wild-type cells with the prion form of Rnq1 cells had multiple small aggregates, in hsp104 or rnq1 deletion strains most cells show only diffused fluorescent staining indicative of soluble 103Q ( and ref. Citation16). In a small fraction of the mutant cells 103Q still aggregated, forming, however, just one large aggregate.Citation16 Based on these findings, it was suggested that Rnq1 prion plays an important role in nucleation of polyQ aggregates.
To address a question whether yeast prions affect association of proteins with 103Q aggregates, we employed the newly-developed method to isolate rare 103Q aggregates from hsp104 and rnq1 deletion strains, and analyze their composition. Since the majority of the mutant cells contained soluble 103Q, the gel filtration step was especially important to separate soluble and aggregated 103Q forms (). After purification proteins associated with aggregates from wild-type and mutant strains were analyzed by 2D gel electrophoresis, and showed similar patterns of the aggregate-associated proteins (data not show). However, more quantitative analysis of the isolated fractions by SDS-PAGE followed by immunoblotting revealed that, compared to the wild-type, aggregates isolated from rnq1 and hsp104 mutants recruited considerably lower amounts of chaperones. In fact, shows that representation of all tested chaperones was significantly decreased in aggregates isolated from the deletion mutants. In particular, Ssa1 (Hsp70) and Ydj1 (Hsp40) are almost undetectable in aggregates from rnq1. In aggregates from hsp104 cells Ssa1 was also present at the very low levels while amounts of associated Ydj1 was decreased twice compared to wild-type. The amounts of Hsp90 and Sis1 associated with polyQ in the aggregates isolated from the mutant cells were also decreased but to a lesser extent. Therefore, Rnq1 protein and apparently its prion status dramatically affect the ability of polyQ aggregates to interact with cellular chaperones, which could play an important role in polyQ toxicity. Importantly, this experiment demonstrates that the newly-developed method of aggregate isolation allows a semi-quantitative analysis of proteins associated with different types of polyQ aggregates.
Discussion
In this report we describe a novel method for isolation of protein aggregates using gel filtration to separate inclusion bodies from soluble polyQ followed by immunoprecipitation, which does not involve attaching of the immunocomplexes to a solid phase. The second step yielded about 50% of aggregated 103Q since we limited the time of the final centrifugation to be certain that no cellular structures besides those recognized by the antibody would be precipitated. This highly reproducible procedure yielded a f raction of polyQ aggregates of diverse size and charge. The isolated aggregates were separated by 2D gel and analyzed by sensitive mass spectrometry methods which uncovered sixteen associated proteins, ten of which are reported for the first time to associate with polyQ, with all assignments having 100% possibility (). In addition to identifying aggregate-associated proteins the described method allows for semi-quantitative comparison of the identified proteins. Large amounts of IgG in the precipitates, did not allow quantifying the degree of purity of polyQ aggregates, and also obscured some proteins in the areas of spots formed by IgG heavy and light chains. This problem is not intrinsic to our isolation procedure and could be solved by using alternative methods of separation of individual proteins in the immunoprecipitates, such as multidimensional separation followed by mass spectrometry analysis.Citation27
As described in the Introduction, isolation of aggregates was previously performed by utilizing the methods not always adequate for identification of the aggregate-associated proteins. The notable advantage of our method is that it avoids exposure of the aggregates to pH and ionic strength extremes as well as to ionic detergents, thus preserving putative weak protein interactions. Furthermore, it provides high yields of heterogeneous aggregates, which are common in inclusion bodies of neurodegenerative diseases and can be used with clinical samples.
Some of the proteins found in polyQ aggregates in this work are homologous to polyQ-interacting proteins previously identified in mammalian systems. These proteins include Hsp70 family members (Ssa1, Ssa2) and Hsp40 (Sis1), that are known to modulate the PolyQ aggregation and toxicity,Citation25 glyceraldehyde-3-phosphate dehydrogenase (GAPDH),Citation21,Citation22 and 14-3-3, protein BMH1.Citation28,Citation29,Citation30 In addition to these proteins, other major chaperones, including a homolog of small heat shock proteins Hsp42 was present in the aggregates, as well as a tetratricopeptide repeat-containing adaptor protein Sgt2, which is involved in formation of multichaperone complexes. Very intriguingly, beside GAPDH, a number of other major glycolytic enzymes including enolase (Eno2), alcohol dehydrogenase (Adh1), fructose bis-phosphatase (Fba1) and phosphoglycerate kinase (Pgk1) were also present in the aggregates. Sequestering of the whole group of glycolytic enzymes in polyQ aggregates may affect glycolysis thus contributing to toxicity. These data are in agreement with the previously reported interactions between glycolytic enzymes including Eno2 and QN-rich domain of Sup35 protein.Citation31,Citation32 In addition, we found Ypr154w and Ynl208w proteins that have prion-forming domains highly enriched in glutamines and asparagines. Their interaction with polyQ aggregates may be important for seeding of aggregates, as was shown for prions Rnq1 and Sup35.Citation31,Citation32
In our experiments, we did not see on 2D gels a prominent spot corresponding to103Q, probably because 103Q amyloid agglomerates could not be solubilized under the SDS-free conditions of the first dimension. We, however, could clearly detect this polypeptide if proteins in isolated samples were analyzed by regular SDS-PAGE. This finding underlines a limitation of separation by 2D gels of proteins that form amyloids.
The advantage of the novel method for isolation and characterization of aggregate-interacting proteins is that it can be applied to almost any type of protein aggregates or other types of heterogeneous very high molecular weight complexes. It could be used not only for identification of associated proteins but also for studies of associated enzymatic activities (e.g., proteasome activity). Eventually it can help to understand the nature of many neurodegenerative disorders.
Figures and Tables
Figure 1 Isolation of 103Q aggregates from yeast cells. (A) Fluorescent micrograph (FITC channel) of yeast wild type cells with GFP-labeled 103Q aggregates after 6 h induction. (B) Scheme of the purification procedure. (C) Fluorescent micrographs of the indicated fractions at x100 magnification; a small portion of the immunoprecipitate (IP) was resuspended in the lysis buffer to obtain the image. (D) SDS-PAGE (Coomassie stained) gel shows approximately equal IgG distribution between IP and supernatant and almost complete purification of the IP from BSA which remains the major band in the non-precipitated fraction, but is almost excluded from the precipitate. Precipitates were reconstituted in the SDS-containing buffer in a volume equal to the volume of the supernatant.
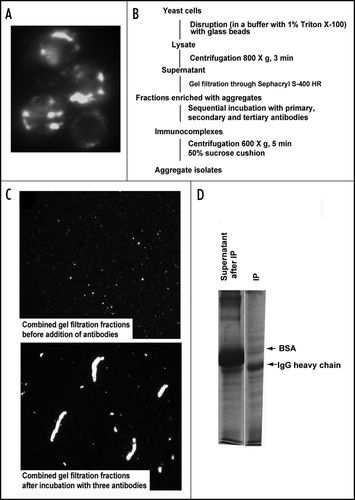
Figure 2 Isolated aggregates and control fraction analyzed by 2-D electrophoresis gels stained with Coomassie blue.
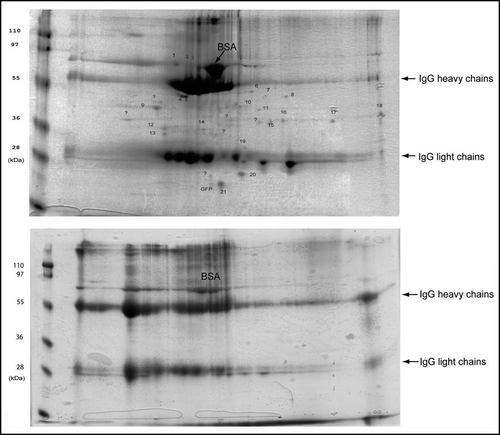
Figure 3 Mass spectra from one of the identified proteins. (A) A representative MALDI-TOF mass spectrum shows matched peptides from the identified protein, Sgt2. The sequence of the Sgt2 protein is shown on the right top corner, identified peptides are underlined. Matched peptides and the monoisotopic m/z values of fragments are indicated above each peak. (B) A representative ESI MS/MS spectrum confirms the identification of proteins by tandem MS/MS of [M+2H]2+ m/z 697.9 matching peptide m/z 1394.Citation7 of the panel a. Matched peptide sequence of Sgt2 protein is shown on the top and resulting b and y ions are indicated.
![Figure 3 Mass spectra from one of the identified proteins. (A) A representative MALDI-TOF mass spectrum shows matched peptides from the identified protein, Sgt2. The sequence of the Sgt2 protein is shown on the right top corner, identified peptides are underlined. Matched peptides and the monoisotopic m/z values of fragments are indicated above each peak. (B) A representative ESI MS/MS spectrum confirms the identification of proteins by tandem MS/MS of [M+2H]2+ m/z 697.9 matching peptide m/z 1394.Citation7 of the panel a. Matched peptide sequence of Sgt2 protein is shown on the top and resulting b and y ions are indicated.](/cms/asset/65b346df-62cf-4aa6-bf80-976fd520ee56/kprn_a_10904440_f0003.gif)
Figure 4 Colocalization of the proteins coprecipitated with 103Q aggregates. Fixed yeast cells with GFP-tagged endogenous proteins (marked on the right) and transformed with mRFP-tagged 103Q were grown in selective medium with raffinose and induced for 6 h with galactose.
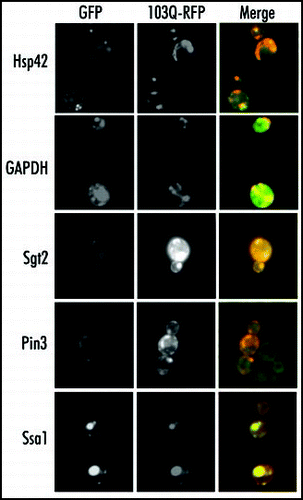
Figure 5 Isolation and analysis of 103Q aggregates from cells without prions. (A) Morphology of 103Q aggregates formed in hsp104 cells differs significantly from those in wild type cells (). (B) Fractions after gel filtration of lysates of hsp104 cells expressing 103Q analyzed by immunoblot with anti-GFP antibody, and quantified by Quantity One software (Bio-Rad). (C) Comparison of chaperones in immunoprecipitates from the wild type and mutant cells. Immunoblot with the respective antibodies (indicated on the right). To detect 103Q, true blot rabbit anti-mouse IgG was used as a secondary antibody. The aggregates were isolated from equal volumes of yeast cultures and solubilized in equal volumes of the Laemmli loading buffer.
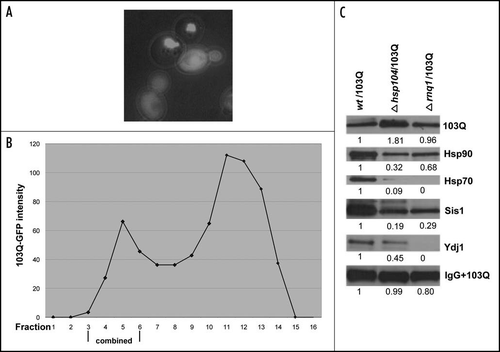
Table 1 List of identified proteins associated with 103Q aggregates
Acknowledgements
We are grateful to Dr. Stephen Farmer for technical support. This research was supported by NIH grants R01 NS047705 (to Michael Y. Sherman) and P41 RR10888 and S10 15942 (to Catherine E. Costello).
References
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci 2000; 23:217 - 247
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997; 90:537 - 548
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997; 277:1990 - 1993
- Perez-Navarro E, Canals JM, Gines S, Alberch J. Cellular and molecular mechanisms involved in the selective vulnerability of striatal projection neurons in Huntington's disease. Histol Histopathol 2006; 21:1217 - 1232
- Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol 2001; 311:173 - 182
- Preisinger E, Jordan BM, Kazantsev A, Housman D. Evidence for a recruitment and sequestration mechanism in Huntington's disease. Philos Trans R Soc Lond B Biol Sci 1999; 354:1029 - 1034
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002; 416:507 - 511
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J Mol Med 2003; 81:678 - 699
- Buxbaum JN. Diseases of protein conformation: What do in vitro experiments tell us about in vivo diseases?. Trends Biochem Sci 2003; 28:585 - 592
- Gutekunst CA, Li SH, Yi H, Ferrante RJ, Li XJ, Hersch SM. The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): Comparison with huntingtin in rat and human. J Neurosci 1998; 18:7674 - 7686
- Huntington JL, Stratton DM, Scattergood TW. Exobiology research on space station freedom. Adv Space Res 1995; 15:135 - 138
- Doi H, Mitsui K, Kurosawa M, Machida Y, Kuroiwa Y, Nukina N. Identification of ubiquitin-interacting proteins in purified polyglutamine aggregates. FEBS Lett 2004; 571:171 - 176
- Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol 2001; 153:283 - 294
- Mitsui K, Nakayama H, Akagi T, Nekooki M, Ohtawa K, Takio K, Hashikawa T, Nukina N. Purification of polyglutamine aggregates and identification of elongation factor-1alpha and heat shock protein 84 as aggregate-interacting proteins. J Neurosci 2002; 22:9267 - 9277
- Meriin AB, Mabuchi K, Gabai VL, Yaglom JA, Kazantsev A, Sherman MY. Intracellular aggregation of polypeptides with expanded polyglutamine domain is stimulated by stress-activated kinase MEKK1. J Cell Biol 2001; 153:851 - 864
- Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol 2002; 157:997 - 1004
- Yates IIIrd JR. Mass spectrometry: From genomics to proteomics. Trends Genet 2000; 16:5 - 8
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature 2003; 422:198 - 207
- Zubarev R, Mann M. On the proper use of mass accuracy in proteomics. Mol Cell Proteomics 2007; 6:377 - 381
- Zhang W, Chait BT. ProFound: An expert system for protein identification using mass spectrometric peptide mapping information. Anal Chem 2000; 72:2482 - 2489
- Burke JR, Enghild JJ, Martin ME, Jou YS, Myers RM, Roses AD, Vance JM. Strittmatter WJ, Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med 1996; 2:347 - 350
- Koshy B, Matilla T, Burright EN, Merry DE, Fischbeck KH, Orr HT, Zoghbi HY. Spinocerebellar ataxia type-1 and spinobulbar muscular atrophy gene products interact with glyceraldehyde-3-phosphate dehydrogenase. Hum Mol Genet 1996; 5:1311 - 1318
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature 2003; 425:686 - 691
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995; 268:880 - 884
- Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci USA 2000; 97:1589 - 1594
- Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell 2001; 106:183 - 194
- Motoyama A, Venable JD, Ruse CI, Yates JR IIIrd. Automated ultra-high-pressure multidimensional protein identification technology (UHP-MudPIT) for improved peptide identification of proteomic samples. Anal Chem 2006; 78:5109 - 5118
- Hsich G, Kenney K, Gibbs CJ, Lee KH, Harrington MG. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med 1996; 335:924 - 930
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 2003; 38:375 - 377
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 2003; 113:457 - 468
- Bailleul PA, Newnam GP. Steenbergen JN, Chernoff YO, Genetic study of interactions between the cytoskeletal assembly protein sla1 and prion-forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae. Genetics 1999; 153:81 - 94
- Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci USA 2004; 101:12934 - 12939