Abstract
It is over 40 years since it was first reported that the yeast Saccahromyces cerevisiae contains two unusual cytoplasmic ‘genetic’ elements: [PSI+] and [URE3]. Remarkably the underlying determinants are protein-based rather than nucleic acid-based, i.e., that they are prions, and we have already learned much about their inheritance and phenotypic effects from the application of ‘classical’ genetic studies alongside the more modern molecular, cellular and biochemical approaches. Of particular value has been the exploitation of chemical mutagens and ‘antagonistic’ mutants which directly affect the replication and/or transmission of yeast prions. In this chapter we describe what has emerged from the application of classical and molecular genetic studies, to the most intensively studied of the three native yeast prions, the [PSI+] prion.
Introduction
Genetic studies with Saccharomyces cerevisiae first revealed the existence of non- Mendelian traits in this species over half a century ago with the discovery of the mitochondrial petite mutation.Citation1,Citation2 A decade after the realization that the yeast cytoplasm contained such a nucleic acid-based genetic determinant, two other cytoplasmic ‘genetic’ elements referred to as [PSI+] and [URE3] emerged from yeast genetic laboratories in the UK and France, respectively.Citation3,Citation4 These new determinants were also originally identified by their non-Mendelian pattern of inheritance, but confounded researchers because they were not associated with mitochondrial DNA nor with twoo other non-Medelian determinants, namely “killer dsRNA” and the 2-micron circle. There followed a period of considerable speculation about the molecular nature of the underlying genetic determinantsCitation5 and it was not until some 30 years later that the ‘genetic’ determinants [PSI+] and [URE3] were shown to be proteinaceous in nature rather than nucleic acid based i.e., they were prions.Citation6 In this chapter we describe what has emerged from the application of classical and molecular genetic studies, to the most intensively studied of the yeast prions, namely the [PSI+] prion.
A Short History of the ‘ψ Factor’, A Novel Non-Mendelian Element
Discovery of the ‘ψ factor’.
As with many of the most important discoveries in science, the ‘ψ factor’ (as the [PSI+] prion was originally known), was discovered by serendipity, emerging from a genetic analysis of mutants of S. cerevisiae that suppressed nonsense mutations. In these studies Brian Cox, then at the University of Oxford, was following the inheritance of SUQ5 (also referred to as SUP16) a dominant nonsense suppressor and was using suppression of the nonsense (UAA-ochre) ade2-1 allele to monitor inheritance of the SUQ5 suppressor.Citation3 Cells carrying the ade2-1 allele usually form red colonies and are adenine auxotrophs (i.e., Ade-) but in the SUQ5-carrying strains being studied by Cox, this allele was suppressed by SUQ5 thereby giving Ade+ cells that gave white colonies. However, in genetic crosses in which SUQ5 was segregating, an unexpectedly high number of haploid spores gave rise to white Ade+ colonies with small red, Ade- sectors. Subsequent genetic analysis of these ‘nonsuppressed’ mutants led to the discovery of the ‘ψ factor’Citation3,Citation7 with the nonsuppressed cells being designated ψ- and the suppressed cells, ψ+.
The key genetic cross that resulted in Cox concluding that the ‘ψ factor’ was a non-Mendelian trait was one in which he crossed cells from one of the red sectors (with the genotype SUQ5 ade2-1 ψ-) to a white suppressed progenitor strain (with the genotype SUQ5 ade2-1 ψ+). The resulting diploid showed the suppressed phenotype i.e., ψ+ was dominant to ψ-, and all the haploid spores emerging from the diploid after meiosis showed the same nonsense suppression phenotype and a 4:0 pattern of inheritance (). As expected, when the SUQ5 ade2-1 ψ+ strain was crossed to an ade2-1ψ- strain lacking the SUQ5 suppressor, each tetrad contained two white Ade+ to 2 red Ade- spores confirming that the SUQ5 mutation was still present in the strain and that the ψ+ mutation was not itself an efficient suppressor of ade2-1 in the absence of SUQ5. Following up on his original observation, Cox went on to demonstrate that a number of different laboratory strains were ψ-, and that the SUQ5 suppressor could also suppress other nonsense mutations (e.g., his5-2, can1-100, lys1-1), but only if the strain carried the ψ+ ‘mutation’. Subsequently it was shown that SUQ5 encoded a mutant form of a serine-inserting tRNA with an altered anticodon sequence such that it can decode the UAA (ochre) codon albeit inefficiently.Citation8 The ade2-1 allele and the other suppressible alleles studied by Cox contained premature ochre mutations within their coding sequences. Certain nonsense alleles (e.g., cyc1-72) could also be suppressed by [PSI+] in the absence of a defined suppressor tRNA.Citation9
The cytoplasmic location of the ψ factor.
While a 4:0 pattern of inheritance is entirely consistent with the ψ factor having a genetic determinant located outside the nucleus, nevertheless additional evidence to support this hypothesis was needed, especially as genetic studies by Young and CoxCitation10 had shown that the ψ+ mutation was inherited independently of mitochondrial genome markers such as erythromycin resistance (ery). The ‘classical’ method for demonstrating transmission of a cytoplasmically-located genetic determinant in fungi is cytoduction, but the formation of heterokaryons is not part of the yeast life cycle. The availability of the karyogamy-defective mutant (the kar1 mutant, ref. Citation11) which blocks the fusion of haploid nuclei from different parents during mating but has no affect on plasmogamy, provided such a genetic tool with which to confirm the cytoplasmic inheritance of the ψ factor. In a kar1 x KAR1+ cross, the parent cells fuse to form a cell containing two haploid nuclei in a mixed cytoplasm and are the equivalent of a dikaryon. New haploid daughter cells can emerge from the binucleate cell, which contain one or other of the parental nuclei, but in a cytoplasm contributed by both parents (). Applying this cytoduction assay to an analysis of the inheritance of the ψ factor showed that all haploid cytoductants segregating in a kar1 x KAR1+ cross were ψ+ irrespective of which parental nucleus was inherited from the binucleate ‘heterokaryon’.Citation12
The unambiguous demonstration that the ψ factor was inherited through the cytoplasm, but was not linked genetically to the mitochondrial genome, presented a new dilemma; what was the molecular nature of the ψ factor? Several other nucleic acid species are found in the cytoplasm of S. cerevisiae with the linear double-stranded (ds) RNA genome of the ‘killer’ virusCitation13 and the circular ds DNA 2 µm plasmidCitation14 being the two major species present. Yet a variety of physical and mutagenesis studies failed to link the ψ factor with any known nucleic acid ‘genome’Citation15 although some intriguing preliminary evidence that ψ- cells could be transformed to ψ+ using a DNA fraction enriched for circular extrachromosomal forms of ribosomal DNA (rDNA) repeats, the so-called 3 µm circles, was subsequently reported by Dai et al.Citation16
Mutagenesis studies on the ψ factor.
Early studies on chemical or physical agents that caused a ψ+ to ψ- mutation proved to be informative albeit with the benefit of hindsight. On the one hand, known DNA active mutagens such as ultraviolet light (UV) and ethyl methane sulphonate (EMS) could induce this mutation but only at a frequency expected of a single nuclear gene.Citation5,Citation17 These data strongly suggested that the ψ factor was dependent in some way on a single nuclear gene and that single hit mutations could result in permanent loss of the ψ factor. We now know that the gene is most likely SUP35.
In contrast to the findings with classical nuclear mutagens, certain non DNA active agents, in particular guanidine hydrochloride (GdnHCl) a protein denaturant, and methanol, were found to efficiently induce the ψ+ to ψ- ‘mutation’ without any apparent underlying mutagenic damage to DNA or RNA.Citation18 One of the possible implications of the discovery of a class of mutagens active against the ψ factor but not DNA was that the phenotype associated with the presence of the ψ factor was the result of an “epigenetic self-sustaining system” that could result in the cells adopting one of two meta-stable states i.e., ψ+ and ψ-.Citation5 The self-sustaining ψ+ state could be perturbed either by mutation of a gene encoding a component of that system or by chemically perturbing the system in some other undefined manner.Citation5 As we now know, that ‘state’ (now more typically referred to as [PSI+]) reflects the presence of altered but self-perpetuating conformers of Sup35p, a cellular protein involved in translation termination, that is encoded by the SUP35 gene.
[PSI+] is the prion form of Sup35p.
In addition to proposing that the non-Mendelian inheritance of the phenotypically unrelated [URE3] determinant could be explained by the prion-like behavior of the Ure2 protein (Ure2p), WicknerCitation6 also suggested that the [PSI+] determinant might be due to a prion. Three independent reports pointed firmly at the product of the SUP35 gene (the Sup35 protein-Sup35p) as the most likely candidate for the prion protein associated with [PSI+]:Citation19 (a) the discovery that mutations in the SUP35 gene could result in cells no longer being able to maintain the [PSI+] determinant i.e., they become [psi-],Citation20,Citation21 (b) that mutations in the SUP35 gene (originally known as sal3 mutants, ref. Citation19) gave the same phenotype as cells carrying [PSI+], and (c) that over expression of the SUP35 gene led to the de novo appearance of [PSI+] strains.Citation22
Within three years of the original Wickner paper,Citation6 a number of independent studies were published that provided important genetic and biochemical data that supported the hypothesis that the [PSI+] determinant (i.e., Cox's mysterious ψ factor) is the prion form of Sup35pCitation23–Citation25 (recent reviews). The definitive proof of a protein-only (prion) based mechanism for [PSI+] came with the demonstration that introduction of a purified recombinant ‘prion-like’ form of Sup35p into a [psi-] cell by ‘protein transformation’ triggers a high rate of conversion of those cells to a stable [PSI+] state.Citation26,Citation27
With the recognition that [PSI+] is the prion form of Sup35p, many if not all of the unusual genetic properties that Cox described for the ψ factor could be accounted for: non-Mendelian inheritance, dependency on a nuclear gene, cytoduction and failure to associate with a known cytoplasmic nucleic acid genome () although many questions still remain unanswered as will be evident from other contributions in this volume.
The [PSI+] Phenotype
As described above, the phenotype of a [PSI+] cell that is most widely exploited is that of nonsense suppression and this phenotype is a direct consequence of a defect in the translation termination machinery in [PSI+] cells. The relative efficiency with which a suppressor tRNA can translate any stop codons be it a natural terminator or a premature stop codon, is directly related to the ability of the tRNA to out-compete the termination machinery in binding to the ribosomal A site where the codon is positioned (). Yeast, as for other eukaryotes, encodes two proteins, eRF1 and eRF3, which constitute the functional release factor that plays the central role in terminating protein synthesis in response to an in-frame stop codon.Citation28,Citation29 eRF1 (encoded by the SUP45 gene in yeast) recognises the stop codon positioned in the ribosomal A site via its N-terminal domain. The central domain of eRF1 interacts with the peptidyl-transferase centre of the ribosome to trigger the hydrolysis of the peptidyl-tRNA ester bond with the concomitant release of the completed polypeptide chain and interacts with eRF3 through its C-terminal domain.Citation30 eRF3 stimulates the termination reaction in a GTP-dependent manner and although the precise role of eRF3 in the termination reaction is not well defined, it may be required for the coupling of stop codon recognition by eRF1 to release of the polypeptide chain from the ribosome.Citation31 Deletion of either the SUP35 or SUP45 gene is lethal although it is conceivable that cell death is because one or both of these proteins may also play essential nontranslational roles in the cell (example in ref. Citation32).
The strength of the nonsense suppression phenotype used to monitor the presence of the [PSI+] prion, is the net result of the interplay between three parameters (). Of paramount importance is the relative efficiency with which a cognate (nonsense suppressor) or near cognate (wild-type) tRNA is able to compete with the translation termination machinery for the target nonsense codon. In a [PSI+] strain, the termination machinery is defective most likely because a high proportion (>90%) of Sup35p is present in the form of prion aggregates,Citation33,Citation34 possibly preventing a functional interaction between Sup35p and Sup45p (eRF1). Mutations in either the SUP35 or SUP45 genes can also give the same termination-defective phenotype. The other two parameters that can influence suppression efficiency are the compatibility of the amino acid inserted in response to the stop codon with function of the encoded polypeptide chain, and the nucleotide sequences immediately 5′ and 3′ to the suppressible stop codon.Citation35,Citation36
In practical terms the most straightforward measure of termination efficiency is via a colony level analysis using strains carrying a nonsense allele of either the ADE1 (e.g., ade1-14UGA) or ADE2 (e.g., ade2-1UAA) gene. In such strains high efficiency nonsense suppression gives white Ade+ colonies, while low efficiency nonsense suppression gives pink colonies showing a weak adenine prototrophy. Nonsuppressed strains will be red and adenine auxotrophs. A more quantitative means of determining the efficiency on nonsense suppression can be achieved through the use of plasmid-borne gene fusions in which two assayable open reading frames (ORFs) are separated by a stop codon; e.g., PGK-stop-lacZ,Citation37 lacZ-stop-luc.Citation38
While the ability of the SUQ5 mutation to suppress ochre alleles is absolutely dependent upon the cells being [PSI+], this is not the case for a large number of other well characterised nonsense suppressors. For example, there are several tyrosine-inserting ochre suppressor tRNAs described e.g., SUP4, that can suppress ade2-1 and other ochre alleles in a [psi-] strain, but are lethal when crossed into a [PSI+] genetic background.Citation39 This lethal phenotype presumably reflects a synergism between the suppressor tRNA and the weakened termination mechanism in the [PSI+] background that also leads to efficient readthrough of naturally-occurring termination codons at the end of ORFs.
Nuclear Genetic Antagonists of [PSI+]
The nonsense suppression phenotype that registers the presence of the [PSI+] prion in a given strain provides a simple yet effective means of screening for mutations that affect the ability of a cell to propagate [PSI+]. Thus starting with either an ade1-14 [PSI+] strain or an SUQ5 ade2-1 [PSI+] strain, mutations that result in loss of the diagnostic white Ade+ phenotype can be readily detected by a visual screen for red Ade- colonies. Alternatively, positive selection of nonsuppressed mutants can be achieved by using the suppressible can1-100 allele; [psi-] cells expressing this allele are resistant to the toxic arginine analogue canavanine whereas in [PSI+] cells the can1-100 allele is suppressed and cells become canavanine sensitive. An alternative positive selectable marker for the loss of [PSI+] has also been developed, based on ura3-14, a nonsense allele of the URA3 gene.Citation40
Using such phenotypic screens, two basic classes of nuclear genetic antagonists of [PSI+] have been identified that have arisen either spontaneously or as a consequence of chemical or UV mutagenesis ():
‘Psi-no more’ (PNM) mutants which lead to a loss in the ability of a cell to propagate the [PSI+] prion and which in turn leads to loss of the [PSI+] phenotype; and | |||||
‘Antisuppressor’ (ASU) mutants which mask the [PSI+] phenotype, but retain the ability to propagate the prion form of Sup35p. | |||||
For both classes of modifier either dominant (gain-of-function) or recessive (loss-of-function) mutants have been described and can be differentiated on the basis of the different patterns of inheritance shown (). Each class of mutant will be described in detail below.
PNM Mutants.
The first ‘PNM’ mutant to be described was designated ‘R’ (red) and carried a dominant chromosomal mutation that eliminated [PSI+] from cells.Citation41 When the R mutant was crossed to a [PSI+] strain not only was the resulting diploid [psi-], but so were all four spores (). However, for a given tetrad only two of the spores retained the dominant PNM character whereas the other two spores gave rise to cells with the normal genetic behavior of a [psi-] strain i.e., were no longer ‘PNM’. The underlying genetic defect in the R mutant was therefore nuclear in nature but resulted in loss of the extrachromosomal [PSI+] determinant. The loss of the [PSI+] determinant was not immediate because if a newly formed [PSI+] x [psi-] PNM zygote was abruptly induced to go into meiosis i.e., without further rounds of cell division, a significant number of [PSI+] spores can be found and the longer the diploid is grown before meiosis is induced, the fewer the number of [PSI+] spores are observed until most if not all spores give rise to [psi-] cells.Citation7,Citation42
To date only two PNM genes (PNM1 and PNM2) have been identified via the genetic analysis of dominant ‘PNM’ alleles. A comprehensive screen for non-suppressing revertants of a [PSI+] strainCitation12 has also identified a large number of recessive pnm mutants although the number of complementation groups these define is not known. In contrast to a dominant PNM mutant, a recessive pnm mutant yields a suppressed [PSI+] diploid when crossed with a [PSI+] strain and the resulting diploid yields suppressed ([PSI+]) and nonsuppressed ([psi-]) spores in various ratios but most typically 2 [PSI+]:2 pnm [psi-].
The PNM1 gene.
The PNM1 gene is allelic to the HSP104 geneCitation43 a finding that is in keeping with the earlier discovery that the product of this gene, the molecular chaperone Hsp104, is essential for the maintenance of the [PSI+] prion.Citation44 Hsp104 is an ATPase that works in combination with the Hsp70 and Hsp40 chaperone to remodel proteins that have aggregated in stressed cells. Hsp104 therefore plays an important role in helping cells recover from temperature stress, but also functions at much lower temperatures to facilitate the propagation of all three yeast prions.Citation45,Citation46
A number of mutations in the HSP104 gene have been described which result in a defect in [PSI+] propagation and these mutations are usually located in one of the two domains implicated in ATP binding and/or hydrolysis () which is to be expected given that this activity is crucial to Hsp104's role as a protein disaggregase. Some of the PNM alleles of HSP104 so far described, including a gene disruption, are recessive with respect to the [PSI+] propagation defect. However other alleles, for example the original PNM1-1 allele (described by Young and Cox, ref. Citation41) and a double mutation that inactivates both nucleotide binding domains (the K218T/K620T),Citation44 both show a dominant phenotype with respect to [PSI+] loss. Sequence analysis of the PNM1-1 allele has revealed mutations in both nucleotide binding domains NBD1 and NBD2.Citation43 A dominant PNM phenotype is probably due to a defect in Hsp104 oligomerisation and/or the generation of nonfunctional Hsp104 hexamers containing both mutant and wild-type Hsp104.
Recent studies have shown that the N-terminal region of Hsp104 (residues 1–147) is dispensable not only for [PSI+] propagation but also protein refolding and thermotoleranceCitation47 indicating that this region of the protein is not required to carry out these functions. In contrast, any mutation in the HSP104 gene that leads to a PNM phenotype impairs the disaggregase function of the chaperone and is consistent with current models for the role of Hsp104 in prion propagation, namely that the chaperone breaks down prion aggregates to generate new prion seeds necessary for continued propagation of the prion state.Citation45,Citation46
The PNM2 gene.
The PNM2 gene is allelic to SUP35Citation21 and the discovery that a mutation in the SUP35 gene could affect the maintenance of the [PSI+] determinant provided a crucial piece of evidence that linked Sup35p with the [PSI+] prion.Citation20,Citation21,Citation23 The PNM2-1 allele sequenced by Doel et al.Citation21 contains a Gly to Asp substitution at residue 58 near the amino terminus of the protein in a region shown by deletion studies to be required for [PSI+] propagation, i.e., in the prion-forming domain (PrD)Citation20,Citation48 (). The mutation was in the second of five oligopeptide repeats located between residues 41 and 97 of Sup35p; the so-called oligopeptide repeat (OPR) region. This region of the Sup35p protein together with the amino terminal Gln/Asn-rich 40 residues (also known as the QN-rich-QNR-region) constitute the prion-forming domain that is essential for the aggregation (the QNR region) and the continued propagation (the OPR region) of the [PSI+] prion.Citation49 Why the mutant Sup35p encoded by the PNM2-1 allele (i.e., Sup35pPNM2-1) has a dominant negative property with respect to prion propagation remains to be established, but it has been noted that the degree of dominance shown by this allele with respect to its ‘PNM’ phenotype is sensitive to the genetic background of the strain or the [PSI+] variant present in the strain in which it is introduced.Citation50
As for the wild-type Sup35p, the Sup35pPNM2-1 protein is able to induce [PSI+] de novo when overexpressed in a [PIN+][psi] strain,Citation50,Citation51 and can also form protein aggregates in vivo and self-seeding amyloid-like aggregates in vitro.Citation49,Citation51 This shows that the mutant Sup35pPNM2-1 protein is able to take up its prion form but once established that form is not efficiently propagated. This in turn leads to a mitotic instability and loss of [PSI+]. In a PNM2-1/+ heterozygote presumably mixed prion aggregates are formed which are impaired in their role in propagation of the prion state.
In addition to the original PNM2-1 allele, several other PNM alleles of SUP35 have been generated by random or site-directed mutagenesis.Citation52,Citation53 Among the mutants described by De Pace et al.Citation52 were PNM alleles which contained single amino acid substitutions located between residues 9 and 33 in the QNR region of the Sup35p-PrD important for protein aggregation, rather than in the OPR region where the PNM2-1 mutation lies. In the QNR region mutants, the soluble mutant Sup35pPNM2-1, molecules are poorly recruited into the prion-like aggregates of Sup35p but, importantly also prevent the generation of new prion seeds required for continued propagation of the [PSI+] prion.Citation52 The PNM mutants reported by KingCitation53 were single amino acid substitutions within the Sup35p-QNR region, but these mutations had differing PNM properties depending on the [PSI+] variant present in the strain (see below).
Surprisingly, even though single amino acid substitutions in either the Sup35p-QNR or Sup35p-OPR regions can lead to a defect in the propagation of the [PSI+] prion, the primary amino acid sequence of the QNR+OPR regions of Sup35p per se, does not seem to be critical for prion formation. This has recently emerged from a study in which the amino acid sequence of Sup35p between residues 3 and 114 were ‘randomized’ and the new ‘scrambled’ sequence used to replace the wild type sequence. That these engineered Sup35p ‘mutants’ were still mostly able to form and propagate the [PSI+] prion suggest that it is the length and/or amino acid composition of the Sup35p-PrD that is critical for the prion-like behaviour of Sup35p, a conclusion that emerged from parallel studies with the Ure2p prion protein.Citation54 The apparent contradiction between these two sets of observations remains to be resolved. Certainly large scale substitutions or deletions within the adjacent OPR region can generate ‘pnm’ alleles of SUP35.Citation20,Citation55,Citation56 What the findings of Ross et al.Citation54 may indicate is that when Sup35p (and Ure2p) form amyloid-like fibres, the interacting prion protein molecules may take up a parallel in-register β-sheet structure rather than an anti-parallel structure.Citation57
Antisuppressor (ASU) mutants.
While studies on the PNM mutants have made important contributions to our understanding of what makes a Sup35p a prion protein and the role of Hsp104 in propagation of the prion state, much less work has been done on antisuppressor mutants which prevent the [PSI+] phenotype from being expressed but without impairing propagation of the [PSI+] prion (). Such mutants are distinct from the recessive antisuppressor mutants originally described by McCready and CoxCitation58 since this latter class (which map to at least eight different loci, designated ASU1–ASU8) are most likely mutations that affect the structure and/or function of the suppressor tRNA. For example, one of the ASU genes (also called MOD5) is required for the synthesis of N6-delta 2-(isopentenyl) adenosine, a modified tRNA nucleoside important for codon-anticodon recognition.Citation59 Another class of yeast antisuppressor described by Chernoff and colleaguesCitation60 were single nucleotide changes in the 18S ribosomal RNA that reduced the efficiency of the nonsense suppression associated with certain sup35 and sup45 alleles that give rise to an omnipotent suppression phenotype that results in suppression and hence readthrough of all three stop codons. The molecular basis for the recessive antisuppression phenotype seen in both the tRNA modification and 18S rRNA mutants is most likely that they reduce the efficiency with which a suppressor tRNA or a naturally-occurring suppressor tRNA competes with the release factor-mediated termination event for the premature nonsense codon (see ). There is no evidence that these recessive antisuppressors affect the propagation of the [PSI+] prion.
Single amino acid substitutions within the Sup35p-PrDCitation52 or deletion of the Sup35p-PrDCitation20,Citation48,Citation55 can lead to a dominant antisuppressor phenotype in a [PSI+] cell. This is because these mutations generate forms of Sup35p that remain largely soluble because the efficiency with which they are seeded by the endogenous [PSI+] Sup35p seeds in vivo and their ability to form amyloid fibrils in vitro are dramatically reduced.Citation52 The soluble Sup35pASU molecules still contain the functional C-terminal region of the Sup35p molecule required for translation termination and their presence in a cell, irrespective of whether or not the cell is [PSI+], results in a shift in the balance towards termination and against nonsense suppression i.e., antisuppression. This contrasts with Sup35pPNM2-1 molecules which can interact with the endogenous [PSI+] seeds but, in so doing, block the ability to generate the new seeds required for continued propagation.Citation49,Citation51,Citation52
Guanidine hydrochloride-induced [psi-] mutants.
Even before the [PSI+] determinant had been identified as a ‘protein-only’ element, a number of chemical agents that were not mutagenic for DNA or RNA-based determinants, had been identified that resulted in efficient elimination of the [PSI+] determinant from growing cells. These agents include methanol, dimethyl sulphoxide,Citation18 high osmolarity,Citation61 the kastellpaolitinesCitation62 and the actin cytoskeleton disruptor, latrunculin A.Citation63
By far the most effective [PSI+] ‘curing agent’ described, and the one we best understand in terms of mode of action, is the protein denaturant guanidine hydrochloride (GdnHCl). When GdnHCl is added to growing [PSI+] cells this results in an essentially prion-free [psi-] culture after 10 to 12 generations of growth with the [psi-] cells appearing only after 4 or 5 generations of growth.Citation18,Citation64 The emergence of [psi-] cells appears to be a consequence of the GdnHCl inhibiting, even at low concentrations (1–5 mM), the key ATPase activity of the molecular chaperone Hsp104.Citation65–Citation67 This in turn results in an inability of the [PSI+] cell to generate the new prion seeds required for continued propagation of the [PSI+] prion. Consequently, the seeds present in the [PSI+] cell at the time the GdnHCl is added, are diluted by cell division and, eventually, seed-free and hence prion-free [psi-] cells emerge. For a detailed discussion of this mechanism and how it can be exploited to gain insights into the nature of the [PSI+].
[psi-] mutants generated by treatment of [PSI+] cells with these chemically diverse collections of compounds do not undergo any permanent change in their nuclear genotype that blocks prion propagation.Citation18 This can be shown by the reintroduction of prion seeds back into the [psi-] mutant by either genetic back crossing or cytoduction () which in both cases reestablishes a stable [PSI+] cell. While the [psi-] mutants induced by most of these agents are able to revert spontaneously to [PSI+] at a frequency of approximately 10-5 to 10-6, those induced by GdnHCl treatment do not revert spontaneously back to [PSI+] at any detectable frequency.Citation68,Citation69 Although at the time this observation was made it was thought that this was due to a physical deletion of the [PSI+] ‘determinant’, we now know that the reason is that the GdnHCl treatment also eliminates all other prions from the yeast cell including the [PIN+] prion that facilitates the de novo formation of [PSI+].Citation70,Citation71 That spontaneous reversion to [PSI+] is seen with other chemically-induced [psi-] mutants would suggest that these ‘mutagens’ act on a different target that only affects the [PSI+] prion and do not generate [pin-] cells.
[PSI+] ‘Variants’: Mutants without Genetic Change
One of the more remarkable properties of prions both in yeast and in animals is that they can exist in different conformational states that modify the associated neuropathology of the disease (for animal PrP) or the associated phenotype (as is the case for all three yeast prions).Citation72 In yeast such prion strains are referred to as ‘variants’ in order not to confuse them with yeast ‘strains’ that may have different phenotypes due to underlying differences in genotypes: yeast prion variants show different phenotypes but the amino acid sequence of the prion protein is identical in the different variants.
Yeast prion variants were first described for [PSI+] by the Liebman group when they noted that [PSI+] strains generated de novo in the same experiment often had different yet stably inherited phenotypes as defined using suppression of the ade1-14 marker i.e., colony color and degree of adenine prototrophy.Citation70,Citation71 Two basic [PSI+] variants have been described and are usually referred to as ‘weak’ and ‘strong’ reflecting directly the efficiency of nonsense suppression: weak variants show low efficiency of suppression (i.e., strong translation termination) while strong variants show efficient suppression.Citation70,Citation71,Citation73 Prion variants also show other readily scorable differences; for example weak variants show reduced mitotic stability and an elevated amount of soluble Sup35p compared with a strong variant.Citation73
The first clue to the physical basis for distinct yeast prion variants came from in vitro seeding studies which showed that the Sup35p aggregates found in a weak [PSI+] variant are less efficient at seeding soluble Sup35p than aggregates from a strong [PSI+] variant.Citation73 This difference in the efficiency of Sup35p conversion would t herefore lead to weak variants having higher levels of soluble Sup35p, which is the case.Citation73 The higher proportion of soluble-and therefore functional-Sup35p in the weak variants would give the characteristic antisuppressor, low efficiency suppression phenotype (see and previous section). Direct conformation that the different [PSI+] variants result from distinct yet heritable conformers of Sup35p came from two groups using novel ‘protein transformation’ assays.Citation26,Citation27 Tanaka et al.Citation26 generated in vitro, distinct amyloid-like forms of a recombinant fragment of Sup35p derived from the N-terminus (Sup35NMp; ), by using different temperatures for the polymerisation. When these different forms of Sup35NMp were introduced into [psi-] cells, they gave rise to distinct [PSI+] variants that were stably propagated over many subsequent generations of growth. For example, Sup35NMp aggregates formed at 4°C gave rise to strong variants whereas aggregates that formed at 37°C gave rise to weak variants. Therefore the Sup35p protein can take up at least three different self-replicating conformational states each of which results in different levels of soluble Sup35p in the cell and hence different [PSI+]-associated phenotypes.
An elegant model to explain the physical basis of prion variants in yeast has recently been presented by Weissman and colleagues.Citation74 In this model—which they validate experimentally—different variants are generated as a consequence of the dynamic interplay between several different parameters including conformation-dependent differences in the rate prion aggregates are formed in the cell and the rate of fragmentation. The Sup35p aggregates in a strong variant grow more slowly than the aggregates found in the weak variants, but the key difference is the fragility of the aggregates formed; in the strong variant these aggregates are much more susceptible to breakage than the faster forming aggregates in the weak variants and so the number of prion seeds was greater in the strong variants.Citation74 It remains to be seen whether this model can also explain different mammalian prion strains.
While most studies on [PSI+] variants compare weak vs. strong, several other [PSI+] variants have also been described. For example KingCitation53 described three different [PSI+] variants that showed distinct phenotypes when introduced into a series of different yeast strains carrying defined sup35 mutations. As with the weak and strong variants, protein aggregates from strains carrying these [PSI+] variants (called [VH], [VK] and [VL]) could be used to infect [psi-] cells and the resulting [PSI+] cells had the phenotype associated with the original variant. Sup35p fibrils formed by these variants also show distinct conformational differences.Citation75
The existence of stable [PSI+] prion variants which have identical nuclear genotypes but which show significant differences in the efficiency with which the translation termination machinery can recognise a stop codon amply illustrates the epigenetic nature of the yeast [PSI+] prion and also the potential and varied impact the prion can have on cell phenotype.
Translation Termination in [PSI+] Strains
The [PSI+] prion provides the yeast cell with a novel epigenetic mechanism with which to modulate translation termination thereby facilitating the translation of in-frame stop codons. This of course raises two important evolutionary questions: “Why would such a mechanism have evolved?” and “What evolutionary benefit—if any—does it confer?” These questions are addressed in the chapters by Wickner et al. and Zhouravleva et al.
As is now well established, [PSI+] strains show a measurable defect in translation termination as defined by their ability to suppress a range of nonsense alleles i.e., in [PSI+] strains the ribosome is able to translate stop codons, albeit inefficiently (see above). Yet it must be remembered that all 6000+ ORFs in the yeast genome are terminated by a stop codon. Any efficient translation of a natural terminator would result in the addition of C-terminal sequences to the encoded polypeptide chain. This in turn could have an adverse effect on function, turnover and/or localisation of that polypeptide. This presumption is well supported by several observations; for example, the reported lethal interactions between certain efficient suppressor tRNAs and [PSI+]Citation39 (see above) and between sal3 alleles of SUP35 and [PSI+].Citation19
Can translation readthrough of a natural terminator actually occur in a [PSI+] strain? Analysis of natural terminators at the 3′ ends of validated yeast ORFs has indicated that there is an over representation of in-frame stop codons 3′ to the authentic terminator. Such ‘tandem stops’ should reduce any potentially detrimental effects of efficient readthrough.Citation76,Citation77 This is not the case for all natural terminators however; for example the yeast genome contains a number of instances of two ORFs separated by a stop codon and in 8/58 cases reported by Namy et al.Citation38,Citation78 there was a high level of readthrough of the terminator separating the two ORFs (efficiency between 3 and 25%). Intriguingly only two of these eight showed a significant reduction in readthrough in a [psi-] strain suggesting that there might also be [PSI+]-independent modulation of termination in yeast. There has also been a report that readthrough of the UAG terminator of the S. cerevisiae PDE2 gene that encodes a high affinity cAMP phosphodiesterase generates a modified form of the protein that is C-terminally extended by 20 residues and is unstable compared to the ‘normal’ Pde2p product.Citation38 In a [PSI+] strain translation readthrough of the ‘natural’ PDE2 stop codon was elevated some 20-fold compared to a [psi-] strain and the consequence of readthrough was an increase in the cellular concentration of cAMP.Citation38 [PSI+] may have its phenotypic effects via changing readthrough of only a small number of ‘natural’ terminators in yeast mRNAs. A further possibility is that the [PSI+]-induced phenotypic variation may also reflect changes in the rate of decay of mRNAs whose natural terminators are being readthrough, so-called ‘nonstop mRNA decay’.Citation79
The very existence of the [PSI+] prion in laboratory strains of S. cerevisiae is therefore something of an evolutionary puzzle, but nevertheless provides researchers with a powerful tool with which rapidly to explore some of the key questions in prion biology: what makes a prion, how is the prion state propagated and how does a cell react to the presence of a prion? Discovering why it exists may be somewhat more of a challenge.
Figures and Tables
Figure 1 The yeast [PSI+] prion is transmitted as an extrachromosomal genetic element. The cytoplasm of [PSI+] cells contains a number of distinct physical entities—propagons—that are necessary for the continued propagation of the [PSI+] state. When a [PSI+] cell is mated to a propagon-free [psi-y] cell (left panel) the diploid is [PSI+] and all four haploid meiotic progeny are also [PSI+]. When a [PSI+] cell is mated to a [psi-] kar1cell that is defective in nuclear fusion (right panel) the resulting unstable heterokaryons generate new haploid cells—cytoductants. At a frequency approaching 100%, such cytoductants are [PSI+] irrespective of the haploid nucleus carried. NB: The nuclei arising from fusion between two parental nuclei are shown with hatches.
![Figure 1 The yeast [PSI+] prion is transmitted as an extrachromosomal genetic element. The cytoplasm of [PSI+] cells contains a number of distinct physical entities—propagons—that are necessary for the continued propagation of the [PSI+] state. When a [PSI+] cell is mated to a propagon-free [psi-y] cell (left panel) the diploid is [PSI+] and all four haploid meiotic progeny are also [PSI+]. When a [PSI+] cell is mated to a [psi-] kar1cell that is defective in nuclear fusion (right panel) the resulting unstable heterokaryons generate new haploid cells—cytoductants. At a frequency approaching 100%, such cytoductants are [PSI+] irrespective of the haploid nucleus carried. NB: The nuclei arising from fusion between two parental nuclei are shown with hatches.](/cms/asset/a6dc9539-fb8a-448e-81b5-7b926ad17dff/kprn_a_10904665_f0001.gif)
Figure 2 The molecular basis of the [PSI+]-associated nonsense suppression phenotype. Left) When a stop codon arrives at the ribosomal A site it is efficiently recognised by the release factor which in yeast and other eukaryotes is comprised of two proteins, eRF1 (Sup45p) and eRF3 (Sup35p). There are some near-cognate tRNAs able to translate termination codons albeit inefficiently and these are only usually detected when the termination machinery is impaired as, for example, is the case in [PSI+] cells or in sal mutants. Translation then proceeds to the next in-frame stop codon, which here is UAA. Right) If the cell encodes a mutant tRNA which is able to recognise a defined stop codon i.e., a cognate nonsense suppressor tRNA such as the SUQ5-encoded tRNASer 8, this tRNA is able to compete more efficiently than a near-cognate tRNA with the release factor for the termination codon at the A site. Again the efficiency with which the cognate suppressor tRNA competes greatly increased if termination machinery is impaired.
![Figure 2 The molecular basis of the [PSI+]-associated nonsense suppression phenotype. Left) When a stop codon arrives at the ribosomal A site it is efficiently recognised by the release factor which in yeast and other eukaryotes is comprised of two proteins, eRF1 (Sup45p) and eRF3 (Sup35p). There are some near-cognate tRNAs able to translate termination codons albeit inefficiently and these are only usually detected when the termination machinery is impaired as, for example, is the case in [PSI+] cells or in sal mutants. Translation then proceeds to the next in-frame stop codon, which here is UAA. Right) If the cell encodes a mutant tRNA which is able to recognise a defined stop codon i.e., a cognate nonsense suppressor tRNA such as the SUQ5-encoded tRNASer 8, this tRNA is able to compete more efficiently than a near-cognate tRNA with the release factor for the termination codon at the A site. Again the efficiency with which the cognate suppressor tRNA competes greatly increased if termination machinery is impaired.](/cms/asset/bfb54d66-e0aa-458f-9e02-c4a6a6f7374b/kprn_a_10904665_f0002.gif)
Figure 3 A genetic cross can differentiate between mutations that eliminate the [PSI+] prion (PNM, ‘PSI-No-More’ mutants) and dominant antisuppressor mutants (ASU). The diploid formed between a PNM mutant and a [PSI+] strain loses the ability to replicate the [PSI+] prion because it can no longer efficiently produce the new propagons required. Consequently all of the haploid meiotic spores do not inherit the ability to propagate the [PSI+] state i.e., are [psi-]. In contrast, in a cross between an ASU mutant and a [PSI+] strain, although the phenotype is the same as the PNM/+ diploid, the ASU/+ diploid still has the ability to generate new propagons. The reason the nonsense suppression phenotype is not expressed is because these cells also produce a significant level of soluble and functional Sup35p which ensures that efficient translation termination occurs. Consequently those meiotic spores that inherit the ASU mutation show a red nonsuppressed phenotype whereas those that do not, show the [PSI+] phenotype.
![Figure 3 A genetic cross can differentiate between mutations that eliminate the [PSI+] prion (PNM, ‘PSI-No-More’ mutants) and dominant antisuppressor mutants (ASU). The diploid formed between a PNM mutant and a [PSI+] strain loses the ability to replicate the [PSI+] prion because it can no longer efficiently produce the new propagons required. Consequently all of the haploid meiotic spores do not inherit the ability to propagate the [PSI+] state i.e., are [psi-]. In contrast, in a cross between an ASU mutant and a [PSI+] strain, although the phenotype is the same as the PNM/+ diploid, the ASU/+ diploid still has the ability to generate new propagons. The reason the nonsense suppression phenotype is not expressed is because these cells also produce a significant level of soluble and functional Sup35p which ensures that efficient translation termination occurs. Consequently those meiotic spores that inherit the ASU mutation show a red nonsuppressed phenotype whereas those that do not, show the [PSI+] phenotype.](/cms/asset/d1266962-72d3-4dd7-9e98-36a8d84f877e/kprn_a_10904665_f0003.gif)
Figure 4 Mutations within the molecular chaperone Hsp104 can give rise to a ‘PSI-No-More’ phenotype. Upper: A schematic representation of the key functional domains of Hsp104 showing the two nucleotide binding domains (ATP; NBD1/NBD2) and the N-terminal (NTD) and C-terminal (CTD) domains plus the linker region between the two NBDs. The proposed functional roles of the different domains are indicated below. See refs. Citation45 and Citation46 for further discussion on the functional organisation of Hsp104. Lower: mutations that have so far been described which give a ‘PNM’ phenotype. The location of the mutations is given together with an indication of whether the mutation in question is dominant or a recessive with respect to the ‘PNM’ phenotype. ND indicates that the dominance/recessive character of the mutant was not reported.Citation1 The single mutations K218T and K620T are also dominant/semi-dominant PNMs in some genetic backgrounds.
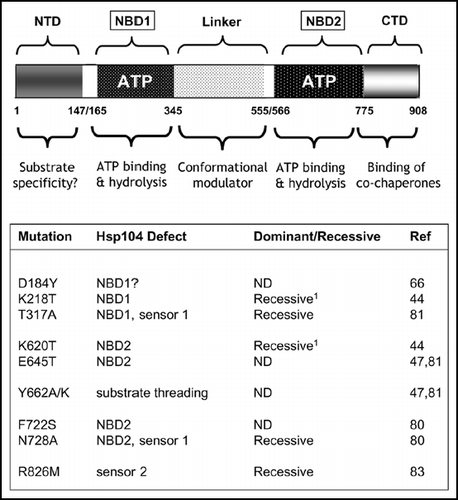
Figure 5 The prion-forming domain of the Sup35p protein. (A) The 685 residue Sup35p protein has three regions defined by the locations of the first three in-frame AUG (Met) codons: the amino terminal N domain (residues 1–123) that is absolutely required for the prion behaviour of the protein; the highly charged middle region (M; residues 124–253) and the carboxyl-terminal domain (C; residues 254–685) which carries the essential release factor activity.Citation20,Citation48 (B) Within the N domain lies a region, the so-called prion-forming domain-which contains a region important for aggregation and a separate but overlapping region important for propagation of the prion state.Citation48 The aggregation element contains a short peptide sequence based around residues 7 to 13, that is highly amyloidogenic. The propagation element contains five imperfect copies of an oligopeptide repeat sequence (R1–R5). (C) The ‘PSI-No-More’ mutant PNM2-1 carries a single amino acid substitution (Gly to Asp) within repeat R2 of the propagation element. This single substitution leads to a form of Sup35p that can still aggregate but can no longer be efficiently propagated in most laboratory strains.Citation20,Citation48,Citation50
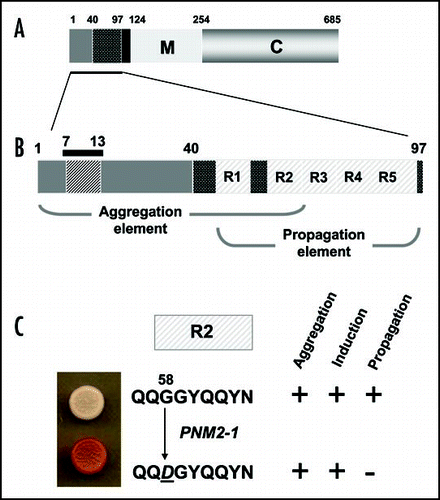
Table 1 Nuclear genetic antagonists of the [PSI+]-associated nonsense suppression phenotype
Acknowledgements
Research on the yeast [PSI+] prion, carried out in the authors' laboratory, is funded by the Biotechnology and Biological Sciences Research Council (BBSRC), the Wellcome Trust, the EC (through the APOPIS project: LSHM-CT-2003-503330) and a personal award to Brian S. Cox by the Leverhulme Trust.
References
- Ephrussi B. . Nucleo-cytoplasmic relations in micro-organisms 1953; Oxford University Press
- Sherman F. Respiration-deficient mutants of yeast. I. Genetics. Genetics 1963; 48:375 - 385
- Cox B. ψ, a cytoplasmic suppressor of super-suppressor in yeast. Heredity 1965; 20:505 - 521
- Lacroute F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J Bact 1971; 106:519 - 522
- Cox BS, Tuite MF, McLaughlin CS. The ψ factor of yeast: A problem in inheritance. Yeast 1988; 4:159 - 178
- Wickner RB. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:566 - 569
- Cox BS. Hall MD, Linder P. Psi Phenomena in Yeast. Early days of yeast genetics 1993; NY Cold Spring Harbor Laboratory Press 219 - 239
- Waldron C, Cox BS, Wills N, Gesteland RF, Piper PW, Colby D, Guthrie C. Yeast ochre suppressor SUQ5-ol is an altered tRNA Ser UCA. Nucleic Acids Res 1981; 9:3077 - 3088
- Liebman SW, Sherman F. Extrachromosomal psi+ determinant suppresses nonsense mutations in yeast. J Bacteriol 1979; 139:1068 - 1071
- Young CS, Cox BS. Extrachromosomal elements in a super-suppression system of yeast. II. Relations with other extrachromosomal elements. Heredity 1972; 28:189 - 199
- Conde J, Fink GR. A mutant of Saccharomyces cerevisiae defective for nuclear fusion. Proc Natl Acad Sci USA 1976; 73:3651 - 3655
- Cox BS, Tuite MF, Mundy CR. Reversion from suppression to nonsuppression in SUQ5 [psi+] strains of yeast: The classification of mutations. Genetics 1980; 95:589 - 609
- Wickner RB. Double-stranded and single-stranded RNA viruses of Saccharomyces cerevisiae. Annu Rev Microbiol 1992; 46:347 - 375
- Velmurugan S, Mehta S, Uzri D, Jayaram M. Stable propagation of ‘selfish’ genetic elements. J Biosci 2003; 28:623 - 636
- Tuite MF, Lund PM, Futcher AB, Dobson MJ, Cox BS, McLaughlin CS. Relationship of the [psi] factor with other plasmids of Saccharomyces cerevisiae. Plasmid 1982; 8:103 - 111
- Dai H, Tsay SH, Lund PM, Cox BS. Transformation of psi- Saccharomyces cerevisiae to psi+ with DNA copurified with 3 micron circles. Curr Genet 1986; 11:79 - 82
- Tuite MF, Cox BS. Ultraviolet mutagenesis studies of [psi], a cytoplasmic determinant of Saccharomyces cerevisiae. Genetics 1980; 95:611 - 630
- Tuite MF, Mundy CJ, Cox BS. Agents that cause a high frequency of genetic change from [psi+] to [psi-] in Saccharomyces cerevisiae. Genetics 1981; 8:691 - 711
- Cox BS. Allosuppressors in yeast. Genet Res 1977; 30:187 - 205
- Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 1994; 137:671 - 676
- Doel SM, McCready SJ, Nierras CR, Cox BS. The dominant PNM2- mutation which eliminates the psi factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 1994; 137:659 - 670
- Chernoff YO, Derkach IL, Inge-Vechtomov SG. Multicopy Sup35 gene induces de novo appearance of Psi-like factors in the yeast Saccharomyces cerevisiae. Curr Genet 1993; 24:268 - 270
- Cox BS. Prion-like factors in yeast. Curr Biol 1994; 4:744 - 748
- Tuite MF, Cox BS. Propagation of yeast prions. Nat Rev Mol Cell Biol 2003; 4:878 - 890
- Shorter J, Lindquist SL. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet 2005; 6:435 - 450
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature 2004; 428:265 - 267
- King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature 2004; 428:319 - 323
- Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, Paushkin SV, Nierras CR, Cox BS, Ter-Avanesyan MD, Tuite MF. The products of the SUP45(eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J 1995; 14:4365 - 4373
- Zhouravleva G, Frolova L, LeGoff X, LeGuellec R, Inge-Vechtomov S, Kisselev L, Philippe M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J 1995; 14:4065 - 4072
- Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF, Hemmings BA, Barford D. The crystal structure of human eukaryotic release factor eRF1—Mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell 2000; 100:311 - 321
- Salas-Marco J, Bedwell DM. GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Mol Cell Biol 2004; 24:7769 - 7778
- Valouev IA, Kushnirov VV, Ter-Avanesyan MD. Yeast polypeptide chain release factors eRF1 and eRF3 are involved in cytoskeleton organization and cell cycle regulation. Cell Motil Cytoskel 2002; 52:161 - 173
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J 1996; 15:3127 - 3134
- Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996; 273:622 - 626
- Bonetti B, Fu L, Moon J, Bedwell DM. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J Mol Biol 1995; 251:334 - 345
- Mottagui-Tabar S, Tuite MF, Isaksson LA. The influence of 5′ codon context on translation termination in S. cerevisiae. Eur J Biochem 1998; 257:249 - 254
- Firoozan M, Grant CM, Duarte JA, Tuite MF. Quantitation of readthrough of termination codons in yeast using a novel gene fusion assay. Yeast 1991; 7:173 - 183
- Namy O, Duchateau-Nguyen G, Hatin I, Hermann-Le Denmat S, Termier M, Rousset JP. Identification of stop codon readthrough genes in Saccharomyces cerevisiae. Nucleic Acids Res 2003; 31:2289 - 2296
- Cox BS. A recessive lethal super-suppressor mutation in yeast and other psi phenomena. Heredity 1971; 26:211 - 232
- Manogaran AL, Kirkland KT, Liebman SW. An engineered nonsense URA3 allele provides a versatile system to detect the presence, absence and appearance of the [PSI+] prion in Saccharomyces cerevisiae. Yeast 2006; 23:141 - 147
- Young CSH, Cox BS. Extrachromosomal elements in a super-suppression system of yeast. I. A nuclear gene controlling the inheritance of the extrachromosomal elements. Heredity 1971; 26:413 - 422
- McCready SJ, Cox BS, McLaughlin CS. The extrachromosomal control of nonsense suppression in yeast: An analysis of the elimination of [psi+] in the presence of a nuclear gene PNM. Mol Gen Genet 1977; 150:265 - 270
- Cox BS, Jones KM, Ho HL, et al. Manuscript in preparation
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995; 268:880 - 884
- True HL. The battle of the fold: Chaperones take on prions. Trends Genet 2006; 22:110 - 117
- Bosl B, Grimminger V, Walter S. The molecular chaperone Hsp104-A molecular machine for protein disaggregation. J Struct Biol 2006; 156:139 - 148
- Hung GC, Masison DC. N-terminal domain of yeast hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by hsp104 overexpression. Genetics 2006; 173:611 - 620
- Ter-Avanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge-Vechtomov SG, Smirnov VN. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two nonoverlapping functional regions in the encoded protein. Mol Microbiol 1993; 7:683 - 692
- Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biology 2004; 2:442 - 451
- Derkatch IL, Bradley ME, Zhou P, Liebman SW. The PNM2 mutation in the prion protein domain of SUP35 has distinct effects on different variants of the [PSI+] prion in yeast. Curr Genet 1999; 35:59 - 67
- Kochneva-Pervukhova NV, Paushkin SV, Kushnirov VV, Cox BS, Tuite MF, Ter-Avanesyan MD. Mechanism of inhibition of Psi+ prion determinant propagation by a mutation of the N-terminus of the yeast Sup35 protein. EMBO J 1998; 17:5805 - 5810
- DePace AH, Santoso A, Hillner P, et al. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 1998; 93:1241 - 1252
- King CY. Supporting the structural basis of prion strains: Induction and identification of [PSI] variants. J Mol Biol 2001; 307:1247 - 1260
- Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc Natl Acad Sci USA 2005; 102:12825 - 12830
- Liu JJ, Lindquist S. Oligopeptide-repeat expansions modulate ‘protein-only’ inheritance in yeast. Nature 1999; 400:573 - 576
- Parham SN, Resende CG, Tuite MF. Oligopeptide repeats in the yeast protein Sup35p stabilize intermolecular prion interactions. EMBO J 2001; 20:2111 - 2119
- Ross ED, Minton A, Wickner RB. Prion domains: Sequences, structures and interactions. Nat Cell Biol 2005; 7:1039 - 1044
- McCready SJ, Cox BS. Antisuppressors in yeast. Mol Gen Genet 1973; 124:305 - 320
- Laten H, Gorman J, Bock RM. Isopentenyladenosine deficient tRNA from an antisuppressor mutant of Saccharomyces cerevisiae. Nucleic Acids Res 1978; 5:4329 - 4342
- Chernoff YO, Newnam GP, Liebman SW. The translational function of nucleotide C1054 in the small subunit rRNA is conserved throughout evolution: Genetic evidence in yeast. Proc Natl Acad Sci USA 1996; 93:2517 - 2522
- Singh A, Helms C, Sherman F. Mutation of the non-Mendelian suppressor, Psi, in yeast by hypertonic media. Proc Natl Acad Sci USA 1979; 76:1952 - 1956
- Bach S, Talarek N, Andrieu T, Vierfond JM, Mettey Y, Galons H, Dormont D, Meijer L, Cullin C, Blondel M. Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nat Biotechnol 2003; 21:1075 - 1081
- Bailleul-Winslett PA, Newnam GP, Wegrzyn RD, Chernoff YO. An antiprion effect of the anticytoskeletal drug latrunculin A in yeast. Gene Expr 2000; 9:145 - 156
- Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 2000; 97:240 - 244
- Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol 2001; 40:1357 - 1369
- Jung GM, Jones G, Masison DC. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA 2002; 99:9936 - 9941
- Grimminger V, Richter K, Imhof A, Buchner J, Walter S. The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J Biol Chem 2004; 279:7378 - 7383
- Lund PM, Cox BS. Reversion analysis of [psi-] mutants in Saccharomyces cerevisiae. Genet Res 1981; 37:173 - 182
- Koloteva-Levin N, Merritt GH, Tuite MF. Manuscript in preparation
- Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996; 144:1375 - 1386
- Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997; 147:507 - 519
- Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 1994; 68:7859 - 7868
- Uptain SM, Sawicki GJ, Caughey B, Lindquist S. Strains of [PSI+] are distinguished by their efficiencies of prion-mediated conformational conversion. EMBO J 2001; 20:6236 - 6245
- Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature 2006; 442:585 - 589
- Diaz-Avalos R, King CY, Wall J, Simon M, Caspar DL. Strain-specific morphologies of yeast prion amyloid fibrils. Proc Natl Acad Sci USA 2005; 102:10165 - 10170
- Williams I, Richardson J, Starkey A, Stansfield I. Genome-wide prediction of stop codon readthrough during translation in the yeast Saccharomyces cerevisiae. Nucleic Acids Res 2004; 32:6605 - 6616
- Liang H, Cavalcanti AR, Landweber LF. Conservation of tandem stop codons in yeasts. Genome Biol 2005; 6:R31
- Namy O, Duchateau-Nguyen G, Rousset JP. Translational readthrough of the PDE2 stop codon modulates cAMP levels in Saccharomyces cerevisiae. Mol Microbiol 2002; 43:641 - 652
- Wilson MA, Meaux S, Parker R, vanHoof A. Genetic interactions between [PSI+] and nonstop mRNA decay affect phenotypic variation. Proc Natl Acad Sci USA 2005; 02:10244 - 10249
- Hattendorf DA, Lindquist SL. Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. EMBO J 2002; 21:12 - 21
- Lum R, Tkach JM, Vierling E, Glover JR. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem 2004; 279:29139 - 29146
- Tkach JM, Glover JR. Amino acid substitutions in the C-terminal AAA+ module of Hsp104 prevent substrate recognition by disrupting oligomerization and cause high temperature inactivation. J Biol Chem 2004; 279:35692 - 35701
- Hattendorf DA, Lindquist SL. Analysis of the AAA sensor-2 motif in the C-terminal ATPase domain of Hsp104 with a site-specific fluorescent probe of nucleotide binding. Proc Natl Acad Sci USA 2002; 99:2732 - 2737