Abstract
The term prion has been used to describe self-replicating protein conformations that can convert other protein molecules of the same primary structure into its prion conformation. Several different proteins have now been found to exist as prions in Saccharomyces cerevisiae. Surprisingly, these heterologous prion proteins have a strong influence on each others’ appearance and propagation, which may result from structural similarity between the prions. Both positive and negative effects of a prion on the de novo appearance of a heterologous prion have been observed in genetic studies. Other examples of reported interactions include mutual or unilateral inhibition and destabilization when two prions are present together in a single cell. In vitro work showing that one purified prion stimulates the conversion of a purified heterologous protein into a prion form, suggests that facilitation of de novo prion formation by heterologous prions in vivo is a result of a direct interaction between the prion proteins (a cross-seeding mechanism) and does not require other cellular components. However, other cellular structures, e.g., the cytoskeleton, may provide a scaffold for these interactions in vivo and chaperones can further facilitate or inhibit this process. Some negative prion-prion interactions may also occur via a direct interaction between the prion proteins. Another explanation is a competition between the prions for cellular factors involved in prion propagation or differential effects of chaperones stimulated by one prion on the heterologous prions.
Introduction
Originally, the term prion was coined to stress the “protein only” nature of an infectious agent causing transmissible spongiform encephalopathies in mammals.Citation1 All variations of this disease in humans and animals were linked to abnormal self-propagating conformations of just one cellular protein, PrP. The intriguing but controversial idea that a protein conformation could be infectious gained considerable support in 1994 when Reed Wickner showed that the prion model could explain the inheritance and behavior of the yeast [URE3] cytoplasmic factor and postulated that another yeast cytoplasmic factor, [PSI+], was also a prion.Citation2 Soon, the discovery that the prion form of [Het-s] has a defined cellular activity in the fungus Podospora anserina, indicated that prions could have functional roles,Citation3,Citation4 whereas the determination that the Hsp104 chaperone is required for the propagation of all known yeast prions showed that cellular machinery has a role in prion replication.Citation5–Citation8 Now the protein only nature of these fungal cytoplasmic determinants has been definitively proven by demonstrating the infectivity of prion-like particles made in vitro from respective recombinant proteins.Citation9–Citation12
The list of prions and the number of prion-carrying species has expanded, but Saccharomyces cerevisiae is the only species in which several prions have been identified: [PSI+], [URE3] and [PIN+] (also known as [RNQ+]). This makes yeast uniquely suited to study interactions between different prions. Here we review the experimental evidence for these interactions, consider models to explain their molecular basis and briefly discuss their functional and evolutionary significance.
Structural Similarity Between Prions Determines the Principle Types of Prion-Prion Interactions
The different proteins known to be capable of forming prions are not homologous. Although the prion forming domains of several prion proteins (Sup35, Ure2, Rnq1) are rich in glutamines (Q) and/or asparagines (N), other prion proteins (Het-s, PrP) are not Q- or N-rich. When not in their prion conformations, these proteins perform very different jobs in the cell. For example, the [URE3]-forming protein, Ure2, regulates the uptake of nitrogen (reviewed in ref. Citation13). Sup35, which can convert into the [PSI+] prion, is normally a GTPase subunit of the translational termination factor responsible for the release of nascent protein chains from the ribosome (reviewed in ref. Citation14). Het-S/[Het-s] is a determinant for heterokaryon incompatibility (reviewed in ref. Citation15) and selective spore killing in Podospora anserina.Citation16 These functions are clearly distinct from the roles assigned to cellular PrP in the central nervous system (see refs. Citation17 and Citation18 and references therein).
However, despite the dissimilarity of their building blocks, the above mentioned prions are strikingly alike (reviewed in ref. Citation19). Their prion aggregates are β-sheet-rich, i.e., prion formation is associated with an increase of β-sheet content relative to non-prion states of the same protein. The joining of new molecules occurs through the formation of inter-molecular interactions between β-strands. Consequently, unlike disordered amorphous aggregates that are generally only held together by hydrophobic interactions, prion aggregates are highly ordered. This explains the poor solubility of prion aggregates in detergents and their resistance to digestion with proteases. Whereas the detailed in vivo architecture of prion aggregates is unknown, the overall arrangement of proteins in prion aggregates strongly resembles that found in amyloid fibers. Indeed, prions bind the Thioflavin T and Congo Red dyes the same way other amyloids do.Citation20,Citation21 It has also been established that larger aggregates, which can be detected by gradient centrifugation and even light microscopy, are composed of smaller SDS-resistant subparticles.Citation22,Citation23 These subparticles could correspond to units of prion propagation and are sometimes referred to as seeds.
In vitro, prion-forming proteins assemble into typical amyloid fibers, a process which can be self-seeded by preformed fibers of the same protein and by extracts of cells bearing the respective prion.Citation8,Citation24–Citation30 Also, both PrP and Sup35 have been shown to form oligomers that have a common structure and that precede the formation of amyloid fibers.Citation31,Citation32 These oligomers are recognized by an antibody that reacts with similar oligomers formed by a wide variety of amyloid-forming proteins including Aβ and polyglutamine.Citation31
Therefore, prions are amyloid aggregates that are heritable and transmissible. Specifically for yeast, this implies that prion-forming proteins have a propensity to form amyloid aggregates that can be partitioned and transmitted during cell divisions. Below we consider how the common properties of these prions may determine the framework and nature of prion-prion interactions.
Direct interaction.
Firstly, since prions are amyloids, it is possible that molecules of one prionogenic protein could join a heterologous prion aggregate. Indeed, the basic structure of all amyloid aggregates is the same (reviewed in ref. Citation33): a ladder of β-strands oriented perpendicular to the fibril axis that may be organized in β-sheets, β-helices, β-nanotubes, etc. The bonds in the β-strands are formed between the peptide backbones, not between the side chains. Thus, prionogenic proteins, prone to form β-strands with amyloids of the same primary sequence, might occasionally attempt to form a β-strand with heterologous amyloids, as long as the side chains do not interfere with the backbone interactions. This would likely block the growth of the fiber to which such a heterologous “cap” was attached, but could simultaneously lead to the de novo formation of a new prion by a cross-seeding mechanism.
Competition for cellular factors.
The critical steps in prion biogenesis and propagation are the same for different amyloidogenic prions. Initially several functional cellular proteins form a prion nucleus or seed. The seed then grows by the addition of new molecules. Finally, the large prion aggregate is broken to create new seeds. At each of these steps the sets of cellular factors that inhibit and promote the formation and propagation of various prions are likely to overlap considerably (reviewed in refs. Citation34 and Citation35) and competition for such factors between heterologous prions is likely. The effects of such competitions might depend upon the stage at which a particular factor affects prion formation and the particular role the factor plays. For example, chaperones that promote the proper folding of nascent polypeptides by eliminating misfolded or partially folded prionogenic intermediates might interfere with the biogenesis of prions. The titration of such factors could promote prion formation. On the other hand, chaperones that break amyloid aggregates into smaller seeds are expected to promote prion propagation. The titration of the chaperones could decrease prion stability. Also, because different prions respond differently to the depletion of various cellular factors,Citation36 their effects on each other may not be reciprocal.
Cellular response to the presence of prions.
Finally, the cell may mount a response to the presence of amyloid or amyloid precursors of prions.Citation36,Citation37 This response is expected to be aimed at prion disaggregation and elimination but may actually facilitate prion transmission, e.g., by breaking prion aggregates into smaller seeds. Such a generic response would affect not only the prion protein that induced it, but other, heterologous prions and prionogenic proteins.
In the following sections we describe known examples, where different prions affect de novo formation, maintenance and phenotypic expression of each other.
Prions Facilitate the de novo Appearance of Heterologous Prions
[PIN+]: The first example of prion-prion interactions.
The idea that the de novo formation of prions is facilitated by preexisting heterologous prion aggregates is linked to the discovery of [PIN+].Citation6,Citation38,Citation39
[PIN+]: A prion interacting with [PSI+]. As prions are alternative self-propagating conformations of cellular proteins, they should occasionally appear in cell populations, and the spontaneous loss or curing of a prion should not preclude the possibility of its reappearance.Citation2,Citation40 Indeed, the rare spontaneous appearance of [URE3] was demonstrated both in [ure0] strains and in derivatives of [URE3] strains treated with the most conventional prion-curing agent, guanidine hydrochloride (GuHCl).Citation2,Citation41 Spontaneous de novo appearance of [PSI+] was also reportedCitation42 but evidence for its reappearance after curing with GuHCl remained controversial.Citation42,Citation43
The issue of the reappearance of [PSI+] after its curing with GuHCl was revisited in the experimental system where the de novo formation of [PSI+] could be induced 100-fold or more by a 5- to 10-fold overproduction of the Sup35 protein.Citation44,Citation45 Such an increase in the rate of [PSI+] prion formation upon overproduction of the prion-forming protein is predicted by the prion model because the accumulation of Sup35 molecules means there is a greater chance that a group of them will misfold and form a prion nucleus.Citation2,Citation40 Also, the dramatic increase in [PSI+] formation can be attributed to the overloading of the protein folding machinery and the imbalance of overexpressed Sup35 relative to its normal binding partners.Citation46,Citation47 To our surprise we eventually established that [PSI+] can be induced (or reappear spontaneously) only in a fraction of GuHCl-cured [psi-] derivatives.Citation6 The proportion of nonrevertible clones increased with increasing GuHCl treatment, but the efficiency of [PSI+] induction in the clones that remained inducible was unchanged. Thus a GuHCl-curable factor was required for [PSI+] formation. The factor was named [PIN+] for [PSI+] induction.Citation6
Genetic analysis of the Pin+ phenotype established that [PIN+] is inherited in a nonmendelian fashion. Diploids resulting from crosses of [PIN+] and [pin-] derivatives were always [PIN+], and in meiosis the segregation was 4 [PIN+]: 0 [pin-].Citation6 Also, [PIN+] was efficiently transmitted by cytoduction,Citation38 an abortive mating where cytoplasms of two cells blend but their nuclei do not fuse resulting in heterokaryons, which bud off haploids with single unmodified parental nuclei and mixed cytoplasm.Citation48 Such inheritance is possible for (1) genetic determinants located on nonnuclear DNA or RNA molecules (plasmids, viruses or mitochondrial DNA) or (2) for prions.Citation2
Elimination of [PIN+] on GuHCl-containing media was compatible with both possibilities since GuHCl both cures prions and induces large deletions in mitochondrial DNA.Citation49,Citation50 However, no link between [PIN+] and mitochondrial DNA was detected.Citation39 Also, [PIN+] reappeared spontaneously following GuHCl curing,Citation38,Citation39 whereas a [pin-] caused by a large deletion would probably not be revertible. On the other hand, disruption of the gene encoding the Hsp104 chaperone previously reported to cure [PSI+] and [URE3]Citation5,Citation7 also eliminated [PIN+].Citation6 Thus, [PIN+] was hypothesized to be a prion, and the effect of [PIN+] on the appearance lof [PSI+] became the first example of a prion-prion interaction.
[PIN+] interacts with [PSI+] at the step of [PSI+] formation. Although essential for the appearance of [PSI+], [PIN+] is dispensable for [PSI+] maintenance: [PSI+][pin-] derivatives (along with [psi-][PIN+] and [psi-][pin-] derivatives) were obtained by growing [PSI+][PIN+] cultures on GuHCl media.Citation39 Furthermore, the loss of [PIN+] generally had no effect on [PSI+] properties such as the size of [PSI+] prion subparticlesCitation23 or the degree of Sup35 aggregation.Citation39 Consequently, the [PSI+] phenotype of nonsense suppression was unaffected.Citation39 This phenotype is caused by depletion of soluble Sup35 available for the termination of translation and is routinely analyzed using the ade1-14 (UGA) or ade2-1 (UAA) reporters with a premature stop codon.Citation43,Citation51 [PSI+] toxicity upon Sup35 overproduction,Citation52,Citation53 which may result from impairment of the cytoskeleton,Citation54 was also unaffected by [PIN+].Citation6 Only the inhibition of growth in [psi-] upon high-level Sup35 overexpression required the presence of [PIN+], but this phenotype is attributed to the induction of [PSI+].Citation6 Taken together these data indicate that [PIN+] specifically facilitates the de novo formation of [PSI+].
Rnq1 is the [PIN+] prion protein. The prion model postulates three approaches for identifying a prion protein: (1) disruption of the gene encoding the prion protein should eliminate the respective prion; (2) overexpression of the prion protein gene should promote de novo prion formation and (3) prion formation should inhibit the normal function of the prion-forming protein.Citation40
Rnq1 was identified as the [PIN+] prion protein using a candidate approach. Since disruption of the gene encoding the [PIN+] protein should cure cells of [PIN+], genes for all known and a few candidate prions were disrupted in a [PIN+] strain. Strains with ure2Δ, new1Δ, pin2Δ and sup35-NMΔ (the latter lacking the prion domain of the otherwise essential SUP35 gene) remained [PIN+].Citation6,Citation38 However disruption of the RNQ1 gene caused the [PIN] strain to become Pin- and all attempts to cytoduce [PIN+] from the rnq1Δ background into a wild-type RNQ1 strain were unsuccessful.Citation38 Newly appearing spontaneous [PIN+]s also could be cytoduced into a wild-type RNQ1 strain but not into rnq1Δ.Citation38 Furthermore, the presence of [PIN+] correlated with an aggregated state of the Rnq1 protein. In [PIN+] strains Rnq1 was insoluble; it became soluble in [pin-] clones obtained on GuHCl and converted back to an aggregated state following spontaneous [PIN+] reappearance.
RNQ1 encoded a prion previously identified in laboratory yeast strainsCitation8 and recently found in wild-type yeast.Citation55,Citation56 It was discovered by Sondheimer and Lindquist,Citation8 who noted a striking similarity between the C-terminal domain of Rnq1 and the prion domain of Sup35. The similarity was manifested by an extremely high frequency of Q/N residues, which gave the gene its name (rich in N and Q). This C-terminal region was sufficient to maintain the prion (aggregated) state of Rnq1 and was thus confirmed to be the prion domain of [RNQ+].Citation8 The same prion was also sufficient for the propagation of [PIN+].Citation38 Thus, [RNQ+] = [PIN+].
A broader ramification of the [PIN+] phenomenon: Various heterologous prions affect each other's appearance.
Other prions also have Pin+ activity. Another approach to identifying the [PIN+] prion protein led to a very different conclusion. Since, according to the prion model, overexpression of the [PIN+] protein gene would promote the de novo formation of [PIN+], a multicopy library of yeast genomic DNA was introduced into a [pin-] derivative to screen for yeast genes that, upon overexpression, caused cells to become Pin+ (i.e., inducible to [PSI+]). Unexpectedly, the screen did not uncover RNQ1 but instead yielded 11 genes encoding functionally unrelated proteins with domains unusually rich in Q/N.Citation38 Among them were URE2 and NEW1, the genes for a well-known prion [URE3] and an artificial prion [NU+], both of which had already been eliminated as the [PIN+] protein encoding genes in the candidate approach (see above). The ability of NEW1 overexpression to make cells Pin+ was simultaneously reported in a separate study.Citation57 Furthermore, the Pin+ phenotype of facilitating the de novo formation of [PSI+] was not only caused by overexpression of Ure2 and New1, but also by the presence of the [URE3] or [NU+] prions.Citation38,Citation57 Thus, [URE3] and [NU+] give cells a Pin+ phenotype. Indeed, to date there is no known yeast prion that does not make cells Pin+.
Positive effects of [URE3], [PSI+] and [PIN+] on each other's appearance. [PIN+] itself is strongly dependant upon other prions for its formation. Indeed, although [PIN+] can appear spontaneously in cultures devoid of any known prions, this is an extremely rare event.Citation39 Also, in the absence of other prions the frequency of fluorescent aggregates caused by overexpression of an Rnq1-GFP fusion construct (indicative of prion formation) was only ∼0.1%, even after prolonged overexpression of the construct. However, the presence of [URE3] or [PSI+] led to up to a 100-fold increase in the formation of bright fluorescent Rnq1-GFP foci.Citation38 Thus, poor induction of [PIN+] by RNQ1 overexpression in prion-less strains explains why RNQ1 was not identified in the library screen for the [PIN+] maintenance gene described above.
[URE3] is similar to [PIN+] in its ability to form in GuHCl-cured cultures devoid of any known prions. And, as in the case of [PIN+], the frequency of [URE3] appearance in such cultures is extremely low: ∼10-5 spontaneously and ∼10-3 following Ure2 overexpression. [PIN+] increases the induction of [URE3] 5 to 500-fold.Citation58
Negative effects of [URE3] and [PSI+] on each other's appearance. While it is clear from the data discussed above that the de novo appearance of prions other than [PSI+] can be promoted by the presence of heterologous prions, this is not always the case. Two groups found that [PSI+] did not promote [URE3] formation but rather inhibited it.Citation36,Citation58 Furthermore, in one study [URE3] had a negative effect on the induction of [PSI+],Citation36 rather than the positive one (described aboveCitation38). Since Bradley et al.Citation58 detected a positive effect of [PIN+] and negative effect of [PSI+] on appearance of [URE3] in the same genetic background, their result cannot be explained by a genomic mutation interfering with all positive prion-prion interactions. It is possible that for some pairs of prions the effect of heterologous facilitation is simply not reciprocal and that [PSI+] never promotes the appearance of [URE3]. On the other hand, it is possible that a particular variant of [PSI+] chosen for these experiments was incapable of promoting the de novo formation of [URE3]. The latter explanation is also compatible with detection of both negative and positive effects of [URE3] on [PSI+].
Conformational variants of prions differ in their propensity to induce heterologous prions. Prions resulting from independent conversions of the same soluble proteins into amyloid aggregates are not all identical. For most prionogenic proteins there is an array of prion states called prion strains, or variants.Citation45,Citation58–Citation60 Prion variants are due to differences in the conformation of prion aggregates (reviewed in ref. Citation19). Apparently, different lengths and arrangements of β-strands and/or different mutual orientations of β-sheets in the amyloid structure,Citation61–Citation64 affect the rate of aggregation and the resistance of amyloid aggregates to fragmentation and, consequently, result in different prion seed sizes.Citation19,Citation22,Citation23
Strong [PSI+] variants have smaller prion subparticles than weak [PSI+] variants but cause more aggregation of Sup35 and therefore cause more efficient nonsense suppression.Citation22,Citation45,Citation65 This suggests that there is a larger number of strong [PSI+] seeds relative to weak [PSI+] seeds per cell. When the effects of strong and weak [PSI+]s on the prionization of Rnq1 were compared, a strong [PSI+] variant caused the appearance of approximately two-fold more Rnq1-GFP foci than a weak [PSI+] variant following either short or prolonged overexpression of Rnq1-GFP.Citation38 However, because only two [PSI+] variants were compared, the correlation between the strength of [PSI+] and its ability to induce [PIN+] cannot be established.
Five [PIN+] variants that differed in the degree of Rnq1 aggregation also had different propensities to promote [PSI+] inductionCitation58 (reviewed in ref. Citation66). There was no correlation between the frequency of [PSI+] induction and the degree of Rnq1 aggregation or the size of [PIN+] subparticles. The best [PSI+] induction was detected in the derivative with the most soluble Rnq1, whereas the derivative with the least soluble Rnq1 was ranked second in the [PSI+] induction test.Citation58 Furthermore, when these [PIN+] variants were screened for their ability to facilitate [URE3] formation, there was no reproducible difference between them, whereas yet another [PIN+] variant from the Wickner lab collection promoted [URE3] appearance much more efficiently.Citation58 Thus, prion variants facilitate the induction of heterologous prions to different extents, and this ability is apparently determined by their conformational differences.
Interactions with non-prion amyloids.
Interactions between polyQ amyloids and prions. Since the prion domains of [PIN+], [PSI+], [URE3] and [NU+] are Q/N-rich and all the proteins identified in the genetic screen described above also contain Q/N-rich domains, it seemed likely that their Pin+ activity resulted from the presence of Q/N-rich sequences. Another type of Q-rich sequences, uninterrupted polyQ stretches, is found in many proteins. When expanded beyond an acceptable limit, polyQ stretches are prone to form β-sheet-rich amyloid aggregates, which are associated with several human diseases including Huntington's disease and MJD. Several groups utilized constructs expressing polyQ-expanded fragments of huntingtin (Ht) and MJD for the analysis of interaction between yeast prions and aggregation-prone polyQ.
As expected, overexpression of constructs carrying the first exon of Ht with expanded polyQ, rendered a fraction of [pin-] cells phenotypically Pin+, and the ability of Ht constructs to promote [PSI+] induction correlated with their ability to aggregate.Citation67 In a reverse experiment [PIN+] and [NU+], but not [PSI+], promoted aggregation of polyQ-expanded MJD.Citation57 Also, [PIN+] was shown to promote aggregation and toxicity of the polyQ-expanded Ht fragments,Citation68 and recent evidence suggests that [PSI+] has the same effect on the Ht toxicity as [PIN+].Citation69 Thus, non-prion polyQ amyloids can engage in interactions with prion proteins at the step of de novo amyloid formation. The fact that MJD aggregation was not stimulated by [PSI+],Citation57 confirms that such interactions are not obligatory and may be nonreciprocal.
Interactions between nonpolyQ amyloids and prions. The question about interactions between Q/N-rich and non-Q/N-rich prions remains open. Perutz explained the high propensity of Q-, N- and Q/N-rich sequences to form amyloid by the involvement of Q and N residues in the stabilization of β-strands.Citation70,Citation71 One possibility is that there are interactions that are specific to Q/N-rich amyloids. Indeed, only Q/N rich proteins were identified as facilitators of the appearance of [PSI+] in the genetic screen (see above). Also, the formation of [PSI+] was not facilitated by non-Q- or N-rich aggregates of the normal and disease variants of the amyloidogenic proteins transthyretin, α-synuclein and synphilin-1.Citation67 However, it should be recalled that even in the case of the Q/N-rich amyloids, not every pair of amyloids can seed each other. Thus it is possible that most amyloidogenic and prionogenic proteins have Q/N-rich domains, and interactions with less common non-polyQ amyloids will be eventually established.
Interactions at the step of the de novo prion formation can be reproduced in vitro.
The assembly of prion proteins into amyloid fibers in vitro is fundamentally similar to prion formation and growth in vivo. Following a lag period, a pure recombinant protein can form fibers in vitro. The lag period can be reduced or eliminated by the addition of preformed fibers made of this protein or by adding extracts from cells bearing a prion form of this protein.Citation8,Citation24–Citation30 Furthermore, amyloid prepared in vitro from pure recombinant proteins can efficiently transform live cells to the prion state.Citation9–Citation12,Citation72,Citation73 Most phenomena associated with prion biogenesis and propagation, including the effects of chaperonesCitation32,Citation74–Citation76 and the existence of strain phenomenon and species barriersCitation62,Citation77–Citation82 have been modeled in vitro using purified recombinant prionogenic proteins and cellular factors.
In the two-step process of amyloid formation in vitro, the lag phase before the ThT or Congo Red-binding fibers are detected is equivalent to the step of the de novo prion formation in vivo. Subsequent rapid growth of fibers models the joining of existing prion aggregates by newly synthesized protein molecules. Accordingly, events equivalent to the prion-prion interactions at the step of the de novo formation should affect the length of the lag. Indeed, addition of preformed Rnq1 fibers to soluble Sup35 shortened the lag before Sup35 amyloid could be detected.Citation67 The effect of Rnq1 fibers was very modest relative to the effect of the addition of homologous, Sup35 fibers, which is consistent with the ability of [PIN+] to promote [PSI+] appearance in only a fraction of the cells in the culture. As recombinant purified Rnq1 and Sup35 were used in these experiments, this result indicates a direct interaction between preexisting prion aggregates and a heterologous prionogenic protein. The non-polyQ amyloids that didn't facilitate [PSI+] formation in vivo also didn't promote Sup35 formation in vitro, which further validates the use of the in vitro model for studies of prion-prion interactions. Interestingly, two non-polyQ amyloids, bovine pancreas insulin and human Ig light-chain amyloid, did stimulate Sup35 conversion in vitro.Citation67 Although these amyloids were not tested in vivo, this finding suggests that the presence of a QN-rich domain is not an absolute requirement for such interactions.
Prions Interfere with Propagation of Heterologous Prions
There are only a few examples of prion-prion interactions that affect the propagation of prions.
[URE3] and [PSI+] negatively affect each other's propagation.
Schwimmer and MasisonCitation36 showed that, at least in the [PIN+] background they used, there are antagonistic interactions between [URE3] and [PSI+]. A negative effect of [URE3] on [PSI+] was detected in an ade2-1 (UAA) SUQ5 strain,Citation51 where [PSI+] restores the red color caused by the ade2-1 mutation to pink or white, depending upon the efficiency of nonsense suppression. [URE3] reduced the suppressor phenotype associated with [PSI+]: [URE3][PSI+] cultures were pink, whereas isogenic [ure0][PSI+] derivatives carrying the same [PSI+] variant formed white colonies. The level of Sup35 aggregation in [URE3][PSI+] cells was also lower than in [ure0][PSI+], which confirmed that [URE3] indeed inhibited [PSI+] aggregation and did not interfere with the termination of translation in some other way. Furthermore, the inhibitory effect of [URE3] toward [PSI+] increased when cells were grown on media selective for [URE3], although even under these conditions [PSI+] remained mitotically stable. [PSI+] exerted a reciprocal inhibitory effect on [URE3], which was detected as reduced de-repression of the DAL5 promoter, due to the appearance of functional Ure2.Citation36 The impairment of [URE3] propagation by [PSI+] was also apparent from the loss of [URE3] in 1% of the mitotically growing [PSI+] derivatives.
Incompatible variants of [PIN+] and [PSI+].
The next example of antagonistic prion-prion interactions came as a surprise. The [PIN+] variant, which was originally detected in a laboratory strainCitation6 and established as a prion essential for the de novo formation of [PSI+], had already been shown not to affect [PSI+] propagation.Citation38 However, Bradley and LiebmanCitation83 found that variants of [PIN+] that appeared spontaneously following prolonged storage of GuHCl-cured [pin-] derivativesCitation38 and were classified as low, medium and very high according to their ability to facilitate [PSI+] formation,Citation58,Citation66 destabilized weak variants of [PSI+]. When weak [PSI+] was introduced into these [PIN+] derivatives by mating or cytoduction, approximately 50% of the cells in the resulting [PIN+] colonies were [psi-].Citation83 Strikingly, these “destabilizing” [PIN+] variants even inhibited the propagation of the same weak [PSI+] variants whose appearance they promoted at the step of induction.
As in the example described for [PSI+] and [URE3] in the previous section, the antagonism of weak [PSI+] and medium destabilizing [PIN+] was reciprocal. This [PIN+] frequently disappeared following the induction of weak [PSI+] and if it was not lost right away, the [PIN+][unstable weak PSI+] derivative soon segregated into two derivatives, either [PIN+][psi-] or [pin-][stable weak PSI+]. On the contrary, low destabilizing [PIN+] was not lost upon [PSI+] induction and clearly won the competition with [PSI+] thereafter. Thus, in the case of antagonistic prion-prion interactions, either one of the prions is eliminated from the population, or the population segregates into derivatives bearing different sets of prions.
How Might Heterologous Prionogenic Proteins Interact and What Might be the Consequences?
Direct interaction of prion aggregates with heterologous prionogenic proteins.
Cross-seeding model. The cross-seeding model () explains the genetic phenomenon of [PIN+] and is compatible with data on the facilitation of the de novo appearance of prions by preexisting heterologous prions or amyloids.Citation38,Citation57 The model postulates that a preexisting aggregate can seed the formation of a new prion by providing a nidus for the assembly and/or conformational conversion during the early stages of prion biogenesis. For example, [PIN+] aggregates are proposed to be used as the site of initial Sup35 assembly and conversion into [PSI+].
A critical argument in favor of this model is the facilitation of the formation of Sup35 amyloid upon the addition of preformed Rnq1 fibers (see above).Citation67 This in vitro result, obtained using pure recombinant proteins, suggests a direct interaction between Rnq1 and Sup35, unmediated by other cellular factors.
The seeding model does not require a permanent interaction between the heterologous amyloids. Indeed, while the growth of amyloid aggregates by the addition of homologous proteins is a very efficient process,Citation84 the binding of a heterologous protein should occur much less frequently and should engage only a small fraction of prion propagons in the cell. Thus, the findings that in a [PIN+][psi-] strain, Sup35 remained monomeric and was not detected within Rnq1 prion subparticles, and that in a [PIN+][PSI+] strain the subparticles of Rnq1 and Sup35 were not intermixedCitation23 do not contradict the model.
The recent detection of mixed aggregates during [PSI+] induction supports the model: when Sup35 was overexpressed in a [PIN+][psi-] derivative, newly forming detergent-insoluble Sup35 aggregates contained some Rnq1.Citation85 Also, using Rnq1 and Sup35 respectively labeled with yellow and cyan fluorescent proteins, we established that all newly forming [PSI+] aggregates partially or completely co-localize with preexisting [PIN+] aggregates.Citation67 This co-localization is hypothesized to be a consequence of the seeding event. Indeed, even though similar co-localization was also detected in [PIN+][PSI+] derivatives propagating two established prions, not all aggregates showed co-localization in these cultures.Citation67 This, latter, finding is compatible with the idea of occasional heterologous interactions between established prions, but can also be explained by the occasional lateral attachment of large amyloid structures or the co-compartmentalization of amyloids.
The fact that mutual induction is detected between proteins with similar Q/N-rich prion domains is compatible with the model. Although amyloid structure is determined by bonds formed by peptide backbones, side chains can either strengthen or weaken the structure.Citation33 One possibility is that Q and N residues form a hydrogen bond “polar zipper” that strengthens the β-spine.Citation71,Citation86 Alternatively, the critical issue could be the interdigitation of side chains in the β-rich structure stabilizing it via hydrophobic interactions and allowing Van der Waals attractions favorable for amyloid formation to be maximized, while avoiding electrostatic repulsions.Citation71,Citation87 When a short peptide derived from the Sup35 prion domain was used to model the structure of the cross-α-spine, Lipfert et al.Citation88 found support for the polar zipper formation, whereas Nelson et al.Citation63 predicted that Q and N side-chains were interdigitated to form a dry, tightly self-complementing steric zipper between two α-sheets. Whether prion amyloids are composed of polar or steric zippers, the positions of specific Q or N residues may be critical for efficient cross-seeding. Indeed, a single substitution in the Sup35 prion domain was reported to block its being seeded with wild-type Sup35 amyloid without seriously interfering with the ability of the mutant to form amyloid in vitro in the absence of seed.Citation89
Another line of support for the cross-seeding model comes from examples of interactions between non-prion amyloids. The mysterious amyloid enhancing factor (AEF), that reduces the time before amyloid protein A (AA) amyloidosis onset in chronic inflammation models, appears to be equivalent to fragments of amyloid fibrils. Strikingly, not only AA fibrilsCitation90,Citation91 but also fibrils of various human amyloids including transthyretin and islet amyloid polypeptide (IAPP),Citation92,Citation93 Sup35,Citation94 or synthetic silk-derived fibrils,Citation95 had this effect. Overall, AA seeding appears to be rather nonspecific. On the contrary, the study of the specificity of seeding of Aβ fibers revealed that only IAPP was an efficient heterologous seeder for this peptide.Citation96 Strikingly, the authors noted the high similarity of the primary structures of Aβ and IAPP amyloidogenic peptides of otherwise nonhomologous proteins and attributed the cross-seeding to this feature.
Capping model. Similar interactions between heterologous proteins could also lead to the curing of a preexisting prion by a heterologous prionogenic protein (). Indeed, binding of a heterologous protein to the growing tip of a prion aggregate could block its rapid growth. Formation of such “caps” on a considerable fraction of prion particles, may lead to a notable reduction in the prion-associated phenotypes and to inefficient transmission to daughter cells.
A similar model was proposed to explain the Pnm ([PSI+] no more) phenotype of Sup35 mutants towards wild-type [PSI+]Citation89 and the curing of [URE3] upon overexpression of Ure2 fragments or GFP fusions from S. cerevisiae and other species.Citation97,Citation98 The authors hypothesized that mutants or fragments would join the growth tip but would not themselves provide a growth point and would thus poison the amyloid crystal. Likewise, the elimination of some [PIN+] variants upon overexpression of Sup35 and the induction of [PSI+] could be explained by this model. However, destabilization of weak [PSI+] by certain [PIN+] variants in the absence of any overexpression is not easily explained by a capping model.Citation83
Extending the seeding model: Role of chaperones and cytoskeleton. Although the in vitro evidence implies that the [PIN+] effect is not strictly chaperone mediated,Citation67 it is still possible that in vivo chaperones bound to preexisting prion aggregates, rather than [PIN+] per se, are mostly responsible for the effect on the de novo induction of [PSI+] ().Citation99
Also, newly appearing Sup35-GFP prion aggregates that appear in [PIN+] cells, with the characteristic ring/ribbon shape that is easily distinguishable from aggregates of “established” prions,Citation38,Citation100 have recently been reported to preferentially associate with actin patches.Citation54 It was thus hypothesized that actin patches provide a scaffold for the formation of large prion aggregates making these areas the sites of direct prion-prion interactions.Citation54
Prion interactions mediated by chaperones or other cellular factors.
Titration model. This model () was originally proposed to explain the [PIN+] phenomenon.Citation38,Citation57,Citation99 The model postulates that cellular factors responsible for the disassembly of aggregates and/or refolding of misfolded proteins are constantly working to dissolve both existing and newly appearing prions. Thus, in the prion-free cells such chaperones efficiently prevent the appearance of new prions (, top). On the contrary, in the presence of another prion, disaggregating factors are titrated away to work on this prion. This allows newly forming prions to escape the protein folding control machinery and achieve the stage of rapid propagation, which is resistant to chaperone curing.
While this model alone does not explain all of the experimental evidence for [PIN+] and especially the in vitro reproduction of this phenomenon (discussion in refs. Citation38, Citation67 and Citation99), the titration and the seeding models are not mutually exclusive. Indeed, the existence of a cellular factor with a weak inhibitory effect towards de novo aggregate formation was hypothesized by Uptain et al.Citation77 to explain their findings that extracts from [psi-][pin-] strains slightly inhibited Sup35 fiber formation in vitro. So far no such inhibitor has been identified. However, Sup35, and specifically [PSI+] aggregates, were shown to bind the Ssa and Ssb chaperones,Citation101 and Ssb1 is a known antagonist of de novo prion formation.Citation47 Also, Rnq1 in the [PIN+] state binds Sis1,Citation102,Citation103 and Ssa1 together with its cochaperones, Ydj1 or Sis1, has recently been shown to inhibit Sup35 fiber formation in vitro.Citation76
As for the antagonistic prion-prion interactions, it is possible that titration of cellular factors by one type of prion aggregate would compromise the propagation of a heterologous prion.
Cellular response model. Schwimmer and MasisonCitation36 proposed this model () to explain the antagonistic interactions of [PSI+] and [URE3]. They found that different prions affect the levels of different chaperones in distinct ways and showed a differential sensitivity of different prions to different chaperones. For example they found that the presence of [PSI+] (and to a lesser extent [URE3]) caused an increase in the expression of Ssa1. Because Ssa1 destabilizes [URE3] but not [PSI+], selective [URE3] destabilization was attributed to the increase in Ssa1 expression.Citation36,Citation37 The simultaneous presence of [PSI+] and [URE3] also resulted in a significant increase in Hsp104 levels, to which [PSI+] is sensitive and [URE3] is not.Citation5,Citation7 The latter could explain the reduction in [PSI+] phenotypic expression. It is also possible, that a chaperone imbalance triggers a more complex cascade of prion-chaperone interactions.Citation104–Citation106
A positive effect of a preexisting chaperone on the formation of another prion is also possible within the framework of this model. For example, excess Ssa could additionally facilitate [PSI+] induction in a [PIN+] background.Citation101
Since the discovery that the prion domains of several proteins have been retained for hundreds of millions of years,Citation107 the question of why these ostensibly functionless regions have been conserved has been raised. Evidence suggests that the unstable nature of [PSI+] and other prions may provide the organism with an evolutionary advantage, appearing when needed and disappearing when no longer advantageous.Citation108–Citation110 Indeed, the special advantage prions might offer generally comes with a price tag of a global effect (e.g., on translation termination in the case of Sup35), and the fact that [PSI+] and [URE3] are not found in the wild can be interpreted to mean that they are diseases.Citation56 One possibility is that prions are retained transiently, when their advantages outweigh their negative effects and until more specific mutations are selected for. In this case the possibility of seeding increases the chance of prion formation, and a benign prion, like [PIN+], which has been found in nonlaboratory yeast isolatesCitation56 would facilitate this process. Then antagonistic prion-prion interactions would facilitate the loss of prions that do not provide selective advantages.
Figures and Tables
Figure 1 Cross-seeding model for [PIN+]. [PIN+] aggregates are proposed to be used as the site of initial Sup35 assembly and conversion into [PSI+] (Rnq1, blue arrows; Sup35, green triangles).
![Figure 1 Cross-seeding model for [PIN+]. [PIN+] aggregates are proposed to be used as the site of initial Sup35 assembly and conversion into [PSI+] (Rnq1, blue arrows; Sup35, green triangles).](/cms/asset/ea743660-764f-4911-841a-6584aa95161e/kprn_a_10904837_f0001.gif)
Figure 2 Capping model. The binding of a heterologous protein (magenta diamonds) to the growing tip of a prion aggregate (blue arrows) could block its rapid growth and lead to its destabilization and loss. At the same time such binding can lead to the formation of a prion by the heterologous protein by a seeding mechanism.
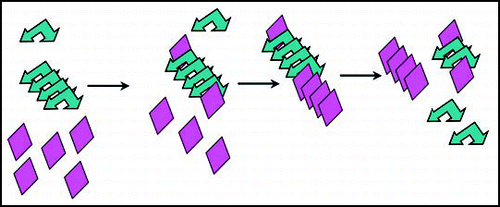
Figure 3 Chaperone-assisted seeding model for [PIN+]. Chaperones (orange sun shapes) bound to preexisting [PIN+] prion aggregates (blue arrows), could be responsible for the enhanced de novo aggregation of Sup35 (green triangles) and thus facilitate [PSI+] formation in vivo.
![Figure 3 Chaperone-assisted seeding model for [PIN+]. Chaperones (orange sun shapes) bound to preexisting [PIN+] prion aggregates (blue arrows), could be responsible for the enhanced de novo aggregation of Sup35 (green triangles) and thus facilitate [PSI+] formation in vivo.](/cms/asset/6ff1b909-100c-4e53-a710-69b2188e3eee/kprn_a_10904837_f0003.gif)
Figure 4 Titration model for [PIN+]. Top: in a [psi-] [pin-] cell, cellular factors (purple shape) keep Sup35 (green triangles) and Rnq1 (blue arrows) from aggregating. Bottom: in a [psi-] [PIN+] cells, much of the factor is bound to the [PIN+] aggregate, so less is available to keep Sup35 from aggregating.
![Figure 4 Titration model for [PIN+]. Top: in a [psi-] [pin-] cell, cellular factors (purple shape) keep Sup35 (green triangles) and Rnq1 (blue arrows) from aggregating. Bottom: in a [psi-] [PIN+] cells, much of the factor is bound to the [PIN+] aggregate, so less is available to keep Sup35 from aggregating.](/cms/asset/52629559-c68f-4031-8620-822aca35b83d/kprn_a_10904837_f0004.gif)
Figure 5 Cellular response model to explain why [PSI+] destabilizes [URE3]. Top: normal propagation of [URE3] (red pentagons). Bottom: [PSI+] (linked green triangles) induces the expression of the Ssa1 chaperone (light blue sickles), which destabilizes [URE3] but not [PSI+].
![Figure 5 Cellular response model to explain why [PSI+] destabilizes [URE3]. Top: normal propagation of [URE3] (red pentagons). Bottom: [PSI+] (linked green triangles) induces the expression of the Ssa1 chaperone (light blue sickles), which destabilizes [URE3] but not [PSI+].](/cms/asset/a1245f12-3858-4dab-9491-305486076c57/kprn_a_10904837_f0005.gif)
Acknowledgements
Work in the authors' laboratories was supported by National Science Foundation Grant 0518482 (Irina L. Derkatch) and National Institutes of Health Grant GM056350 (Susan W. Liebman). We thank Yakov Vitrenko, Vidhu Mathur, Andrew O'Dell, Michele Kadnar, Catherine Potenski and N. Kaye Horstman for helpful comments on the manuscript.
Notes
This manuscript has been previously published: Derkatch IL, Liebman, SW. Prion-Prion Interactions. In: Protein-Based Inheritence. Chernoff, Y ed. Austin and New York: Landes Bioscience and Kluwer Academic Press, 2007; 39–46.
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Wickner RB. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:566 - 569
- Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA 1997; 94:9773 - 9778
- Wickner RB. A new prion controls fungal cell fusion incompatibility. Proc Natl Acad Sci USA 1997; 94:10012 - 10014
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [PSI+]. Science 1995; 268:880 - 884
- Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997; 147:507 - 519
- Moriyama H, Edskes HK, Wickner RB. [URE3] prion propagation in Saccharomyces cerevisiae: Requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol 2000; 20:8916 - 8922
- Sondheimer N, Lindquist S. Rnq1: An epigenetic modifier of protein function in yeast. Mol Cell 2000; 5:163 - 172
- Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci USA 2002; 99:7402 - 7407
- King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature 2004; 428:319 - 323
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature 2004; 428:323 - 328
- Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: Amyloid of Ure2p is infectious. EMBO J 2005; 24:3082 - 3092
- Magasanik B. The transduction of the nitrogen regulation signal in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 2005; 2:16537 - 16538
- Inge-Vechtomov S, Zhouravleva G, Philippe M. Eukaryotic release factors (eRFs) history. Biol Cell 2003; 95:195 - 209
- Saupe SJ. Molecular genetics of heterokaryon incompatibility in filamentous ascomycetes. Microbiol Mol Biol Rev 2000; 64:489 - 502
- Dalstra HJ, van der Zee R, Swart K, Hoekstra RF, Saupe SJ, Debets AJ. Nonmendelian inheritance of the HET-s prion or HET-s prion domains determines the het-S spore killing system in Podospora anserina. Fungal Genet Biol 2005; 42:836 - 847
- Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci USA 2006; 10:3416 - 3421
- Zhang CC, Steele AD, Lindquist S, Lodish HF. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc Natl Acad Sci USA 2006; 103:2184 - 2189
- Chien P, Weissman JS, DePace AH. Emerging principles of conformation-based prion inheritance. Annu Rev Biochem 2004; 73:617 - 656
- Bendheim PE, Barry RA, DeArmond SJ, Sites DP, Prusiner SB. Antibodies to a scrapie prion protein. Nature 1984; 310:418 - 421
- Kimura Y, Koitabashi S, Fujita T. Analysis of yeast prion aggregates with amyloid-staining compound in vivo. Cell Struct Funct 2003; 28:187 - 193
- Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem 2003; 278:49636 - 49643
- Bagriantsev SN, Liebman SW. Specificity of prion assembly in vivo: [PSI+] and [PIN+] form separate structures in yeast. J Biol Chem 2004; 279:51042 - 51048
- Baskakov IV. Autocatalytic conversion of recombinant prion proteins displays a species barrier. J Biol Chem 2004; 279:7671 - 7677
- Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ. Lindquist, Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997; 89:811 - 819
- Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 1999; 283:1339 - 1343
- Thual C, Komar AA, Bousset L, Fernandez-Bellot E, Cullin C, Melki R. Structural characterization of Saccharomyces cerevisiae prion-like protein Ure2. J Biol Chem 1999; 274:13666 - 13674
- Schlumpberger M, Wille H, Baldwin MA, Butler DA, Herskowitz I, Prusiner SB. The prion domain of yeast Ure2p induces autocatalytic formation of amyloid fibers by a recombinant fusion protein. Protein Sci 2000; 9:440 - 451
- Dos Reis S, Coulary-Salin B, Forge V, Lascu I, Begueret J, Saupe SJ. The HET-s prion protein of the filamentous fungus Podospora anserina aggregates in vitro into amyloid-like fibrils. J Biol Chem 2002; 277:5703 - 5706
- King CY, Tittmann P, Gross H, Gebert R, Aebi M, Wuthrich K. Prion-inducing domain 2–114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci USA 1997; 94:6618 - 6622
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Hall JE, Glave CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300:486 - 489
- Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 2004; 304:1793 - 1797
- Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol 2004; 15:3 - 16
- Chernoff YO. Amyloidogenic domains, prions and structural inheritance: Rudiments of early life or recent acquisition?. Curr Opin Chem Biol 2004; 8:665 - 671
- Jones GW, Tuite MF. Chaperoning prions: The cellular machinery for propagating an infectious protein?. Bioessays 2005; 27:823 - 832
- Schwimmer C, Masison DC. Antagonistic interactions between yeast [PSI+] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol Cell Biol 2002; 22:3590 - 3598
- Jung G, Jones G, Wegrzyn RD, Masison DC. A role for cytosolic hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress. Genetics 2000; 156:559 - 570
- Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: The story of [PIN+]. Cell 2001; 106:171 - 182
- Derkatch IL, Bradley ME, Masse SV, Zadorsky SP, Polozkov GV, Inge-Vechtomov SG, Liebman SW. Dependence and independence of [PSI+] and [PIN+]: A two-prion system in yeast?. EMBO J 2000; 19:1942 - 1952
- Wickner RB. Prions of yeast and heat-shock protein 104: ‘Coprion’ and cure. Trends Microbiol 1995; 3:367 - 369
- Aigle M, Lacroute F. Genetical aspects of [URE3], a nonmitochondrial, cytoplasmically inherited mutation in yeast. Mol Gen Genet 1975; 136:327 - 335
- Lund PM, Cox BS. Reversion analysis of [psi-] mutations in Saccharomyces cerevisiae. Genet Res 1981; 37:173 - 182
- Tikhodeev ON, Gwetmanova EV, Tikhomirova VL, et al. Shestakov SV, Tarasov VA. Ambiguity of translation in yeast. Molecular Mechanisms of Genetic Processes 1990; Moscow Nauka 218 - 228
- Chernoff YO, Derkach IL, Inge-Vechtomov SG. Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr Genet 1993; 24:268 - 270
- Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996; 144:1375 - 1386
- Derkatch IL, Bradley ME, Liebman SW. Overexpression of the SUP45 gene encoding a Sup35p-binding protein inhibits the induction of the de novo appearance of the [PSI+] prion. Proc Natl Acad Sci USA 1998; 95:2400 - 2405
- Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD. Evidence for a protein mutator in yeast: Role of the Hsp70-related chaperone ssb in formation, stability, and toxicity of the [PSI] prion. Mol Cell Biol 1999; 19:8103 - 8112
- Conde J, Fink GR. A mutant of Saccharomyces cerevisiae defective for nuclear fusion. Proc Natl Acad Sci USA 1976; 73:3651 - 3655
- Tuite MF, Mundy CR, Cox BS. Agents that cause a high frequency of genetic change from [PSI+] to [psi-] in Saccharomyces cerevisiae. Genetics 1981; 98:691 - 711
- Juliani MH, Da Costa SO. Induction of rho- mutants in Saccharomyces cerevisiae by guanidine hydrochloride. II. Conditions that prevent rho- induction. Mutat Res 1975; 30:335 - 342
- Cox BS. Psi, a cytoplasmic suppressor of super-supression in yeasts. Heredity 1965; 20:505 - 521
- Dagkesamanskaya AR, Ter-Avanesyan MD. Interaction of the yeast omnipotent suppressors SUP1 (SUP45) and SUP2 (SUP35) with nonmendelian factors. Genetics 1991; 128:513 - 520
- Chernoff YO, Inge-Vechtomov SG, Derkach IL, Ptyushkina MV, Tarunina OV, Dagkesamanskaya AR, Ter-Avanesyan MD. Dosage-dependent translational suppression in yeast Saccharomyces cerevisiae. Yeast 1992; 8:489 - 499
- Ganusova EE, Ozolins LN, Bhagat S, Newnam GP, Wegrzyn RD, Sherman MY, Shernoff YO. Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol Cell Biol 2006; 26:617 - 629
- Resende CG, Outeiro TF, Sands L, Lindquist S, Tuite MF. Prion protein gene polymorphisms in Saccharomyces cerevisiae. Mol Microbiol 2003; 49:1005 - 1017
- Nakayashiki T, Kurtzman CP, Edskes HK, Wichner RB. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci USA 2005; 102:10575 - 10580
- Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell 2001; 106:183 - 194
- Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion “strains” in yeast. Proc Natl Acad Sci USA 2002; 99:16392 - 16399
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 1968; 78:301 - 311
- Schlumpberger M, Prusiner SB, Herskowitz I. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol 2001; 21:7035 - 7046
- Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 2005; 435:765 - 772
- Tanaka M, Chien P, Yonekura K, Weissman JS. Mechanism of cross-species prion transmission: An infectious conformation compatible with two highly divergent yeast prion proteins. Cell 2005; 121:49 - 62
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005; 435:773 - 778
- Liebman SW. Structural clues to prion mysteries. Nat Struct Mol Biol 2005; 12:567 - 568
- Zhou P, Derkatch IL, Uptain SM, Patino MM, Lindquist S, Liebman SW. The yeast nonmendelian factor [ETA+] is a variant of [PSI+], a prion-like form of release factor eRF3. EMBO J 1999; 18:1182 - 1191
- Liebman SW, Bagriantsev SN, Derkatch IL. Biochemical and genetic methods for characterization of [PIN+] prions in yeast. Methods 2006; 39:23 - 34
- Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-poly-Q amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci USA 2004; 101:12934 - 12939
- Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol 2002; 157:997 - 1004
- Gokhale KC, Newnam GP, Sherman MY, Chernoff YO. Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. J Biol Chem 2005; 280:22809 - 22818
- Perutz MF. Glutamine repeats and neurodegenerative diseases: Molecular aspects. Trends Biochem Sci 1999; 24:58 - 63
- Perutz MF, Pope BJ, Owen D, Wanker EE, Scherzinger E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid beta-peptide of amyloid plaques. Proc Natl Acad Sci USA 2002; 99:5596 - 5600
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science 2004; 305:673 - 676
- Collin P, Beauregard PB, Elagoz A, Rokeach LA. A nonchromosomal factor allows viability of Schizosaccharomyces pombe lacking the essential chaperone calnexin. J Cell Sci 2004; 117:907 - 918
- DebBurman SK, Raymond GJ, Caughey B, Lindquist S. Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc Natl Acad Sci USA 1997; 94:13938 - 13943
- Inoue Y, Taguchi H, Kishimoto A, Yoshida M. Hsp104 binds to yeast sup35 prion fiber but needs other factor(s) to sever it. J Biol Chem 2004; 279:52319 - 52323
- Krzewska J, Melki R. Molecular chaperones and the assembly of the prion Sup35p, an in vitro study. EMBO J 2006; 25:822 - 833
- Uptain SM, Sawicki GJ, Caughey B, Lindquist S. Strains of [PSI+] are distinguished by their efficiencies of prion-mediated conformational conversion. EMBO J 2001; 20:6236 - 6245
- Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell 2000; 100:277 - 288
- DePace AH, Weissman JS. Origins and kinetic consequences of diversity in Sup35 yeast prion fibers. Nat Struct Biol 2002; 9:389 - 396
- Chien P, Weissman JS. Conformational diversity in a yeast prion dictates its seeding specificity. Nature 2001; 410:223 - 227
- Hara H, Nakayashiki T, Crist CG, Nakamura Y. Prion domain interaction responsible for species discrimination in yeast [PSI+] transmission. Genes Cells 2003; 8:925 - 939
- Vanik DL, Surewicz KA, Surewicz WK. Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell 2004; 14:139 - 145
- Bradley ME, Liebman SW. Destabilizing interactions among [PSI+] and [PIN+] yeast prion variants. Genetics 2003; 165:1675 - 1685
- Satpute-Krishnan P, Serio TR. Prion protein remodelling confers an immediate phenotypic switch. Nature 2005; 437:262 - 265
- Salnikova AB, Kryndushkin DS, Smirnov VN, Kushnirov VV, Ter-Avanesyan MD. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its nonheritable amyloids. J Biol Chem 2005; 280:8808 - 8812
- Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci USA 1994; 91:5355 - 5358
- Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delglio F, Tycho R. A structural model for Alzheimer's beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA 2002; 99:16742 - 16747
- Lipfert J, Franklin J, Wu F, Doniach S. Protein misfolding and amyloid formation for the peptide GNNQQNY from yeast prion protein Sup35: Simulation by reaction path annealing. J Mol Biol 2005; 349:648 - 658
- DePace AH, Santoso A, Hillner P, Weissman JS. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 1998; 93:1241 - 1252
- Niewold TA, Hol PR, van Andel AC, Lutz ET, Gruys E. Enhancement of amyloid induction by amyloid fibril fragments in hamster. Lab Invest 1987; 56:544 - 549
- Lundmark K, Westermark GT, Nystrom S, Murphy CL, Solomon A, Westmark P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci USA 2002; 99:6979 - 6984
- Ganowiak K, Hultman P, Engstrom U, Gustavsson A, Westmark P. Fibrils from synthetic amyloid-related peptides enhance development of experimental AA-amyloidosis in mice. Biochem Biophys Res Commun 1994; 199:306 - 312
- Johan K, Westermark G, Engstrom U, et al. Acceleration of amyloid protein A amyloidosis by amyloid-like synthetic fibrils. Proc Natl Acad Sci USA 1998; 95:2558 - 2563
- Lundmark K, Westermark GT, Olsen A, Westermark P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc Natl Acad Sci USA 2005; 102:6098 - 6102
- Kisilevsky R, Lemieux L, Boudreau L, Yang DS, Fraser P. New clothes for amyloid enhancing factor (AEF): Silk as AEF. Amyloid 1999; 6:98 - 106
- O'Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem 2004; 279:17490 - 17499
- Edskes HK, Gray VT, Wickner RB. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci USA 1999; 96:1498 - 1503
- Edskes HK, Wickner RB. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full-length protein. Proc Natl Acad Sci USA 2002; 99:16384 - 16391
- Osherovich LZ, Weissman JS. The utility of prions. Dev Cell 2002; 2:143 - 151
- Zhou P, Derkatch IL, Liebman SW. The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI+] and [PIN+]. Mol Microbiol 2001; 39:37 - 46
- Allen KD, Wegrzyn RD, Chernova TA, et al. Hsp70 chaperones as modulators of prion life cycle: Novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. Genetics 2005; 169:1227 - 1242
- Sondheimer N, Lopez N, Craig EA, Lindquist S. The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J 2001; 20:2435 - 2542
- Lopez N, Aron R, Craig EA. Specificity of class II Hsp40 Sis1 in maintenance of yeast prion [RNQ+]. Mol Biol Cell 2003; 14:1172 - 1181
- Song Y, Masison DC. Independent regulation of Hsp70 and Hsp90 chaperones by Hsp70/Hsp90-organizing protein Sti1 (Hop1). J Biol Chem 2005; 280:34178 - 34185
- Song Y, Wu YX, Jung G, Murpy PJ. Role for Hsp70 chaperone in Saccharomyces cerevisiae prion seed replication. Eukaryot Cell 2005; 4:289 - 297
- Jones G, Song Y, Chung S, Masison DC. Propagation of Saccharomyces cerevisiae [PSI+] prion is impaired by factors that regulate Hsp70 substrate binding. Mol Cell Biol 2004; 24:3928 - 3937
- Nakayashiki T, Ebihara K, Bannai H, Nakamura Y. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell 2001; 7:1121 - 1130
- Eaglestone SS, Cox BS, Tuite MF. Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism. EMBO J 1999; 18:1974 - 1981
- True HL, Berlin I, Lindquist SL. Epigenetic regulation of translation reveals hidden genetic varn provides a mechanism for genetic variation and phenotypic diveiation to produce complex traits. Nature 2004; 431:184 - 187
- True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000; 407:477 - 483