Abstract
The absence of specific immune response is a hallmark of prion diseases. However, in vitro and in vivo experiments have provided evidence that an anti-PrP humoral response could have beneficial effects. Prophylactic passive immunization performed at the time of infection delayed or prevented disease. Nonetheless, the potential therapeutic effect of PrP antibodies administered shortly before the clinical signs has never been tested in vivo. Moreover, a recent study showed the potential toxicity of PrP antibodies administered intracerebrally. We aimed at evaluating the effect of a prolonged intracerebral anti-PrP antibody administration at the time of neuroinvasion in BSE infected Tg20 mice. Unexpectedly, despite a good penetration of the antibodies in the brain parenchyma, the treatment was not protective against the development of BSE. Instead, it led to an extensive neuronal loss, strong astrogliosis and microglial activation. Since this effect was observed after injection of anti-PrP antibodies as whole IgGs, F(ab')2 or Fab fragments, the toxicity was directly related to the ability of the antibodies to recognize native PrP and to the intracerebral concentration achieved, and not to the Fc portion or the divalence of the antibodies. This experiment shows that a prolonged treatment with anti-PrP antibodies by the intracerebral route can induce severe side-effects and calls for caution with regard to the use of similar approaches for late therapeutic interventions in humans.
Introduction
Transmissible spongiform encephalopathies (TSE) or prion diseases are fatal neurodegenerative disorders that can be infectious, genetic or sporadic in origin.Citation1,Citation2 Examples of these diseases include different forms of Creutzfeldt-Jakob disease (CJD) in humans, scrapie in sheep and goat, bovine spongiform encephalopathy (BSE) in cattle and chronic wasting disease in cervids.
The hallmark of prion diseases is the accumulation in the central nervous system of a misfolded form, PrPSc or PrPRes, of an endogenous protein, PrPC. PrPSc presents abnormal physicochemicals properties such as insolubility, ability to form amyloid fibrils and protease resistance.Citation3 According to the “protein only” theory,Citation4,Citation5 this misfolded conformer is the primary component of the infectious entity, called prion, and acts as a template to convert non pathogenic PrPC into pathogenic PrPSc. Since this conformational switch is a key event in the pathogenesis, most therapeutic strategies against prion diseases are focused on blocking the interaction of PrPSc with its PrPC substrate.Citation6–Citation8
In a cell free conversion system, PrP-specific antibodies were able to inhibit the conversion of PrPC into PrPRes.Citation9 In vitro, scrapie-infected cell cultures are cured by anti-PrP antibodies.Citation10–Citation14 Several mechanisms have been proposed, including the inhibition of the PrPC/PrPSc interaction,Citation14 the increase of the sequestration of PrPC at the cell surface, the release of PrPC into the extracellular compartmentCitation15 or the acceleration of PrPC degradation.Citation16,Citation17 In vivo, expression of 6H4 anti-PrP light fragment IgM in B lymphocytes of transgenic mice protects against the disease after peripheral infection.Citation18 This experiment provided the proof of principle of the relevance of using anti-PrP antibodies in pre-exposure prophylaxis.
Classical immunizations against homologous PrPRec, monomeric or dimeric, lead to significant levels of serum anti-PrP antibodies but interfere only weakly with prion pathogenesis, regardless of the immunization route.Citation19–Citation25 The modesty of this effect may be related to the lack of recognition of native PrP due to immune tolerance toward PrPC.Citation23,Citation26 One way to overcome this hurdle is the administration of selected antibodies by passive immunization. When injected within four weeks after intraperitoneal inoculation with the RML scrapie strain, anti-PrP antibodies interfere successfully with lymphoinvasion and prevent subsequent neuroinvasion.Citation27
However, most of the patients are diagnosed once the first clinical symptoms have appeared and the infectious agent is already in the central nervous system, out of reach of the bulk of peripheral antibodies. A tempting therapeutic approach would consist of the direct intraventricular antibody administration with a cannula linked to an Ommaya reservoir. However, a strong warning has been raised by the study of Solforosi et al. showing that some PrP antibodies induce neuronal death when injected into the hippocampus of non-infected mice.Citation28
In the present study, we therefore wanted to evaluate the safety and potential therapeutic effect of a prolonged intracerebral anti-PrP antibody administration. Tg20 mice were inoculated intraperitoneally with the 6PB1 mouse-adapted BSE strain. The 4H11 monoclonal anti-PrP antibody was selected for its efficiency to recognize native PrPC and to cure scrapie N2a cells. Mice were injected with total IgG or the F(ab′)2 fragment of the 4H11 antibody by intraventricular route continuously during two weeks, starting at the early stage of neuroinvasion (85 dpi). The effect of the treatment on the brain homeostasis and on prion replication was analyzed immediately after removal of the pumps in one group of mice. The therapeutical effect was assessed in another group of mice. The potential toxic effect of the aforementioned antibody preparations, together with that of Fab frament, was also analyzed on non-infected mice.
Despite an extensive diffusion of the antibodies in the brain, the treatment did not significantly increase the survival of the animals. However, it induced modifications of the animal's behavior. The histological analysis of the brains revealed extensive neuronal loss, astrogliosis and microglial recruitment in the regions surrounding the ventricle, mostly in the animals treated with the F(ab′)2 fragments. Similar toxicity was observed in non-infected and infected animals, and also with Fab fragments.
This study points out the possible adverse effects of PrP antibody treatments and calls for further studies before any such treatment could be envisaged in humans affected by CJD.
Methods
Animals.
Eight-week-old Tg20 miceCitation29 were inoculated by intraperitoneal route with 100 µl of 2% brain homogenate prepared from either 6PB1Citation30 BSE-infected C57BL/6 mice at the terminal stage of disease (titrated as 107.2 50% LD50/g of brain) or healthy C57BL/6 mice. In each group, three or four animals were sacrificed immediately at the end of the treatment (animals were treated from 85 to 100 dpi) and five to nine animals at the terminal stage of the disease. Animals infected with BSE developed the first clinical signs of the disease, such as tail rigidity, kyphosis and abnormal gait at about 105 days after inoculation. At the terminal stage, animals suffered from bradykinesia, loss of weight and dehydration and were sacrificed. Left brain hemispheres were frozen to perform biochemical analyses. Right hemispheres were fixed in Carnoy's fluid (10% vol/vol acetic acid, 30% vol/vol chloroform, 60% vol/vol 100% ethanol) for 24 hours, then transferred into butanol-1 until embedding and sectioned (5 µm sections) to perform immunohistological analyses.
One-way analysis of variance (ANOVA) with Tukey's post test was done to compare the means between the different groups. Statistical analyses were performed with GraphPad InStat version 3.01 for Mac (GraphPad Software, San Diego, California).
The toxicity of different forms of 4H11 antibody was also evaluated in non-infected mice. Three mice were infused with the whole IgG 4H11, four mice with the F(abµ)2 4H11 and two mice with the Fab 4H11 during 15 days.
Preparation of antibodies and stereotaxical implantation of osmotic pumps.
Antibody treatments started at 85 dpi until 100 dpi. Antibodies were diluted in “artificial cerebrospinal fluid” (Harvard Apparatus, France) under sterile conditions. The concentration of the anti-PrP 4H11 and irrelevant IgG2a control antibodies was about 10 mg/ml, allowing a daily administration of 67 µg of antibody in the brain. The concentration of the F(ab′)2 and the Fab fragments was adjusted to 6.7 mg/ml allowing a daily administration of 45 µg of antibody fragments. This difference in the amount of antibodies and F(ab′)2 was calculated in order to inject the same quantity of paratopes. Mock treated mice were infused with artificial CSF only, control mice were not infused. Alzet osmotic pumps 1002, Durect, Charles River, France) were filled with 100 µl of antibody or control solutions and connected to the cannula (Brain Infusion Kit I, Durect, Charles River, France) following the provider's instructions. Mice were anaesthetized with avertin (-2, -2, -2 tribromoethanol and tertiary amylic alcohol, W:V 1:1, Sigma, France 2,5% in saline solution) at a dose of 17 µl/g of mouse. The delivery system was implanted stereotaxically in the right lateral ventricle (coordinates adapted to Tg20 from the mouse brain atlas by K. Franklin and G. Paxinos, 1997, Academic Press, AP: 0, L: -1, V: -3) and worked continuously for two weeks. At the end of the treatment, the infusion system was removed using the same anaesthesia procedure as that used for the implantation.
PrPSc enrichment using g5p and detection by Western blot.
PrP precipitation was performed using g5p conjugated to tosyl-activated superparamagnetic beads (previously described in ref. Citation31). Briefly, 10 µl of 10% brain homogenate clarified by centrifugation at 3000 g for ten minutes was incubated overnight with 50 µl of g5p beads and 940 µl of binding solution (2% Tween 20, 2% Igepal 630 in 1x PBS at pH 7.4). The beads were then rinsed three times in the same buffer and incubated at 37°C for one hour in 20 ml lysis 1x buffer with or without proteinase K 0.1 µg/ml final. Samples were then subjected to western blotting. PrP was detected with the anti-PrP D18 antibody at a 1:1000 dilution.Citation32
PrPSc purification and detection by ELISA.
Purification of PrPSc was performed from 100 µl of 5% brain homogenate by using the TeSeE Bovine Detection kit (Biorad) according to the manufacturer's instructions. An increase in the concentration of proteinase K, from 20 µg/ml to 100 µg/ml, was required in order to eliminate the excess of PrPC. PrPSc was then detected by sandwich ELISA. Antibodies SAF53 and 11C6-G4 were used as capture and detection antibodies, respectively. Adding Ellman's reagent and measuring optical density at 415 nm revealed acetylcholinesterase activity.
Immunohistochemical labelings.
After deparaffinization, the slides were placed in 3% H2O2 in milliQ pure water for ten minutes and rinsed in PBS with Triton 0.1% (PBS-T). For PrPSc detection and labeling of microglial cells with the F4/80 antibody, sections were pretreated by a digestion with Proteinase K (Eurobio, France) respectively at 2 or 20 µg/ml in PBS, for 20 minutes at 37°C. Then, sections were saturated for 20 min with 20% normal horse serum (Sigma, France) in PBS-T. Incubations with the primary antibodies were performed for two hours at room temperature at the following concentrations: Anti-Fab (Sigma, France) 1:100; Anti-GFAP (DAKO, France) 1:2000; biotinylated anti-PrP SAF34, 1:200; NeuN (Chemicon, France) 1:250; Anti-Keratan sulfate (Calbiochem, France) 1:100; biotinylated anti-F4/80 (Serotec, France) 1:100. Then, slides were rinsed three times in PBS-T and incubated for 30 minutes with horseradish peroxidase (HRP) complexes: Envision rabbit (Dakocytomation, France), Envision mouse (K4001, Dakocytomation, France) or Streptavidine-HRP complex (Vector Lab., France). Labelings were revealed with the Novared system (Vector Lab., France) and then the sections were counterstained with Mayer's hematoxylin (Vector Lab., France) before dehydration and mounting. Control reactions were performed by omission of specific primary antibodies and showed the absence of non-specific peroxidase reaction products.
Immunofluorescence analysis.
ApoBrdU TUNEL assay was performed according to the manufacturer's directions (Molecular probes, France). Sections were incubated for one hour in DNA labeling solution and for 30 minutes in anti-BrdU antibody solution. They were stained with DAPI before mounting.
For double immunostainings, the cell-specific labelings were performed before the TUNEL apoptosis procedure. Slides were treated as describe above. Primary antibodies were added at the following concentrations: Anti-GFAP (DAKO, France) 1:500; Anti-F4/80 (Serotec, France) 1:20. Then, slides were washed three times and stained according to the ApoBrdU Kit procedure. To reveal the anti-GFAP or anti-F4/80 antibodies, anti-rabbit-Alexa 568 1:250 (Molecular Probes, France) or streptavidine-Alexa 568 1:250 (Molecular Probes, France), respectively, were added to the solution of secondary antibody of the ApoBrdU kit.
Multiple immunofluorescence was visualized using the Axio Imager A1 microscope (Carl Zeiss, Germany). Images were acquired sequentially using the AxioCam MRc camera with the green (FITC), red (CY3) and blue (DAPI) filters, and analysed with the AxioVision 4.5 software (Carl Zeiss, Germany).
Results
Distribution of antibodies after a two-week intracerebroventricular injection.
Since prolonged administration of anti-PrP antibodies by intracerebroventricular route has never been described, we assessed the biodistribution of the 4H11 and the control antibodies at the end of the continuous 15 days infusion.
We used an anti-mouse Fab antibody to detect both F(ab′)2 fragments and whole IgGs. The circulation of the CSF in the ventricle system and in the subarachnoid space led to a distribution in the regions close to the lateral, the third and the fourth ventricles and at the surface of the brain. The antibodies crossed the ependymal layer adjoining the ventricular cavities and spread in the brain parenchyma with a decreasing gradient from the regions close to the ventricles (cortex, hippocampus, striatum) to farther regions (hypothalamus, pons, cerebellum, olfactory bulb) (, red color).
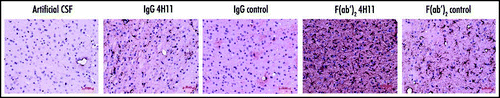
All antibodies were retained in the brain (compare antibodies versus artificial CSF in ), but to a variable extent depending on the antibody. Observation of whole brains (, top row) showed a wider distribution and a stronger labeling by the F(ab′)2 fragments when compared to the IgGs, and of the 4H11 anti-PrP antibodies versus their respective F(ab′)2 and Ig controls.
In the control animals infused with the artificial CSF only, the red color was restricted to the vascular system and to the glial scar, and was due to the recognition of endogenous immunoglobulins.
The observation of the thalamic parenchyma at a higher magnification (, bottom row) revealed the presence of the injected antibodies in the neuropile and within cells harbouring a neuronal morphology. The intensity of this cell staining correlated positively with the local concentration of antibodies. In addition, the F(ab′)2 4H11-treated mice, which presented the highest concentrations of anti-PrP fragments, displayed a strong perturbation of the parenchyma, characterized by an abnormal histoarchitecture, the disappearance of the blood vessels, and the presence of numerous cells harboring a pycnotic nucleus.
Neurological effects of the intracerebroventricular delivery of PrP antibodies.
Tg20 mice were inoculated intraperitoneally with the mouse adapted BSE strain 6PB1 and were treated by intracerebroventricular administration of anti-PrP antibodies, control antibodies or artificial CSF (used as the vehicle solution for the antibodies) when 60% of the time to death had elapsed (corresponding to 85 dpi), which is the time of onset of neuroinvasion in mouse models.Citation21 The intraperitoneal route of inoculation was chosen as a standard route to mimick natural or accidental peripheral infections with prions. The antibodies were administered centrally to overcome pharmacokinetic problems mainly due to their poor ability to cross the blood-brain barrier, and because we wanted to investigate the possibility to implement a therapeutical intervention once the infectious agent has already reached the central nervous system. Clinical monitoring revealed that a few days after implantation of the osmotic pumps, mice injected with the anti-PrP antibodies or the anti-PrP F(ab′)2 fragments displayed neurological signs such as nervousness and scratching as well as perturbation of the circadian rythms. The animals treated with the PrP specific antibodies where much more active than the animals from any control group, with shorter sleeping phases. They also had a reduced social behavior. For example, they slept scattered in the cages rather than gathered.
Another group of non-infected mice was treated by intracerebroventricular administration of the same panel of antibodies in a way similar to that of the BSE-infected mice. Two mice from this non-infected group, one treated with anti-PrP whole IgG and one with the anti-PrP F(ab′)2 fragment, displayed such strong behavioral changes and alteration of condition that they were humanely sacrificed after 14 days of treatment.
At the end of the fifteen days of injection, four mice per group were killed for the evaluation of the effects of the treatment on PrPSc levels and CNS homeostasis (see below).
Effects of the intracerebroventricular delivery of PrP antibodies on the course of BSE.
Another set of animals (5–9 per group) was kept until they developed BSE. Mice started to display the first clinical signs of BSE at 105 days post infection. Mice treated with the whole anti-PrP 4H11 died at 140 ± 8 dpi against 143 ± 10 for the control whole IgG. Mice treated with the F(ab′)2 fragment 4H11 or control F(ab′)2 died respectively at 143 ± 14 and 144 ± 11 dpi. The mock-infused mice died at 145 ± 2. One-way analysis of variance (ANOVA) with Tukey's post-test shows that there was no difference in the survival between the treatments. However, there was a larger variability in the incubation period of the animals treated with antibodies when compared to the animals injected with artificial CSF ( and ).
Effect of the treatment on PrP levels.
An analysis of PrPSc accumulation was performed by g5p precipitation () or by the Biorad SAF purification method followed by a sandwich ELISA (). At the end of the treatment, PrPSc levels were below the detection threshold in all groups of mice. At the terminal stage of BSE, all animals displayed detectable levels of PrPSc with no statistical difference between antibody-treated and non treated animals.
Anti-PrP antibodies trigger neuronal death.
In the occipital cortex, the hippocampus, the thalamus or the striatum, as well as in regions close to the lateral ventricle of 4/7 F(ab′)2 4H11- and 1/8 whole IgG 4H11-treated mice, hematoxylin-erythrosin staining revealed an extensive perturbation of the parenchyma with a disorganization of the neuropile and the presence of shrunk cells with pycnotic nuclei, indicative of apoptosis. The example of the hippocampus is shown in .
NeuN labeling revealed an extensive loss of neurons in the region where pycnotic nuclei were observed, correlating with the staining for apoptosis ( compare top and bottom rows).
Double labeling for apoptosis and GFAP or F4/80 revealed the presence of non-apoptotic, activated astrocytes ( top row, red labeling) and microglial cells (, bottom row, red labeling) in the vicinity of dying neurons. The two non-infected, Fab 4H11-treated mice also displayed apoptosis (data not shown).
Gliosis.
Astroglial activation was assessed immediately after the end of the treatment by GFAP labeling. In PrP-antibody treated mice, reactive astrocytes were found in the vicinity of the ventricle and to a lower extent in other parts of the brain. The intensity of this astrocytic response followed the gradient of the antibody concentration achieved in the brain: F(ab′)2 4H11 > IgG 4H11 ≥ F(ab′)2 control > IgG control > artificial CSF (). Even if a slight astocytic activation could be observed in F(ab′)2 control-treated mice, this was insufficient to induce neuronal death or treatment-related neurological signs.
F4/80 staining revealed amoeboid microglial cells in association with the neuronal death specifically in the PrP antibody-treated groups but was not associated with amyloid deposits at the terminal stage of the disease. On the contrary, keratan sulfate positive microglial cells of stellar and amoeboid morphology were not specifically associated with the anti-PrP antibodies administration but were associated with amyloid deposit at the terminal stage. F4/80 and keratan sulfate labelings were mutually exclusive showing that different types of microglial cells respond to the injury resulting from antibody administration versus PrP deposits (data not shown).
Discussion
The development of a therapy against prion diseases remains a major goal. Several compounds have shown an anti-prion activity (reviewed in ref. Citation6). However, the activity of most of them has been evaluated at early stages of the disease during the lymphoinvasion phase and few are suitable for a late stage treatment because of their poor ability to cross the blood brain barrier. As to now, this hurdle can only be overcome by evaluating the therapeutical efficiency of good candidates by direct intraventricular injection. Pentosane polysulfate is the only compound which has been evaluated by intraventricular injection in the brain of patients affected with Creutzfeldt-Jakob disease. Apart from one report of a prolonged survival in a patient affected with variant Creutzfeldt-Jacob disease,Citation33 these trials have so far met with limited success.Citation34,Citation35
Several experiments have already demonstrated the efficiency of PrP antibodies both in vitro and in vivo at the early times of infection, emphasizing their therapeutic potential.Citation15,Citation18,Citation27 In addition, other approaches using antibodies directed against the receptor LRP/LR revealed an effect in the reduction of peripheral PrPsc propagation.Citation36 However, the limited ability of the antibodies to cross the blood brain barrier, the immune tolerance towards homologous PrP, the possibility that immunization be ineffective after the neuroinvation phase, and the possible neurotoxic effects of PrP antibodies in the brain are all major hurdles to overcome in immunotherapeutical approaches for patients at the preclinical or clinical stages. We tackled this issue by starting to investigate the effects of the direct and prolonged intracerebral delivery of PrP antibodies in a mouse model of BSE infection.
Selection of the 4H11 anti-PrP antibody.
The PrP antibody needed to fulfill three major requirements:
First, it should recognize native PrPC and/or PrPSc.Citation23 The 4H11 antibody was raised against a recombinant dimeric form of PrP designed to mimic a PrPCto PrPSc conversion intermediate assumed to be relevant in the pathogenic process.Citation37 4H11 recognized native PrPC exposed at the surface of N2a cells by FACS analysis (Supplemental Fig. 1). It did so even better than two other PrP antibodies, the Bar 214Citation15 and the SAF34 used to cure scrapie-infected cells.Citation17
Secondly, it should recognize at least one epitope critical for prion replication. Previous experiments showed that the targeting of three different epitopes leads to the inhibition of PrPSc accumulation in infected cell cultures: the octarepeat region (amino acids 59–89) recognized by the antibody SAF34,Citation17 the intermediary region (aa 97–102) recognized by the antibodies D13Citation14 and ICSM35Citation16 and the central region of the helix1 (aa 130–160) recognized by the 6H4, the D18, the ICSM18 and the ICSM19 antibodies.Citation11,Citation14,Citation16
However, one of these regions (aa 95–105) is associated with neuronal death when the divalent antibodies are injected intracerebrallyCitation28 and is therefore not a good target region for therapeutical studies. Our epitope mapping experiment showed that the 4H11 recognizes the octarepeat region of PrP.
Third, the antibody should be able to cure scrapie-infected cells in culture. The 4H11 displayed a strong ability to clear ScN2a cells from PrPSc after five days of treatment (Supplemental Fig. 2). The IC50 of the 4H11 IgG and F(ab′)2 fragment were about 5 and 3 nM, respectively, compared with 9 nM for SAF34Citation17 and the Fab fragment D18.Citation14 Consequently, we undertook to assess the ability of the 4H11 antibody to impede prion progression in the CNS in vivo. Tg20 mice were used despite their 8-times overexpression of PrP, because they exhibit shorter incubation times than wild-type mice, and therefore the duration of the treatment proportionally to the duration of the incubation period would be longer. BSE infected Tg20 mice, which exhibit clinical signs after about 105 days following i.p. inoculation, were treated at the onset of the estimated phase of neuroinvasion (60% throughout the incubation period, i.e., 85 dpi) during 15 days, by continuous pump delivery. Longer intracerebroventricular treatments were excluded due to technical constraints and the onset of neurological symptoms in some mice. Since the Fc fragment of antibodies can induce an unspecific activation of astrocytes or microglial cells and has been shown to be responsible for clearance of the antibodies from the brain parenchymaCitation38 we injected both whole IgGs and F(ab′)2 fragments (lacking the Fc fragment) of the anti-PrP antibodies and of the irrelevant controls.
Biodistribution and availability of the antibodies in the brain.
The antibodies diffused in the brain to an extent which was beyond our expectation. This was particulartly striking in the anterior parts of the brain. However, the antibodies did not reach such high concentrations in the caudal regions of the brain where nuclei involved in the vital functions are targets for the infectious agent.
We observed a better distribution of the F(ab′)2 fragments when compared to the whole IgGs. This could be due to the lower molecular weight of F(ab′)2 fragments, allowing a higher diffusion rate, and/or to the mechanisms of Fc specific clearance which decrease dramatically the half life of brain IgGs. This mechanism involves a Fc-like receptor transporting the antibodies from the brain parenchyma to the blood compartment.Citation38 Anti-PrP antibodies were more abundant than their respective controls, probably because their binding to PrP slows down their elimination.
Biological effect of the treatment.
The two weeks of continuous intracerebral administration of PrP antibodies did not delay the onset of the disease. This might be due to the PrPC overexpression by Tg20 mice, which diminished the probability of antibodies effectively reducing the levels of PrP, or to the insufficient antibody concentration in relevant clinical target areas. However, mice treated with anti-PrP antibodies displayed neurological signs during the treatment. It was highly unlikely that these symptoms be linked with the CSF infusion itself, because the pumps delivered a total of 6 µl per day, corresponding to about 5% of the total CSF volume in a mouse. Moreover, these symptoms were specifically associated with the treatment by anti-PrP antibodies and were not observed in mice treated with artificial CSF or control antibodies. Hence, this observation prompted us to investigate the effect of the treatments on the different cell populations of the brain.
We found that the animals treated with PrP antibodies displayed an apoptotic neuronal death. The extent of this effect was directly correlated with the antibody concentration, as it was more pronounced in the F(ab′)2 4H11- than in IgG 4H11-treated mice. This observation expands the observations by Solforosi et al. that crosslinking of cellular PrP can trigger neuronal apoptosis.Citation28 In our experiment, we show that this effect is not restricted to antibodies recognizing the 95–105 region of PrP but can also be induced by antibodies binding the octarepeat region. Moreover, we found that monovalent Fab 4H11 fragments were also neurotoxic. These results suggest that PrP crosslinking is probably not the exclusive neurotoxic pathway triggered by PrP-antibodies. “Coating” the cell surface PrP with antibodies or fragments of antibodies may induce other toxic signals, such as abnormal PrP internalization or preventing the transduction of the physiological signal by PrPC (in the same way as it has been hypothesized in case of binding of truncated PrP or doppel to the PrP ligandCitation39,Citation40).
We also observed an astrocytic reaction related to the treatment with PrP antibodies. It may have been due to a direct effect of the antibodies on astrocytic surface PrP, or to the release of proinflammatory components by dying neurons. In addition, numerous activated F4/80 positive microglial cells displaying an amoeboid shape were found in close association with apoptotic neurons. They could represent scavenger cells recruited because of the presence of dead cell debris.
Conclusions and Perspectives
In conclusion, this study is the first attempt to evaluate the therapeutic potential of anti-PrP antibodies by a prolonged infusion in the brain of infected animals at the preclinical stage. It led to several observations bearing immediate implications for the development of a passive immunotherapy for presymptomatic or symptomatic patients.
We showed that antibodies do diffuse well in the anterior part of the mouse brain but fail to reach efficiently posterior regions, which are also relevant to the pathogenesis of prion diseases. The development of molecules with a higher diffusion ability or modifications of the delivery system (capacity, site of implantation) should be considered. PrP-binding molecules which cross the blood brain barrier and could be administered by peripheral route would still constitute the preferred alternative.
We also showed that the neurotoxicity of anti-PrP antibodies is not restricted to those directed against the intermediate region of PrP (around aa 95–105). Thus the selection of an anti-PrP antibody for in vivo usage should be epitope driven, assuming there are regions of PrP that can be targeted without triggering harmful side effects. Antibodies recognizing exclusively PrPSc or toxic aggregates of PrP, but not cellular PrP may be the key to avoid toxicity. In any case a better understanding of the toxic pathways induced by PrP binding molecules is required before passive immunization assays be considered in humans.
Of note, active immunizations of patients suffering from Alzheimer's disease have led to accidental meningoencephalitis in 6% of the patientsCitation41 and it still remains to be determined if passive immunization represents a safer approach (reviewed in ref. Citation42).
Overall, our study points out strategic areas of investigation for the development of passive immunotherapy of human prion diseases and calls for caution regarding the implementation of such strategies before such work has actually been performed.
Abbreviations
| PrP | = | prion protein |
| PrPSc/PrPRes | = | abnormal isoform of PrP |
| PrPC | = | normal-cellular isoform of PrP |
| BSE | = | bovine spongiform encephalopathy |
| TSE | = | transmissible spongiform encephalopathy |
| CJD | = | Creutzfeldt-Jakob disease |
| 6PB1 | = | BSE mouse-adapted prion strain |
| RML | = | Rocky Mountain Laboratories ovine prion strain |
| CSF | = | cerebrospinal fluid |
| GFAP | = | glial fibrillary acidic protein |
| TUNEL | = | TdT-mediated dUTP nick end labeling |
| i.p. | = | intraperitoneal |
| ICV | = | intracerebroventricular |
| PK | = | proteinase K |
Figures and Tables
Figure 1 Distribution of the antibodies in the brain. The antibodies were detected in the brain of the infected animals after a continuous 15 days treatment during the neuroinvasion (85–100 dpi). Red-brown labeling of the antibodies in the encephalon (first row, bar = 2.5 mm) highlights the greater diffusion rate of the F(ab′)2 fragments (columns 4 and 5) when compared to the corresponding IgGs (columns 2 and 3). Higher magnification microphotographs of the thalamus (second row, bar = 50 µm) reveal the distribution of the antibodies in the neuropile and in cell bodies. The mice treated with 4H11 F(ab′)2 fragments displayed a strong perturbation of the neuropile associated with shrunk cells and pycnotic nuclei (column 4).
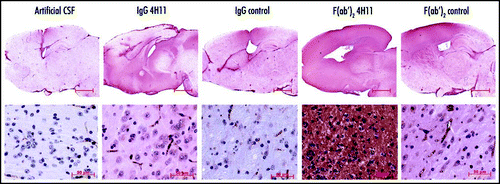
Figure 2 Survival of 6PB1 infected mice. Grey circles: mice infused with the artificial CSF. Black triangle: mice treated with the PrP and control IgGs (left panel). Black squares: mice treated with PrP and control F(ab′)2 fragments (right panel).
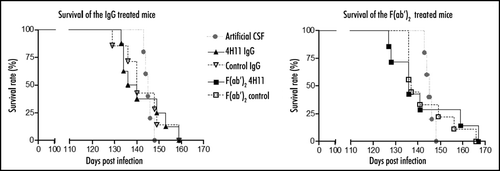
Figure 3 PrPSc accumulation in the brain of treated animals. (A) PrPSc was enriched from brain homogenates by precipitation with g5p and digested or not with PK (-/+ lanes). PrPSc was evaluated at the end of the treatement (lanes 1–10) or at the terminal stage of disease (lanes 11 and 12). (B) PrPSc was purified from brains of animals at the end of treatments (left set of bars), or at terminal stage (right set of bars) by the purification technique described in the methods, and then detected by sandwich ELISA.
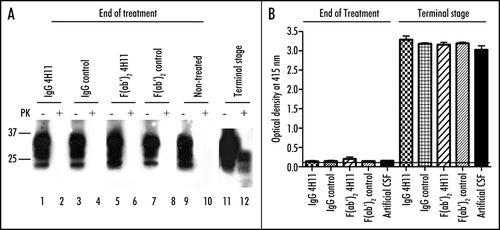
Figure 4 Effect of the antibody treatment on neurons and glial cells in infected animals at the end of the treatment. (A) Neuron specific NeuN labeling highlighting the neuronal loss in the regions CA1 and CA2 of the hippocampus of the animals treated with 4H11 F(ab′)2 fragments, while the animals from all the other groups did not show neuronal loss (row 1). TUNEL labeling showing apoptotic neurons in the same region of the hippocampus of the animals treated with 4H11 F(ab′)2 fragments (row 2). Bar = 500 µm. (B) Higher magnification and labeling for gliosis of the hippocaampus of the 4H11 F(ab′)2 treated mouse shown in (A) (lines show the corresponding panel A). Immunofluorescence analysis revealed the presence of GFAP positive reactive astrocytes (row 1, red labeling) and F4/80 positive microglial cells (row 2, red labeling) in the vicinity of the apoptotic neurons labelled by TUNEL (2nd and 4th columns, green) in the hippocampus of 4H11 F(ab′)2 fragments treated animals. Bar = 50 µm.
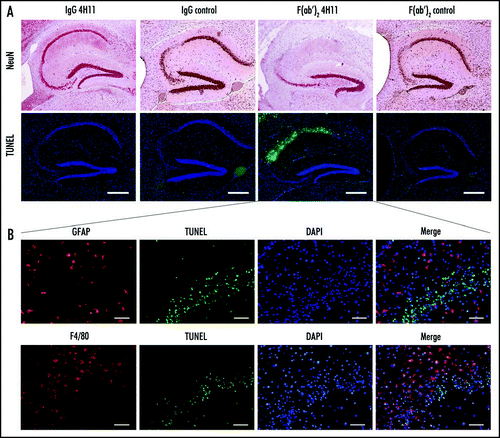
Figure 5 Effect of the antibody treatment on the astroglial reaction. Reactive astrocytes were detected at the end of the treatment by labeling for GFAP (brown). (Thalamus, Bar = 50 µm).
Additional material
Download Zip (188 B)Acknowledgments
This work was financially supported by the Commissariat à L'Energie Atomique (CEA, Fontenay-aux-Roses, France), the Agence Française de Sécurité Sanitaire des Aliments (AFSSA, Lyon, France), the Scripps Research Institute (Jupiter, Florida, USA) and by the European Community (grant ≠ QLK3-CT-2001-00283). We are indebted to Drs. G. Kneale and J. McGeehan (University of Portsmouth, UK) for having kindly provided recombinant g5p protein. We are grateful to J. Comte, B. Dufresnois, J. Desmercière and M.C. Mondon for helpful technical assistance. We gratefully acknowledge the critical reading of the manuscript by C. Weissmann.
References
- Aguzzi A, Weissmann C. Prion diseases. Haemophilia 1998; 4:619 - 627
- Lasmézas CI, Weiss S. Cary JW, Linz JE, Bhatnagar D. Molecular biology of prion diseases. Microbial foodborne diseases 2000; Lancaster, USA Technomic Publishing Company, Inc. 495 - 537
- Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem 1998; 67:793 - 819
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Weissmann C, Aguzzi A. Approaches to therapy of prion diseases. Annu Rev Med 2005; 56:321 - 344
- Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain 2006; 129:2241 - 2265
- Vana K, Zuber C, Nikles D, Weiss S. Novel aspects of prions, their receptor molecules, and innovative approaches for TSE therapy. Cellular and Molecular Neurobiology 2007; 27:107 - 128
- Horiuchi M, Caughey B. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J 1999; 18:3193 - 3203
- Donofrio G, Heppner FL, Polymenidou M, Musahl C, Aguzzi A. Paracrine inhibition of prion propagation by anti-PrP single-chain Fv miniantibodies. J Virol 2005; 79:8330 - 8338
- Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci USA 2001; 98:9295 - 9299
- Gilch S, Wopfner F, Renner-Muller I, Kremmer E, Bauer C, Wolf E, Brem G, Groschup MH, Schatzl HM. Polyclonal anti-PrP auto-antibodies induced with dimeric PrP interfere efficiently with PrPSc propagation in prion-infected cells. J Biol Chem 2003; 278:18524 - 18531
- Miyamoto K, Nakamura N, Aosasa M, Nishida N, Yokoyama T, Horiuchi H, Furusawa S, Matsuda H. Inhibition of prion propagation in scrapie-infected mouse neuroblastoma cell lines using mouse monoclonal antibodies against prion protein. Biochem Biophys Res Commun 2005; 335:197 - 204
- Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 2001; 412:739 - 743
- Feraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C, Vilette D, Lehmann S, Grassi J. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem 2005; 280:11247 - 11258
- Beringue V, Vilette D, Mallinson G, Archer F, Kaisar M, Tayebi M, Jackson GS, Clarke AR, Laude H, Collinge J, Hawke S. PrPSc binding antibodies are potent inhibitors of prion replication in cell lines. J Biol Chem 2004; 279:39671 - 39676
- Perrier V, Solassol J, Crozet C, Frobert Y, Mourton-Gilles C, Grassi J, Lehmann S. Anti-PrP antibodies block PrPSc replication in prion-infected cell cultures by accelerating PrPC degradation. J Neurochem 2004; 89:454 - 463
- Heppner FL, Musahl C, Arrighi I, Klein MA, Rulicke T, Oesch B, Zinkernagel RM, Kalinke U, Aguzzi A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 2001; 294:178 - 182
- Bade S, Baier M, Boetel T, Frey A. Intranasal immunization of Balb/c mice against prion protein attenuates orally acquired transmissible spongiform encephalopathy. Vaccine 2006; 24:1242 - 1253
- Ishibashi D, Yamanaka H, Yamaguchi N, Yoshikawa D, Nakamura R, Okimura N, Yamaguchi Y, Shigematsu K, Katamine S, Sakaguchi S. Immunization with recombinant bovine but not mouse prion protein delays the onset of disease in mice inoculated with a mouse-adapted prion. Vaccine 2007; 25:985 - 992
- Kimberlin RH, Walker CA. Pathogenesis of mouse scrapie: Dynamics of agent replication in spleen, spinal cord and brain after infection by different routes. J Comp Pathol 1979; 89:551 - 562
- Magri G, Clerici M, Dall'Ara P, Biasin M, Caramelli M, Casalone C, Giannino ML, Longhi R, Piacentini L, Della Bella S, Gazzuola P, Martino PA, Della Bella S, Pollera C, Puricelli M, Servida F, Crescio I, Boasso A, Ponti W, Poli G. Decrease in pathology and progression of scrapie after immunisation with synthetic prion protein peptides in hamsters. Vaccine 2005; 23:2862 - 2868
- Polymenidou M, Heppner FL, Pellicioli EC, Urich E, Miele G, Braun N, Wopfner F, Schatzl HM, Becher B, Aguzzi A. Humoral immune response to native eukaryotic prion protein correlates with anti-prion protection. Proc Natl Acad Sci USA 2004; 101:14670 - 14676
- Schwarz A, Kratke O, Burwinkel M, Riemer C, Schultz J, Henklein P, Bamme T, Baier M. Immunisation with a synthetic prion protein-derived peptide prolongs survival times of mice orally exposed to the scrapie agent. Neurosci Lett 2003; 350:187 - 189
- Sigurdsson EM, Brown DR, Daniels M, Kascsak RJ, Kascsak R, Carp R, Meeker HC, Frangione B, Wisniewski T. Immunization delays the onset of prion disease in mice. Am J Pathol 2002; 161:13 - 17
- Gregoire S, Bergot AS, Feraudet C, Carnaud C, Aucouturier P, Rosset MB. The murine B cell repertoire is severely selected against endogenous cellular prion protein. J Immunol 2005; 175:6443 - 6449
- White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003; 422:80 - 83
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, Williamson RA. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004; 303:1514 - 1516
- Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 1996; 15:1255 - 1264
- Lasmézas CI, Deslys JP, Demaimay R, Adjou KT, Hauw JJ, Dormont D. Strain specific and common pathogenic events in murine models of scrapie and bovine spongiform encephalopathy. J Gen Virol 1996; 77:1601 - 1609
- Zou WQ, Zheng J, Gray DM, Gambetti P, Chen SG. Antibody to DNA detects scrapie but not normal prion protein. Proc Natl Acad Sci USA 2004; 101:1380 - 1385
- Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR. Mapping the prion protein using recombinant antibodies. J Virol 1998; 72:9413 - 9418
- Parry A, Baker I, Stacey R, Wimalaratna S. Long term survival in a patient with variant Creutzfeldt-Jakob disease treated with intraventricular pentosan polysulphate. J Neurol Neurosurg Psychiatr 2007; 78:733 - 734
- Todd NV, Morrow J, Doh-ura K, Dealler S, O'Hare S, Farling P, Duddy M, Rainov NG. Cerebroventricular infusion of pentosan polysulphate in human variant Creutzfeldt-Jakob disease. J Infect 2005; 50:394 - 396
- Whittle IR, Knight RS, Will RG. Unsuccessful intraventricular pentosan polysulphate treatment of variant Creutzfeldt-Jakob disease. Acta Neurochir (Wien) 2006; 148:677 - 679
- Zuber C, Knackmuss S, Rey C, Reusch U, Rottgen P, Frohlich T, Arnold GJ, Pace C, Mitteregger G, Kretzschmar HA, Little M, Weiss S. Single chain Fv antibodies directed against the 37kDa/67kDa laminin receptor as therapeutic tools in prion diseases. Mol Immunol 2008; 45:144 - 151
- Jansen K, Schafer O, Birkmann E, Post K, Serban H, Prusiner SB, Riesner D. Structural intermediates in the putative pathway from the cellular prion protein to the pathogenic form. Biol Chem 2001; 382:683 - 691
- Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J Neuroimmunol 2001; 114:168 - 172
- Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, Hood L, Westaway D, DeArmond SJ, Tremblay P. Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci USA 2001; 98:15288 - 15293
- Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, Rulicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998; 93:203 - 214
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003; 61:46 - 54
- Lemere CA, Maier M, Jiang L, Peng Y, Seabrook TJ. Amyloid-beta immunotherapy for the prevention and treatment of Alzheimer disease: Lessons from mice, monkeys, and humans. Rejuvenation Res 2006; 9:77 - 84