Abstract
Among the large family of diseases caused by protein aggregation and misfolding is that associated to the Prion Protein (PrP). This is a small extracellular membrane-anchored molecule of yet unknown function. Understanding how PrP folds both into its cellular and pathological forms is thought to be crucial for explaining protein misfolding in general and the specific role of PrP in disease. Since the first structure determination, an increasing number of structural studies of PrP have become available, showing that the protein is formed by a flexible N-terminal region and a highly conserved globular C-terminal domain. We review here the current knowledge on PrP structure. We focus on vertebrate PrPs and analyse in detail the similarities and the differences among the coordinates of the C-terminal domain of PrP from different species, in search for understanding the mechanism of disease-causing mutations and the molecular bases of species barrier.
Introduction
A family of rare but all fatal neurodegenerative diseases which affect not only humans but also various animal species is related to the prion protein (PrP).Citation1–Citation4 In humans, the pathologies connected with PrP include kuru, Creutzfeldt-Jakob and Gerstmann-Straussler-Scheinker diseases and fatal familial insomnia.Citation5 In animals, they are known in several domestic and wild mammals, such as for instance sheep,Citation6 cattleCitation7 and cervids,Citation8 where the pathologies take specific names such as scrapie, bovine spongiform encephalopathy (BSE) and chronic wasting disease respectively. PrP related diseases may arise spontaneously, be inherited, or be acquired by infection. In the latter case, they are known as transmissible spongiform encephalopathies (TSEs). Under still unknown conditions, TSEs can some time assume epidemic proportions, which could potentially cross species barriers,Citation9 as it is the case for the BSE epidemic spread out amongst cattle in the United Kingdom around the 90's.Citation10
PrP diseases are thought to be part of the larger family of pathologies which is caused by protein misfolding and aggregation.Citation11–Citation15 Characteristic symptoms are brain vacuolation, astrogliosis and neuronal apoptosis, associated to accumulation in the central nervous system of extracellular protein deposits that may have or have not the properties of amyloid fibrils.Citation16–Citation19 These formations have been shown to contain PrP proteins although in a form apparently different from that observed in healthy individuals: although identical in their primary structure, PrP aggregates are protease resistant and have a β-enriched secondary structure,Citation20–Citation22 thus strongly suggesting that they are misfolded PrP species.Citation23–Citation30 It is now common to distinguish between the two forms and refer to the cellular non-pathological PrP as PrPC, while the misfolded form is named PrPSc.Citation31
The most unusual feature of TSEs, which makes this disease unique among the pathologies caused by protein misfolding, is the nature of the pathogenic agent in infectivity. It has been long debated whether the pathogen could be a virus, nucleic acids, a protein or another infective agent. While the involvement of nucleic acids has been definitely ruled out by recent studies,Citation32,Citation33 it is widely accepted that the infective agent is PrP itself according to the so-called “protein-only hypothesis.”Citation34–Citation37 According to this hypothesis, the infectious pathogen is the misfolded PrPSc, which, by forming aggregates observed as cerebral accumulations in different TSEs, acquires new properties, such as toxicity and protease K resistance, which are absent in the native protein.Citation38,Citation39 The aberrant conformer PrPSc is believed to act as a structural template which induces conversion of other PrPC molecules into the pathological form. Since this model implies that proteins rather than viruses or nucleic acids may be infectious and carry heritable information, the protein-only hypothesis represents a new paradigm of molecular biology.Citation7,Citation36 This hypothesis is now supported by several independent lines of evidence. Injecting the recombinant mouse PrP (mPrP) in the form of amyloid-like fibrils into mice that express the endogenous protein generated neurological dysfuncyion.Citation40 Consistently, in vitro prion replication is inhibited in PrPC deficient mouseCitation41 and cattle.Citation42 Protein-based inheritance has been observed both in yeast and in fungi.Citation43–Citation47 The occurrence of self-perpetuating conformations,Citation48 called PrP strains, and the recently accomplishment of in vitro PrPSc propagationCitation15,Citation49–Citation52 provide further support to the protein-only hypothesis.
Since pathology and infectivityCitation53 are so intimately related to a protein that may exist in at least two different conformational states, large interest has been paid over the last 15 years to the structure of PrP both in its nativeCitation54 and pathological forms.Citation55 Structure could also help to elucidate the cellular role of this protein, which although highly abundant in neurons and in other tissues,Citation56 remains to the date elusive. The scope of this review is that of summarizing the state of art of the structural field which has much advanced since the last exhaustive summary.Citation54 Because of the huge plethora of literature in the PrP field, we are forced to focus our interests to specific aspects. We shall mainly cover the experimental three-dimensional structures of full-length PrP from vertebrates and of its structured domains and compare the knowledge acquired both for the folded PrPC with the much more limited description of the misfolded PrPSc form. We shall exclude from our analysis the structures of peptides and studies of metal binding properties and of unfolding/misfolding pathways. We hope that our overview may provide a valuable reference for further studies.
Overview of the PrP Sequences and Structure
The human PrP (huPrP) gene is found in chromosome 20 and encodes a proto-protein 253 amino acids long before processing (). In the mature form, the first 22 residues are cleaved after translation, whereas the last 23 amino acid residues are cleaved prior to the addition of a glycosyl phosphoinositol (GPI) anchor to Ser230. PrP are extracellular proteins normally attached to the outer surface of the cellular membrane by means of the GPI anchor. They have also two N-linked glycosylation sites at residues Asn181 and Asn197.Citation57,Citation58 They are highly conserved amongst mammals:Citation59,Citation60 huPrP has 94.9%, 99.2% and 92.8% sequence identity with the protein from sheep, chimpanzee and cow respectively. More distant but still highly homologous orthologues (30% identity and 50% similarity) are present in reptiles and amphibians.
PrPC is divided into two regions with distinct structural and dynamical properties.Citation54 In mammals, the N-terminus hosts a variable number (depending on the organism) of octapeptide PHGGSWGQ repeats. Each octarepeat is able to bind divalent metals such as copper and others,Citation61 although the physiological significance of this interaction remains unclear. The octarepeats in mammalian PrPs are hexarepeats in birds and reptiles and a not readily apparent repeat pattern in frogs.
The N-terminus, up to residue ∼120, is flexibly disordered at pH 4.5.Citation62 The HGGGW and GWGQ segments of the octarepeats, which span residues 61–84, were shown to adopt a loop and a β-turn-like conformation respectively at pH 6.2.Citation63 Therefore, an increase in the population of transient secondary or tertiary structure is observed in this region at pH 6.2 relative to pH 4.5. Studies of full-length sheep PrP (shPrP) (25–233) at pH 5.5 by vibrational Raman optical activity and CD spectroscopy have also suggested that the shPrP N-terminus (25–93) adopts predominantly a polyproline II conformation.Citation64 This motif, which has the peculiar feature of lacking intra-helical backbone hydrogen bonds, is known to be involved in molecular recognition and is a typical target of Src homology 3 (SH3) domains.Citation65 Recognition of the C-terminal SH3 domain of the Grb2 protein was indeed found but it was mapped in the region 100–109.Citation66
The C-terminus of PrP is structured and presents a globular fold of three α-helices (H1, H2 and H3) and a short, double-stranded, antiparallel β-sheet (S1, S2)Citation54 (). A disulfide bridge between Cys179 and Cys214 links H2 and H3.
Since the first NMR structure of the C-terminal domain of mPrP, solved in 1996,Citation68 the number of PrP entries in the PDB database has increased continuously. Most of them were solved by NMR, having in solution either the full-length or truncated forms of PrP, while the X-ray structures are few and restricted to the C-terminal domains of the human and ovine proteins. This could suggest an intrinsic tendency of these proteins to elude crystallization, possibly due to intrinsic heterogeneity or, more likely, to local flexibility.
Hereafter, we review in detail the available structures of the globular C-terminal domain of vertebrate PrP, first dividing them according to the technique used and then comparing the results to gain an overall picture.
NMR Structures of the C-Terminal Domain of PrP
Almost all the sets of NMR coordinates available (with only a few exceptions) come from the same laboratory and were solved under very similar experimental conditions, i.e., at pH 4.5, 293 K and 10–50 mM acetate buffer (). They cover different species from humans to reptiles. All together, the NMR structures provide a wealth of information which results in a picture of PrPC rich of structural details.
HuPrP structures.
There are currently nine structures of huPrP, which cover different variants (). Constructs of different lengths were studied, some spanning the full-length mature PrP, others containing only the C-terminal domain (). They all agree well with an average r.m.s.d. of 0.9 Å, as obtained by superposing the backbone atoms of the secondary structure elements. Small differences were however observed in the chemical shifts of H2 and H3 when comparing the values for the full-length protein and for shorter constructs. These differences were first attributed to small pH variationsCitation69 and then reinterpreted as due to transient interactions between the N-terminal tail and the C-terminal domain.Citation70 This interpretation suggests that the N-terminus has a small but detectable stabilising effect on H2 and H3, as further supported by following data.Citation63,Citation71,Citation72
The positions of the secondary elements are in optimal agreement in all structures, with S1 and S2 positioned at residues 129–130 and 162–163. The sheet is in some structures prolonged by a β-bulge between residues 131–133 and 160–161. H1, H2 and H3 span, on average, residues 144–152, 173–194 and 200–227. In some of the structures however H2 and especially H3 are interrupted into two regions, one forming a regular α-helix, the other forming a 310 helix. The maximal variability both within each NMR bundle and among the different structures is observed at H3 and at the loop between S2 and H2 (residues 167–171), which is in a conformational slow exchange between two or more conformations in the millisecond time scale. It has been suggested that these dynamical properties may have relevance for PrP infectivity since these regions have been implicated in immune response and in species-specific contacts with a still unidentified ‘protein X’ which was suggested to mediate the transition from PrPC to PrPSc.Citation73
Variability could therefore be related to differences in the amino acid composition which leads to a higher predisposition to infectivity of certain variants. The structure which shows the largest differences with the other human PrP coordinates is 1fkc, which corresponds to the E220K variant.Citation71
Other mammalians PrPs.
After the structure determination of the C-terminal domain and full-length mPrP,Citation68,Citation83–Citation86 PrPs from several different mammalian species have been studied and compared in the attempt to understanding both the relation to disease-causing mutations and the molecular bases of species barrier (, and references therein). The information obtained by this comparison confirms, reiterates and reinforces what observed in the huPrP variants, with the structures being in excellent agreement with one another. The Cα atoms of the structures of mPrPCitation68 (1xyx) and of hamster PrPCitation69 (1b10), for instance, superpose with a 1.9 Å r.m.s.d, having been solved at different pH values and by different groups. The most diverse structure is that from elk (ePrP). In ePrP(121–231), the loop between residues 166–175 connecting S2 and H2, which is disordered in all other mammalian PrPs, is well structured and relatively rigid, as directly supported by the presence of sharp resonances from this region in the NMR spectra.Citation78 The authors put forward the hypothesis that the loop is part of the hypothetical ‘protein X.’Citation78 Structure determinations of two mPrP variants, mPrP[N174T] and mPrP[S170N,N174T] in which the mouse residues were substituted by the elk ones, show that the loop conformation is determined by the co-presence of only two amino acids (170 and 174), so that mPrP[S170N,N174T] exactly mimics ePrPC.Citation78 The non-regular but well-defined secondary structure formed by residues 166–175 is stabalized by two separate, local H-bond networks. The N-terminal region of the loop in mPrP[S170N,N174T] forms a 310-helical turn comprising residues 165–169. A second network of H-bonds is formed by the side chains of Asn171 and Thr174. This observation has suggested an explanation for the ease of transmission observed among the free-range elk and deer herds, but not with other species.
Birds and reptiles.
The NMR structures of the C-terminal domain of PrPs from chicken [chPrP(121–225)], turtle [tPrP(121–225)] and frog [xlPrP(90–222)] have also been solved, thus adding more evolutionarily distant examplesCitation82 (). Despite the relatively low sequence identity (30%), the structures are very similar, showing that sequence diverges faster than structure: the backbone atoms of H2, H3 and of the β-sheet are superposable both with each others and with mammalian PrPs with r.m.s.d. values of 1.1 Å or less. When superposing these secondary structure elements, however, some of the loops and the orientation of H1 show significant variations which may be regarded as ‘structural signatures’ for PrPs from the different evolutionary subgroups. Loop 166–173, which shows dynamic disorder in most of the mammalian PrPs except for ePrPCitation78 is more precisely defined in the avian and reptilian proteins. This loop is stabilized by a long-range H-bond between Val171 and Tyr222 in tPrP, by insertion of a proline in chPrP, and by a two-amino acids insertion in xlPrP. The polyThr stretch present at the C-terminus of H2, which consists of a tetrathreonine segment in huPrP, is quite different from the capped helices of chPrP and tPrP, and the kinked helix of xlPrP. An additional “structural signature” for chPrP is provided by an insertion between the H2 and H3, which forms a flexibly disordered loop and an N-terminal elongation of H3 in chPrP(121–225). This feature is conserved in all known avian PrP sequences.Citation87
X-Ray Structures of the PrP C-Terminal Domain
The first X-ray structure of huPrP was reported in 2001.Citation88 Since a swapped dimer was found in the crystal, domain swapping was proposed as a possible model for oligomerization in PrP propagation. Two more papers, published in 2004, described the shPrP structure both as a monomerCitation89 and as antibody bound complexes.Citation90 Interestingly, although larger constructs (approximately from residue 90 to 231) had been originally considered for X-ray studies, the crystallized proteins resulted to be smaller proteolytic fragments (see below).
Ovine PrP.
In sheep, a polymorphism at positions 136, 154 and 171 (shPrP numbering) confers different susceptibility to scrapie. The genotype Ala136-Arg154-Arg171 (ARR) is resistant to scrapie, whereas the Val136-Arg154-Gln171 (VRQ) and Ala136-Arg154-Gln171 (ARQ) variants have high and medium susceptibility, respectively. How these mutations affect the pathology is unknown. Polymorphisms also occur in other species; for example Met/Val polymorphism at codon 129 of the huPrP gene has been related to Creutzfeldt-Jakob disease.Citation91 Genotypes, however, that protect from the disease—as in sheep—have not been observed so far. This renders shPrP an excellent candidate for multifaceted studies in relation to disease susceptibility.
The structures of the C-terminal domain of shPrP are available for the free ARQ variant (residues 94–234, PDB entry code 1uw3)Citation89 and for three variants in complex with Fab fragments which cross react with both PrPC and PrPSc (PDB entry codes 1tpx, 1tqb and 1tqc for the ARQ, VRQ and ARR variants respectively).Citation90 The structures are very similar one another, with r.m.s.d. of 0.2–0.3 Å among the Fab complexes and a r.m.s.d. between 1tpx—taken as the representative structure of antibody bound shPrP—and the free ARQ variant (1uw3) of 1.31 Å ( and ), the largest difference being in the S1-H1 segment. The free ARQ variant used for crystallization trials of free shPrP contains a naturally occurring Arg151Cys mutation (shPrP numbering which corresponds to Arg148 in huPrP), in which the additional cysteine is protected by a mixed disulphide with glutathione. In the crystal, a truncated 119–231 fragment was observed by mass spectrometry, even though the region 119–120 is not visible in the electron density map and the fragment 121–135 is one of the most disordered regions.
A similar truncation was observed in the three antibody bound structures, where, although the constructs spanned residues 103–234, the crystals were shown to contain the sequence 114–234 due to proteolytic cleavage. Residues 114–126 and 229–234 are also not visible in the electron density, being disordered. These three structures provide not only subtle differences amongst the variants but also structural details about antibody recognition. The interaction resides in the stretch 188–199 that encompasses the C-terminus of H2 and the initial part of the loop connecting H2 and H3. From a comparative analysis it emerges that the antibody does not affect the three-dimensional structure, apart from limiting the flexibility of its epitope.Citation90 The mutated residues at positions 136, 154 and 171 are on the protein surface and therefore solvent exposed. The mutations induce conformational changes in the side chains of these residues. The authors noticed that, with respect to ARQ taken as a reference, VRQ is stabilised by an additional H-bond between Arg139 and the side chain carbonyl of Asn162. ARR, on the contrary, looses the H-bond between Arg167 and Gln171 and is therefore destabilised with respect to ARQ. Additional H-bonds are found in the region S1-H1-S2 in ARQ and VRQ with respect to the scrapie resistant variant ARR. The authors propose that correlation of the sequence to the scrapie resistance could be attributed to the different stability of the three variants. Other hypotheses cannot however be ruled out.Citation90
Human PrP.
Of the recombinant huPrP protein (90–231) used for crystallization, only the sequence 119–226 was found in the crystal. In the structure, due to a closer packing, the N-terminus is more ordered and visible in the electron density with respect to the other X-ray structures. Therefore, the structural features of this segment are known only from huPrP.
Unexpectedly, the crystal structure determination of huPrP revealed the occurrence of a covalent homo-dimer built up from domain swapping.Citation88 The r.m.s.d. between the composite monomer of huPrP and shPrP (1uw3) is 0.9 Å (). It was suggested that in solution there is equilibrium between the monomeric and dimeric forms, but only the latter is found in the crystal. The dimer is stabilized by two inter-subunit disulphide bridges between Cys179 and Cys214. The structural features are dominated by the swapping phenomenon. The H3 helix is forced to pack against H2 of the other subunit rather than packing against H2 of its own monomer by the covalent disulphide bond. Apart from the switch region and differences in the short loop connecting H2 to H3, the interactions between secondary structure elements are substantially conserved. In the swapped form, novel β strands, which amyloid fibrils have a high content of, are formed in the hinge loop. Therefore, domain swapping, which is a well known mechanism for dimerization/oligomerization, was suggested as a plausible mechanism in PrP amyloid generation.Citation92
Detailed Structure Analysis
The overall fold and the secondary structure elements of the crystallographic structures are very similar to those described in the NMR section. The two sets superpose with r.m.s.d. which vary between 1.3–4.2Å. These values are well within what is expected from species variability within the structure resolution and precision (). In the following sections, we describe a detailed comparison, trying to analyze subtle differences which might be correlated to the PrP pathology. Because they require high resolution, our analysis has been inferred from the X-ray structures, although most of our observations hold, when appropriate, also for the structures in solution. We analyse hierarchically the secondary structure, the interactions that direct and stabilise the tertiary structure and describe the interactions between adjacent molecules in the crystals.
Interactions stabilising the secondary and tertiary structure.
A schematic representation of the PrPC fold with the most important ionic interactions is shown in . The two short S1 and S2 β-strands, which form an antiparallel β-sheet, are strictly conserved in all structures. Each strand is two-residues long, with S1 being exposed on the surface. The sheet is stabilized by H-bonds between Met129 and Tyr163; Gly131 and Val161.
H1, 8–10 residues long, is always followed by three residues in a 310 helical conformation. This sequence contains several charged residues which make favorable sequential ionic interactions: the region comprising H1 and the 310 helix contains up to two Asp, two Glu and three Arg; the latter at positions 148, 151 and 154 respectively (huPrP numbering). A careful comparative analysis of the X-ray structure reveals subtle differences in the salt bridge pattern which stabilises H1. In huPrP, Arg148 and Glu152 are connected by a salt bridge. This interaction, which cannot be formed in 1uw3 because of the Arg148Cys mutation, is present in 1tqc and 1tqb (2.6 and 4.1 Å respectively), but missing in 1tpx, where Arg151 (corresponding to Arg148 in 1i4m and 1uw3) is engaged in a salt bridge with chain B of the antibody (see par. on interfaces). In both human and non-complexed ovine PrP (1uw3), Arg151 (corresponding to Arg154 in shPrP numbering) forms a salt bridge with Asp147, whereas in the antibody-bound shPrPs, studied at pH 6.3, the latter residue interacts with His140.
One of the major determinants of the tertiary structure of PrP is the disulphide bridge between Cys179 and Cys214, which anchors H2 to H3. Apart from this, H2 does not make significant interactions at the tertiary structure level. The only exception is a salt bridge between Arg167 and Asp181 in the antibody-bound scrapie-resistant ARR variant, suggesting a role of this interaction in disease resistance. In huPrP, H2 is broken into two regions (referred as H2 and H2') separated by a nearly β-patch; H2 ends to residue 188, whereas the segment Val189-Phe198 crosses the dimer interface and links each monomer to H3 (). The H2-H2' loop adopts an anti-parallel β-sheet structure (residues 190–194) with strands belonging to different chains. This region has been suggested to contain a ‘frustrated’ or ‘discordant’ sequence which, in the monomeric structure, assumes a helical conformation, instead of the predicted most favored β-structure.Citation93,Citation94 Conversion of the tetraThr stretch (residues 190–193) to a β-structure would relieve ‘frustration’.
H3 contains several charged residues. Arg208 and 220, and Glu211 and 221 are engaged in a cluster of charge interactions (distances in the range 5.7–6.8 Å) along the helix. Electrostatic interactions link H3 to other structural elements, stabilizing the tertiary structure. Apart from huPrP, where neither H1 nor H3 of the same subunit are involved, in all shPrPs, H1 and its terminus in a 310 conformation are anchored to the region encompassing the H2–H3 loop and H3. In particular, Glu146 is salt bridged to Lys204, and Arg156 is linked to both Glu196 and Asp202 (). It is impressive to notice the conservation and the high number of contacts engaged by the guanidinium moiety of Arg156. As a result, the relative orientation between H1 and H3 is conserved in all the ovine structures and is ∼-70°.
The H1–H2 loop is (internally) stabilized by a salt bridge between Arg164 and Glu168 only in huPrP. The H2–H3 loop is stabilized by a salt bridge between Lys194 and Glu196 only in shPrP (1uw3). This strong interaction is not found in either huPrP, in which this region is involved in the hinge of the swapped dimer, or antibody-bound shPrPs, being this region the antibody epitope.
Interfaces.
The crystal state of a protein or of its complexes offers the opportunity to examine the interface between adjacent symmetry related molecules and hence to study how the molecule can interact not only with small ligands but also with macromolecules. In PrP, the dimer-oligomer formation is a crucial step towards formation of the pathological scrapie form. A careful analysis of the structural features of the interfaces between neighboring PrP molecules may thus be useful for understanding the oligomerization process. We took into account only the interfaces with an area ≥380 Å2. A partial list is reported in .
H2, the following H2–H3 loop and the C-terminus of H3 form the most extensive interfaces in all X-ray structures. They are the regions most prone to interact with other molecules. In huPrP, distinct contacts are also formed between the H2-H2' loop (). This segment forms two distorted β-strands, suggesting a tendency of the C-terminus of H2 and especially of the polyThr stretch residues 190–194) to adopt an extended conformation.
The C-terminus of H2, the H2–H3 loop and the N-terminus of H3 contribute to the largest contact interface in the crystal of 1uw3 (815 A2). The two molecules, related by a dyad axis, face each other and form an extended network of interactions (). In the shPrP-antibody complexes, the C-terminus of H2 and the H2–H3 loop are extensively involved in contacts with the heavy chain of the antibody.Citation90 Binding to the antibody is further stabilized by a salt bridge between H2 Lys197 and Asp29 belonging to the antibody light chain (CitationFig. 5).
Numerous intermolecular contacts are formed between H1 and H3. In 1uw3, hydrogen bonds and salt bridges (Asp144 and Asp 147 linked to Arg228) stabilise the inter-helix interaction (). The largest interface between H1 (including 310) and H3 is located at the dimer interface of the huPrP structure. As H3 protrudes out towards the other subunit, the stabilizing contacts observed in the monomeric shPrP structures between these two helices are preserved in the huPrP swapped dimer ().
In 1uw3, the S1 strand forms an anti-parallel β-sheet with S1 of a symmetry-related molecule. The elongated sheet is stabilized by two intermolecular backbone H-bonds between Tyr128 and Leu130 (). As a result, a four stranded β-sheet is formed across the crystallographic dimer (382 A2). The authors speculate that, despite the small number of contacts, this intermolecular β-sheet is one of the loci for fibre formation.
As a comparison, in all shPrP-antibody complexes, S1 makes contacts with a β-strand of the light chain of the antibody, generating an intermolecular β-sheet (). In this interface, also the side chains of S1-H1 and S2 participate to stabilize the interface by forming H-bonds with the antibody (). The interface is further stabilized by a H-bond between the antibody and H3 (not in figure), suggesting a high tendency of S1 to elongate the β-sheet by interacting also with macromolecules other than PrP. This tendency is part of a general mechanism observed in protein oligomerization.Citation95
Role of non-bulky water molecules.
The PrP crystal structures contain structured water molecules participating in H-bond networks either with the protein or with other water/ligand molecules. We analysed these H-bond patterns in four crystal structures. The structure of the shPrP variant ARR (1tqc) was ignored because its lower resolution and the fewer water molecules—mainly bound to the antibody—with respect to the other structures.
Despite the different crystal packing, the crystal structures contain a few conserved sites where water molecules are bound, indicating a high tendency of these regions to be hydrated.Citation97 One site in particular deserves some comments (water #1, 8, 4 and 62 in the PDB files 1uw3, 1tpx, 1tqb and 1i4m respectively). This water molecule acts as a donor to the side chain oxygen of Ser132 present at the C-terminus of S1 and to OE1 atom of Gln217 in H3 and as an acceptor from the amide nitrogen of Val161 at the N-terminus of S2 (). In this way, the water molecule bridges three secondary structure elements which are distant in the sequence (S1, S2 and H3) and may therefore contribute to stabilising the tertiary structure of the PrP fold. Other studies have been carried out on PrP hydration, but a detailed discussion of the results is beyond the scope of this review.
Validity of Structural Studies of Recombinant Proteins as Models for the Disease
All available PrPC structures were expressed in simplified host organisms such as E. coli, leading to overexpressed recombinant exogenous proteins. This is a common practice in most of the structural and biochemical laboratories. Correctly, however, some concern was put into demonstrating that recombinant PrP is a relevant model of the native PrPC. This point could in principle be particularly tricky and the in vitro studies have no validity for PrP which is a glycoprotein anchored to the lipid membrane. A study on PrPC extracted from healthy calf brain demonstrated that native bovine PrP exhibits the same circular dichroism and 1H-NMR spectra as the recombinant protein.Citation98 Another report showed that the anchoring of recombinant PrPC to a lipid membrane does not affect its fold.Citation99 All together this evidence suggests that in vitro PrPC adopts substantially the same three-dimensional structure of its recombinant form and legitimated the available structures as reliable models of the native protein.
What Does Structure Tell Us About PrP Function?
Despite the impressive number of available studies, the physiological role of PrP remains unknown.Citation41,Citation100 An approach that has proven useful in such cases is that which relies on the knowledge that structure is retained through evolution longer than sequence similarity.Citation101 Identification of structural similarities could therefore suggest new hypotheses about function. Unfortunately, the PrP fold is relatively unique. High structural similarity was nevertheless detected between the PrP C-terminal domain and the doppel (Dpl) proteinCitation102 despite their distinct origins.Citation60 The two proteins share ∼25% identityCitation60 and their structures superpose with an r.m.s.d. of ∼3.8–4.0 ÅCitation103,Citation104 (). In knock-out mice studies, it was noticed that while some PrP0/0 lines were viable and appeared phenotypically normal,Citation105 others developed late-onset ataxia related to degeneration of Purkinje neurons.Citation106 Although not normally expressed in the central nervous system, Dpl was found to be upregulated in the PrP0/0 lines that develop ataxia.Citation102 This phenotype could be rescued by crossing the null mice with those overexpressing wild-type mPrP, thus suggesting that Dpl is able of replacing PrPC in vivo.Citation107 While very tempting, this hypothesis still provides relatively little advancement to understanding the function of PrP, since even less (if possible) is known about the Dpl function. Forthcoming studies on the recently discovered PrP-like protein, namely Shadoo, may add new light to understanding the PrP protein family.Citation108,Citation109
The Structure of PrPSc in Amyloid Fibres
While not directly the aim of this review, we shall now summarize briefly the current knowledge on the structure of PrPSc. Its physicochemical properties greatly differ from the cellular form. While PrPC is a monomeric and easily degradable protein linked to the out-membrane, PrPSc, as isolated from infected brains, is aggregated and at least partially protease resistant.Citation20–Citation22 Only the N-terminal 23–89 fragment can be cleaved, generating a well defined resistant core of an apparent mass of 27–30 kDa, termed PrP27–30.Citation110 PrP27–30 retains infectious character and can further polymerize to form fibres.Citation20,Citation111 In addition, PrPSc can differ depending on many factors, i.e., diverse PrPSc are formed with or without the GPI anchor.Citation17,Citation18 Particularly intriguing is the phenomenon of the PrP strains.Citation48,Citation112,Citation113
As opposed to PrPC, very little is known about the PrPSc structure due to its fibrillar nature and its insolubility, which have hampered structural studies by classical high resolution methods and demanded the use of alternative approaches. FT-IR spectroscopy using both conventional and synchrotron sources has indicated that PrPSc and PrP27–30 contain a higher amount of β-structure and a lower α-helical content.Citation21,Citation114,Citation115 The amyloid nature of the PrP27–30 fibres was supported by the presence of a cross-β structure, as observed by X-ray fibre diffraction.Citation116 Electron diffraction data on two-dimensional crystals of PrP27–30 allowed building a model, in which the region 89–175 forms left-handed β-helices that associate in turn to form trimers (). The remaining sequence of the C-terminal domain is supposed to maintain its native α-helical structure (H2 and H3).Citation55,Citation117,Citation118 Sugars, labelled with monoamino nano-gold, were also localised at the periphery of the oligomers. Chemical cross-linking experiments support this model.Citation119
A β-helix is however inconsistent with mass spectrometry and with H/D backbone amide exchange data obtained from fibrillar samples of the recombinant proteinCitation120 which suggest that the sequence covering residues ∼169–221 contributes to the core of the β-sheet, whereas residues 90–168 do not form H-bonded structures. A way to explain these differences is to assume that they arise from the different nature of the specimens used.
Further information on the PrPSc structure could potentially come from solid state NMR studies,Citation121 which have been successfully used to characterize other amyloidogenic proteins.Citation122
Finally, it is worth mentioning the recent work by the Eisenberg's group on short peptides, among which are the MIHFGND, NNQNTF and SNQNNF sequences from mPrP, ePrP and huPrP respectively, which are known to form fibrils.Citation123,Citation124 Their X-ray structures at atomic resolution have revealed that the peptides exhibit a cross-β spine as a common structural feature. Specifically, two interdigitated β-sheets pack against each others generating a complementary dry ‘steric zipper’ (). This motif has been surmised to be the universal structure of amyloid. The polymorphism of the steric zipper observed among various crystals is consistent with PrP polymorphism and strain. Future studies along this direction will almost certainly provide a further contribution to our knowledge of the amyloid structure in general and of PrPSc in particular.Citation125
Conclusion
The structural description of PrPC as reviewed in this paper provides an important prerequisite for our further comprehension of the nature of the structural conversion from a healthy protein to a fatal pathological agent observed in PrP diseases and to provide a model of how infectivity may take place. The available data provide precious details onto the subtle differences between highly homologous proteins from quite different species. How this knowledge can now be exploited to identify the cellular function of PrP and extended to characterize the structure of PrPSc remains an important challenge for the future. There is anyway no doubt about the central role that structural biology plays in our attempt to undertake the long journey which will hopefully leads eventually to a deep comprehension of the pathological mechanisms of PrP diseases.
Abbreviations
| PrP | = | prion protein |
| PrPC | = | cellular PrP |
| PrPSc | = | scrapie PrP |
| shPrP | = | sheep PrP |
| huPr | = | human PrP |
| mPrP | = | mouse PrP |
| ePrP | = | elk PrP |
| chPrP | = | chicken PrP |
| tPrP | = | turtle PrP |
| xlPrP | = | frog PrP |
| H1 | = | helix 1 |
| H2 | = | helix 2 |
| H3 | = | helix 3 |
| S1 | = | strand 1 |
| S2 | = | strand 2 |
Figures and Tables
Figure 1 Overview of the PrP sequence. (A) Block diagram of the PrP architecture. The residue numbering refers to huPrP. The positions of the glycosilation sites are indicated at Asn181 and Asn197. Cys179 is covalently bound to Cys214. The number of octarepeats depends on the species considered. (B) Charge distribution along the sequence of huPrP. Only the charged residues are indicated. The N-terminus contains only positively charged residues (11 charges), whereas the C-terminus contains both types, with a slight excess of negative residues.
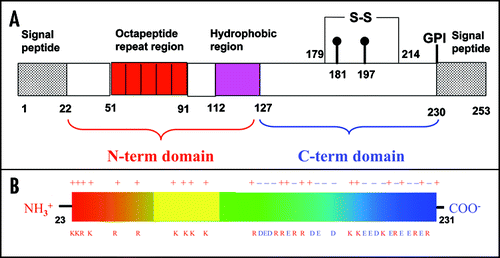
Figure 2 Sequence alignment and overall fold of PrP. (A) Sequence alignment of a subset of PrP precursor sequences chosen as the ones for which the structure of the C-terminal domain is available (boxed in blue). The alignment was obtained and color coded by Clustalx.Citation67 Stars, semicolons and dots refer to conserved and partially conserved residues, according to Clustalx convention. (B) Ribbon representation of the C-terminal domain of mPrP.Citation68 The secondary structure elements and the N- and C-termini are labelled. The sulphur bridge is indicated in yellow.
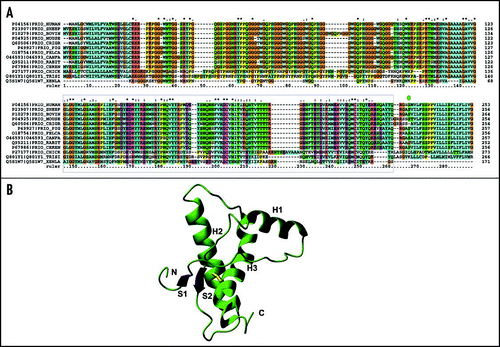
Figure 3 Bundles of the NMR structures. (A) Superposition of the nine huPrP structures. The backbone atoms of the secondary structure elements (residues 129–130, 145–152, 162–163, 173–190, 200–222) were arbitrarily superposed on the coordinates of 1qm0 (shown in red). The minimised structure was used when available. Otherwise, the first structure of the bundle was selected. (B) Superposition of the PrP structures from other mammals. The best pairwise superposition according to Dali was found arbitrarily using the coordinates of mPrP (shown in red). (C) The coordinates of the C-terminal domains of bird and reptile PrPs. Left, turtle; middle, frog; right, chicken. The same orientation as in Figure 2B was chosen.
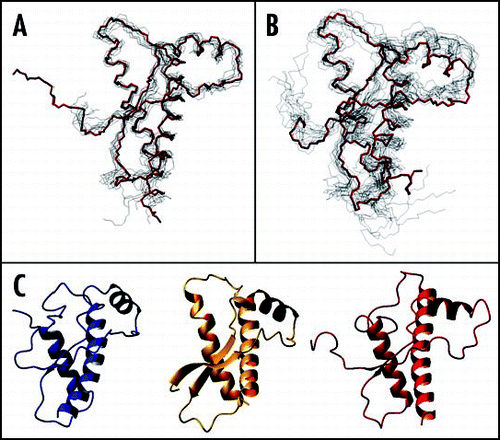
Figure 4 Comparison between the X-ray structures of the PrP C-terminal domain. (A) Superposition of shPrP (1uw3, in red) from Leu125 to Ala230 (huPrP numbering) with shPrP (1tpx, in blue) from Gly127 to Tyr228. (B) Superposition of shPrP (1uw3, in red) from Leu125 to Ala230 (huPrP numbering) with huPrP (1i4m, chain A in cyan, chain B in magenta) from residue Gly119A to Thr191A and from residue Glu196B to Tyr226B.
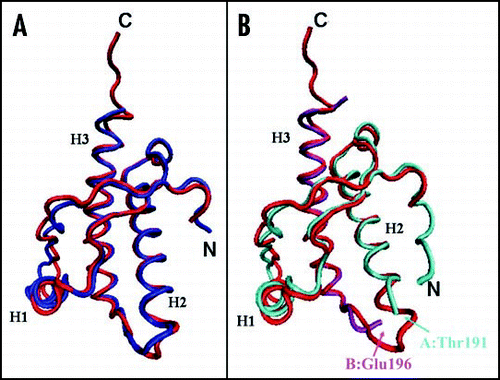
Figure 5 Schematic representation of the fold of the PrP C-terminal domain, indicating the most relevant ionic interactions (within 3.8 Å) as observed in the X-ray structures. The disulfide bridge connecting H2 to H3 is indicated in yellow. Residue numbering, sequence (apart from the mutation R148C) and secondary structure assignment refer to huPrP and are consistent with 1uw3 (the sequence of the antibody-bound shPrP differs by +3). Ellipses represent residues in α-helix; green rectangles represent residues in β-strand; diamonds represent residues in 310 helix; transparent rectangles represent charged residues in loops. Red, blue, green, purple and cyan lines refer to interactions observed in the 1u3w, 1tpx, 1tqb, 1tqc and 1i4m structures, respectively. Interactions within the dimer were considered for 1i4m. Inter-molecular interactions between adjacent molecules in the crystal state are shown by red dotted lines.
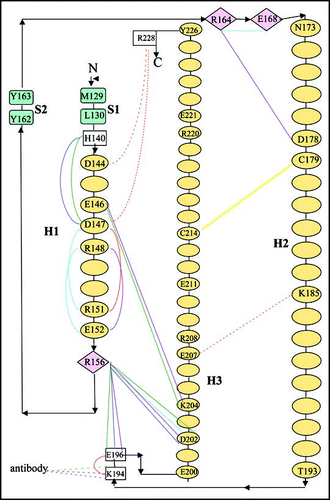
Figure 6 Interfaces observed in the crystal structures (see also ). (A) Dimeric swapped structure of huPrP (1i4m). Chains A and B are drawn in cyan and magenta respectively. The extended surface partly encompasses H1, H2, H2' and H3. A detailed view of the hinge region between the two subunits is shown in the inset. The newly formed β-strand encompasses the polyThr stretch (190–194). (B) shPrP structure (1uw3). The two symmetry axis related molecules are drawn in red and green. The contact surface encompasses the C-terminus of H2, the connecting loop, and the N-terminus of H3. The ionic interaction between the side chain of Lys185 and Glu207 is indicated. H-bonds close to the surface are omitted for clarity. (C) Close up view of shPrP 1uw3 showing how the β-sheet elongates through interaction between adjacent molecules. The side chains of selected residues are indicated. Met129 and Leu130 are labelled only once. Only backbone H-bonds are drawn, with N and O atoms indicated by spheres. (D) β-sheet elongation in the 1tpx shPrP structure (in blue). The antibody C chain is drawn in green. The contact surface encompasses PrP S1 and the short edge strand belonging to the four-stranded β-sheet, named I, of the antibody C chain. Only two strands of sheet I are drawn.
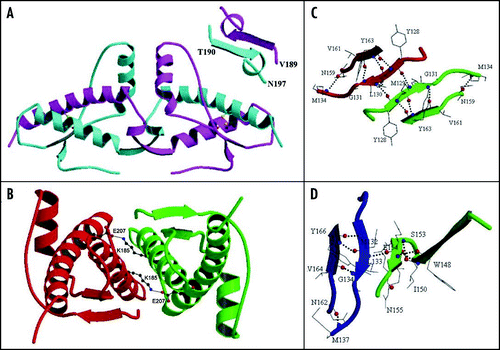
Figure 7 Conserved hydration site in the crystal state. The figure refers to shPrP (1uw3), as an example. Distances are given in Å.
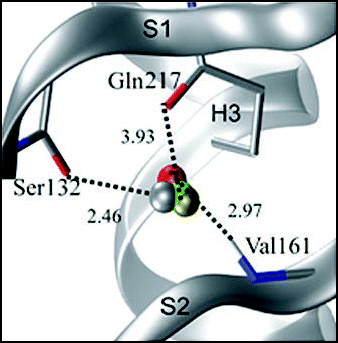
Figure 8 Structural comparison between the C-terminal domain of PrP and Dpl. The structures of (A) mPrP (1ag2), (B) hamster PrP (1b10), (C) mouse Dpl (1i17) and (D) human Dpl (1lg4) were superposed by the Dali server and then displaced. The two Dpl structures superpose with an r.m.s.d. of 2.0 Å over 97 residues (Z-score 13.45), whereas the two mouse paralogs superpose with an r.m.s.d. of 3.8 Å over 87 residues (Z score 5.0).
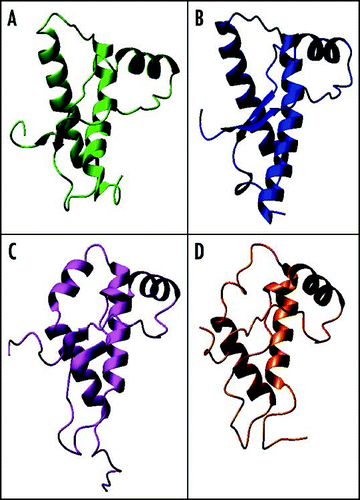
Figure 9 Current models of PrPSc. (A) Trimeric model of PrP27–30 after Govaerts et al., 2004.Citation55 (B) Crystal structure of the PrP SNQNNF peptide (PDB code 2ol9).Citation124
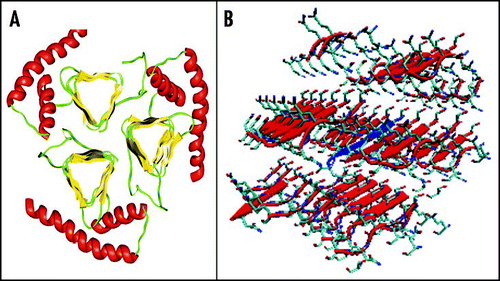
Table 1 Summary of relevant information about the NMR structures
Table 2 Summary of relevant information about the X-ray structures
Table 3 R.m.s.d. (Å) between the X-ray and the NMR structures; the number of the Cα atoms superposed is given in parentheses
Table 4 Classification of a few selected interfaces observed in the crystal state; only some interfaces with an area ≥380 Å2 were listed
Acknowledgements
We apologize to those colleagues whose work could not be cited due to space limitation. We thank CNISM for supporting A.P. as a visiting scientist at the University of Naples.
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 144
- Prusiner SB. Prions are novel infectious pathogens causing scrapie and Creutzfeldt-Jakob disease. Bioessays 1986; 5:281 - 286
- Prusiner SB. The prion diseases. Brain Pathol 1998; 8:499 - 513
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Prusiner SB. Human prion diseases and neurodegeneration. Curr Top Microbiol Immunol 1996; 207:1 - 17
- Huang Z, Prusiner SB, Cohen FE. Scrapie prions: A three-dimensional model of an infectious fragment. Fold Des 1995; 1:13 - 19
- Campbell PN. Bovine spongiform encephalopathy—some surprises for biochemists. IUBMB Life 2005; 57:273 - 276
- Sigurdson CJ, Aguzzi A. Chronic wasting disease. Biochim Biophys Acta 2007; 1772:610 - 618
- Beringue V, Andreoletti O, Le Dur A, Essalmani R, Vilotte JL, Lacroux C, Reine F, Herzog L, Biacabe AG, Baron T, Caramelli M, Casalone C, Laude H. A bovine prion acquires an epidemic bovine spongiform encephalopathy strain-like phenotype on interspecies transmission. J Neurosci 2007; 27:6965 - 6971
- Aguzzi A, Polymenidou M. Mammalian prion biology: One century of evolving concepts. Cell 2004; 116:313 - 327
- Baldwin MA, James TL, Cohen FE, Prusiner SB. The three-dimensional structure of prion protein: Implications for prion disease. Biochem Soc Trans 1998; 26:481 - 486
- Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem 1998; 67:793 - 819
- Harrison PM, Bamborough P, Daggett V, Prusiner SB, Cohen FE. The prion folding problem. Curr Opin Struct Biol 1997; 7:53 - 59
- Baskakov IV, Legname G, Gryczynski Z, Prusiner SB. The peculiar nature of unfolding of the human prion protein. Protein Sci 2004; 13:586 - 595
- Soto C, Estrada L, Castilla J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem Sci 2006; 31:150 - 155
- Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. Amyloid: Toward terminology clarification. Report from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 2005; 12:1 - 4
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 1439
- Aguzzi A. Cell biology: Prion toxicity: All sail and no anchor. Science 2005; 308:1420 - 1421
- Aguzzi A, Heikenwalder M. Pathogenesis of prion diseases: Current status and future outlook. Nat Rev Microbiol 2006; 4:765 - 775
- McKinley MP, Meyer RK, Kenaga L, Rahbar F, Cotter R, Serban A, Prusiner SB. Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. J Virol 1991; 65:1340 - 1351
- Sunde M, Blake CC. From the globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Q Rev Biophys 1998; 31:1 - 39
- Leffers KW, Wille H, Stohr J, Junger E, Prusiner SB, Riesner D. Assembly of natural and recombinant prion protein into fibrils. Biol Chem 2005; 386:569 - 580
- Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci 1999; 24:329 - 332
- Vendruscolo M, Zurdo J, MacPhee CE, Dobson CM. Protein folding and misfolding: A paradigm of self-assembly and regulation in complex biological systems. Philos Transact A Math Phys Eng Sci 2003; 361:1205 - 1222
- Dobson CM. Protein folding and misfolding. Nature 2003; 426:884 - 890
- Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol 2004; 15:3 - 16
- Dobson CM. Experimental investigation of protein folding and misfolding. Methods 2004; 34:4 - 14
- Dobson CM. Structural biology: Prying into prions. Nature 2005; 435:747 - 749
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 2006; 75:333 - 366
- Dobson CM. Protein aggregation and its consequences for human disease. Protein Pept Lett 2006; 13:219 - 227
- Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem 2002; 277:21140 - 21148
- Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, Riesner D. Search for a prion-specific nucleic acid. J Virol 2005; 79:10796 - 10806
- Caughey B, Baron GS. Prions and their partners in crime. Nature 2006; 443:803 - 810
- Griffith JS. Self-replication and scrapie. Nature 1967; 215:1043 - 1044
- Prusiner SB. Scrapie prions. Annu Rev Microbiol 1989; 43:345 - 374
- Soto C, Castilla J. The controversial protein-only hypothesis of prion propagation. Nat Med 2004; 10:63 - 67
- Abid K, Soto C. The intriguing prion disorders. Cell Mol Life Sci 2006; 63:2342 - 2351
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J Mol Med 2003; 81:678 - 699
- Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J Biol Chem 2006; 281:13828 - 13836
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science 2004; 305:673 - 676
- Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: A phenotype under challenge. Prion 2007; 2:83 - 93
- Richt JA, Kasinathan P, Hamir AN, Castilla J, Sathiyaseelan T, Vargas F, Sathiyaseelan J, Wu H, Matsushita H, Koster J, Kato S, Ishida I, Soto C, Robl JM, Kuroiwa Y. Production of cattle lacking prion protein. Nat Biotechnol 2007; 25:132 - 138
- Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 2005; 435:765 - 772
- Chernoff YO. Stress and prions: Lessons from the yeast model. FEBS Lett 2007; 581:3695 - 3701
- Surewicz WK. Protein science: Discriminating taste of prions. Nature 2007; 447:541 - 542
- Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T. Prions of fungi: Inherited structures and biological roles. Nat Rev Microbiol 2007; 5:611 - 618
- Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T, Engel A, McCann L, Kryndushkin D. Yeast prions: Evolution of the prion concept. Prion 2007; 2:83 - 93
- Tessier PM, Lindquist S. Prion recognition elements govern nucleation, strain specificity and species barriers. Nature 2007; 447:556 - 561
- Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell 2005; 121:195 - 206
- Castilla J, Saa P, Morales R, Abid K, Maundrell K, Soto C. Protein misfolding cyclic amplification for diagnosis and prion propagation studies. Methods Enzymol 2006; 412:3 - 21
- Saa P, Castilla J, Soto C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem 2006; 281:35245 - 35252
- Deleault NR, Harris BT, Rees JR, Supattapone S. From the cover: Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 2007; 104:9741 - 9746
- Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, Ghetti B, Harris DA. Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol 2003; 77:7611 - 7622
- Wuthrich K, Riek R. Three-dimensional structures of prion proteins. Adv Protein Chem 2001; 57:55 - 82
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Natl Acad Sci USA 2004; 101:8342 - 8347
- Heikenwalder M, Julius C, Aguzzi A. Prions and peripheral nerves: A deadly rendezvous. J Neurosci Res 2007; 85:2714 - 2725
- Rudd PM, Endo T, Colominas C, Groth D, Wheeler SF, Harvey DJ, Wormald MR, Serban H, Prusiner SB, Kobata A, Dwek RA. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc Natl Acad Sci USA 1999; 96:13044 - 13049
- Rudd PM, Wormald MR, Wing DR, Prusiner SB, Dwek RA. Prion glycoprotein: Structure, dynamics, and roles for the sugars. Biochemistry 2001; 40:3759 - 3766
- Wopfner F, Weidenhofer G, Schneider R, von Brun A, Gilch S, Schwarz TF, Werner T, Schatzl HM. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol 1999; 289:1163 - 1178
- Rivera-Milla E, Oidtmann B, Panagiotidis CH, Baier M, Sklaviadis T, Hoffmann R, Zhou Y, Solis GP, Stuermer CA, Malaga-Trillo E. Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. Faseb J 2006; 20:317 - 319
- Millhauser GL. Copper and the prion protein: Methods, structures, function, and disease. Annu Rev Phys Chem 2007; 58:299 - 320
- Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, Prusiner SB, Wright PE, Dyson HJ. Structure of the recombinant full-length hamster prion protein PrP(29–231): The N terminus is highly flexible. Proc Natl Acad Sci USA 1997; 94:13452 - 13457
- Zahn R. The octapeptide repeats in mammalian prion protein constitute a pH-dependent folding and aggregation site. J Mol Biol 2003; 334:477 - 488
- Blanch EW, Gill AC, Rhie AG, Hope J, Hecht L, Nielsen K, Barron LD. Raman optical activity demonstrates poly(L-proline) II helix in the N-terminal region of the ovine prion protein: Implications for function and misfunction. J Mol Biol 2004; 343:467 - 476
- Rath A, Davidson AR, Deber CM. The structure of “unstructured” regions in peptides and proteins: Role of the polyproline II helix in protein folding and recognition. Biopolymers 2005; 80:179 - 185
- Lysek DA, Wuthrich K. Prion protein interaction with the C-terminal SH3 domain of Grb2 studied using NMR and optical spectroscopy. Biochemistry 2004; 43:10393 - 10399
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 2003; 31:3497 - 3500
- Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121–321). Nature 1996; 382:180 - 182
- Liu H, Farr-Jones S, Ulyanov NB, Llinas M, Marqusee S, Groth D, Cohen FE, Prusiner SB, James TL. Solution structure of Syrian hamster prion protein rPrP(90–231). Biochemistry 1999; 38:5362 - 5377
- Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 2000; 97:145 - 150
- Lopez Garcia F, Zahn R, Riek R, Wuthrich K. NMR structure of the bovine prion protein. Proc Natl Acad Sci USA 2000; 97:8334 - 8339
- Viles JH, Donne D, Kroon G, Prusiner SB, Cohen FE, Dyson HJ, Wright PE. Local structural plasticity of the prion protein: Analysis of NMR relaxation dynamics. Biochemistry 2001; 40:2743 - 2753
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83:79 - 90
- Zahn R, Guntert P, von Schroetter C, Wuthrich K. NMR structure of a variant human prion protein with two disulfide bridges. J Mol Biol 2003; 326:225 - 234
- Calzolai L, Zahn R. Influence of pH on NMR structure and stability of the human prion protein globular domain. J Biol Chem 2003; 278:35592 - 35596
- Calzolai L, Lysek DA, Guntert P, von Schroetter C, Riek R, Zahn R, Wuthrich K. NMR structures of three single-residue variants of the human prion protein. Proc Natl Acad Sci USA 2000; 97:8340 - 8345
- Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sonnichsen FD. Solution structure of the E200K variant of human prion protein: Implications for the mechanism of pathogenesis in familial prion diseases. J Biol Chem 2000; 275:33650 - 33654
- Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci USA 2005; 102:646 - 650
- Lysek DA, Schorn C, Nivon LG, Esteve-Moya V, Christen B, Calzolai L, von Schroetter C, Fiorito F, Herrmann T, Guntert P, Wuthrich K. Prion protein NMR structures of cats, dogs, pigs, and sheep. Proc Natl Acad Sci USA 2005; 102:640 - 645
- Li J, Mei FH, Xiao GF, Guo CY, Lin DH. 1H, 13C and 15N resonance assignments of rabbit prion protein (91–228). J Biomol NMR 2007; 38:181
- James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang H, Donne DG, Kaneko K, Groth D, Mehlhorn I, Prusiner SB, Cohen FE. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci USA 1997; 94:10086 - 10091
- Calzolai L, Lysek DA, Perez DR, Guntert P, Wuthrich K. Prion protein NMR structures of chickens, turtles, and frogs. Proc Natl Acad Sci USA 2005; 102:651 - 655
- Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231). FEBS Lett 1997; 413:282 - 288
- Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wuthrich K. Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci USA 1998; 95:11667 - 11672
- Billeter M, Riek R, Wider G, Hornemann S, Glockshuber R, Wuthrich K. Prion protein NMR structure and species barrier for prion diseases. Proc Natl Acad Sci USA 1997; 94:7281 - 7285
- Glockshuber R, Hornemann S, Riek R, Wider G, Billeter M, Wuthrich K. Three-dimensional NMR structure of a self-folding domain of the prion protein PrP(121–231). Trends Biochem Sci 1997; 22:241 - 242
- Yang JM, Zhao DM, Liu HX, Li N, Hao YX, Ning ZY, Qin XH. Comparative analysis of the prion protein open reading frame nucleotide sequences in peacock and parakeet. Virus Genes 2005; 30:193 - 196
- Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol 2001; 8:770 - 774
- Haire LF, Whyte SM, Vasisht N, Gill AC, Verma C, Dodson EJ, Dodson GG, Bayley PM. The crystal structure of the globular domain of sheep prion protein. J Mol Biol 2004; 336:1175 - 1183
- Eghiaian F, Grosclaude J, Lesceu S, Debey P, Doublet B, Treguer E, Rezaei H, Knossow M. Insight into the PrPC—>PrPSc conversion from the structures of antibody-bound ovine prion scrapie-susceptibility variants. Proc Natl Acad Sci USA 2004; 101:10254 - 10259
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996; 39:767 - 768
- Bennett MJ, Sawaya MR, Eisenberg D. Deposition diseases and 3D domain swapping. Structure 2006; 14:811 - 824
- Kallberg Y, Gustafsson M, Persson B, Thyberg J, Johansson J. Prediction of amyloid fibril-forming proteins. J Biol Chem 2001; 276:12945 - 12950
- Dima RI, Thirumalai D. Exploring the propensities of helices in PrP(C) to form beta sheet using NMR structures and sequence alignments. Biophys J 2002; 83:1268 - 1280
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 2004; 22:1302 - 1306
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol 2007; 372:774 - 797
- De Simone A, Dodson GG, Verma CS, Zagari A, Fraternali F. Prion and water: Tight and dynamical hydration sites have a key role in structural stability. Proc Natl Acad Sci USA 2005; 102:7535 - 7540
- Hornemann S, Schorn C, Wuthrich K. NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep 2004; 5:1159 - 1164
- Eberl H, Tittmann P, Glockshuber R. Characterization of recombinant, membrane-attached full-length prion protein. J Biol Chem 2004; 279:25058 - 25065
- Hu W, Kieseier B, Frohman E, Eagar TN, Rosenberg RN, Hartung HP, Stuve O. Prion proteins: Physiological functions and role in neurological disorders. J Neurol Sci 2007;
- Irving JA, Whisstock JC, Lesk AM. Protein structural alignments and functional genomics. Proteins 2001; 42:378 - 382
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AF, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE, Westaway D. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 1999; 292:797 - 817
- Mo H, Moore RC, Cohen FE, Westaway D, Prusiner SB, Wright PE, Dyson HJ. Two different neurodegenerative diseases caused by proteins with similar structures. Proc Natl Acad Sci USA 2001; 98:2352 - 2357
- Luhrs T, Riek R, Guntert P, Wuthrich K. NMR structure of the human doppel protein. J Mol Biol 2003; 326:1549 - 1557
- Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356:577 - 582
- Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Kataoka Y, Houtani T, Shirabe S, Okada H, Hasegawa S, Miyamoto T, Noda T. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 1996; 380:528 - 531
- Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, Erpel SP, Nakaoke R, Atarashi R, Houtani T, Torchia M, Sakaguchi S, DeArmond SJ, Prusiner SB, Katamine S. A mouse prion protein transgene rescues mice deficient for the prion protein gene from purkinje cell degeneration and demyelination. Lab Invest 1999; 79:689 - 697
- Watts JC, Drisaldi B, Ng V, Yang J, Strome B, Horne P, Sy MS, Yoong L, Young R, Mastrangelo P, Bergeron C, Fraser PE, Carlson GA, Mount HT, Schmitt-Ulms G, Westaway D. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J 2007; 26:4038 - 4050
- Watts JC, Westaway D. The prion protein family: Diversity, rivalry, and dysfunction. Biochim Biophys Acta 2007; 1772:654 - 672
- Turk E, Teplow DB, Hood LE, Prusiner SB. Purification and properties of the cellular and scrapie hamster prion proteins. Eur J Biochem 1988; 176:21 - 30
- McKinley MP, Prusiner SB. Biology and structure of scrapie prions. Int Rev Neurobiol 1986; 28:1 - 57
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274:2079 - 2082
- Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ, Lantos P. The same prion strain causes vCJD and BSE. Nature 1997; 389:448 - 450 526
- Kneipp J, Miller LM, Joncic M, Kittel M, Lasch P, Beekes M, Naumann D. In situ identification of protein structural changes in prion-infected tissue. Biochim Biophys Acta 2003; 1639:152 - 158
- Wang Q, Kretlow A, Beekes M, Naumann D, Miller L. In situ characterization of prion protein structure and metal accumulation in scrapie-infected cells by synchrotron infrared and X-ray imaging. Vibrational Spectroscopy 2005; 38:61 - 69
- Nguyen JT, Inouye H, Baldwin MA, Fletterick RJ, Cohen FE, Prusiner SB, Kirschner DA. X-ray diffraction of scrapie prion rods and PrP peptides. J Mol Biol 1995; 252:412 - 422
- Wille H, Prusiner SB. Ultrastructural studies on scrapie prion protein crystals obtained from reverse micellar solutions. Biophys J 1999; 76:1048 - 1062
- Wille H, Michelitsch MD, Guenebaut V, Supattapone S, Serban A, Cohen FE, Agard DA, Prusiner SB. Structural studies of the scrapie prion protein by electron crystallography. Proc Natl Acad Sci USA 2002; 99:3563 - 3568
- Onisko B, Fernandez EG, Freire ML, Schwarz A, Baier M, Camina F, Garcia JR, Rodriguez-Segade Villamarin S, Requena JR. Probing PrPSc structure using chemical cross-linking and mass spectrometry: Evidence of the proximity of Gly90 amino termini in the PrP 27–30 aggregate. Biochemistry 2005; 44:10100 - 10109
- Lu X, Wintrode PL, Surewicz WK. Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc Natl Acad Sci USA 2007; 104:1510 - 1515
- Tycko R. Molecular structure of amyloid fibrils: Insights from solid-state NMR. Q Rev Biophys 2006; 39:1 - 55
- Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer‘s beta-amyloid fibrils. Biochemistry 2006; 45:498 - 512
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005; 435:773 - 778
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007; 447:453 - 457
- Eisenberg D, Nelson R, Sawaya MR, Balbirnie M, Sambashivan S, Ivanova MI, Madsen AO, Riekel C. The structural biology of protein aggregation diseases: Fundamental questions and some answers. Acc Chem Res 2006; 39:568 - 575