Abstract
In vivo amyloid formation is a widespread phenomenon in eukaryotes. Self-perpetuating amyloids provide a basis for the infectious or heritable protein isoforms (prions). At least for some proteins, amyloid-forming potential is conserved in evolution despite divergence of the amino acid (aa) sequences. In some cases, prion formation certainly represents a pathological process leading to a disease. However, there are several scenarios in which prions and other amyloids or amyloid-like aggregates are either shown or suspected to perform positive biological functions. Proven examples include self/nonself recognition, stress defense and scaffolding of other (functional) polymers. The role of prion-like phenomena in memory has been hypothesized. As an additional mechanism of heritable change, prion formation may in principle contribute to heritable variability at the population level. Moreover, it is possible that amyloid-based prions represent by-products of the transient feedback regulatory circuits, as normal cellular function of at least some prion proteins is decreased in the prion state.
Introduction: Prions as the Second Order Templates
The central dogma of molecular biologyCitation1 provides a specific mechanism for the previously postulatedCitation2 template principle in biology. DNA and RNA can be considered as the first order templates, that is, linear or sequence templates, either for each other or for polypeptides. Discovery of infectious proteins (prions),Citation3 and especially of prion mechanism of inheritanceCitation4 introduced templates of another type, structural or conformational templates, which could be designated as second order templates.Citation5 According to the current view,Citation4,Citation6,Citation7 the process of propagation of the amyloid-based prions begins with a conformational change in the protein, and is followed by “linear crystallization,” producing amyloid fibers. The new rounds of prion multiplication may be initiated or seeded with preexisting amyloid fragments. Transmission of these fragments in cell divisions results in the inheritance of the prion state in yeast and fungal systems. Physiochemical studies of elementary amyloid particles uncovered the β-rich structure,Citation8–Citation10 in some examplesCitation11 held together by the intermolecular parallel β-sheets. Variations of this structure apparently determine patterns of the specific variants, or “strains” of a given prion protein.Citation12
Conformation templating of a yeast prion can be reproduced in vitro,Citation13,Citation14 resulting in generation of infectious prion particles, faithfully reproducing the variant-specific patterns upon transformation into the yeast cells. Yeast and fungal prions known to date are described in detail in other reviews (see refs. Citation4 and Citation6). Patterns of the mammalian prion protein PrP have also been reviewed recently (refs. Citation15 and Citation16).
Prion propagation is a highly sequence-specific process. Domains forming an axis of the amyloid fiber should be identical to each other at the level comparable to that required for the complementary interaction of nucleic acid sequences. However, aggregating proteins of different sequences can facilitate aggregation of each other in certain assays. For example, de novo appearance of the prion conformation of the yeast protein Sup35, containing a QN-rich prion domain, is facilitated in the presence of the prion isoform of another QN-rich protein, Rnq1,Citation17–Citation19 reflecting the existence of a prion network. Molecular mechanism of this interaction is still unclear, and it does not seem to involve a template-like component.
Amyloid is probably an ancient fold, as almost any protein can form an amyloid in vitro depending on conditions.Citation20,Citation21 Moreover, second order templating is not restricted to prions. Other examples of “structural inheritance” involve inheritance of preformed structures in Protozoa.
As prions and similar phenomena appear to be widespread, the question arises whether these phenomena play a biological role. Two possible models of the biological role of prions were proposed in literature. One model, designated here and further as “prion pathology” model, states that prion (or amyloid) formation is a pathological process, while conservation of amyloid-forming potential in evolution is due to other adaptive functions of prion-forming proteins, which are not necessarily related to prion formation per se (example in ref. Citation22). Another model, designated here and further as model of “adaptive prionization,” suggests that prion formation by itself could be an adaptive process, so that certain prions are responsible for adaptive traits (example in ref. Citation23).
“Prion Pathology” Model
Mammalian prion diseases and other aggregation-related diseases.
Examples of the “prion diseases” are well known and include various infectious neurodegenerative diseases in mammals.Citation15,Citation16 According to the “protein only” concept, which is now accepted by the majority of experts, the PrP protein in its prion form (PrPSc) is the sole component of a “transmissible particle” that is responsible for the genesis and transmission of a disease. Usually, there is a correlation between the disease and cerebral accumulation of PrP.Citation3,Citation24 The properties of PrP are very similar to those seen in various noninfectious amyloidoses and neural inclusion disorders, a large and heterogeneous group including more than 20 human diseases, among them Alzheimer's, Huntington's and Parkinson's diseases,Citation25 resulting from conversion of certain proteins or their fragments from the normally soluble form to insoluble fibrils or plaques.
Although protein-destabilizing mutations can confer the ability to form amyloids in vivo even to such commonly known proteins as lysozyme,Citation26 usually disease-related aggregation depends on the presence of the specific elements of the primary structure. One feature frequently associated with aggregation is the presence of regions within proteins that comprise a single homopolymeric tract of a particular amino acid and are called homopeptide repeats, or SSR (single sequence repeats).Citation27 It has been shown that uncontrolled genetic expansions of SSR regions lead to the development of some neurodegenerative disorders, for example Huntington's disease, associated with the expanded poly-Q tract in the protein called huntingtin.Citation28 Several other diseases involve different proteins with poly-Q tracts but exhibit a similar mechanism of pathology. It was also demonstrated that some SSRs not linked to the specific disease are toxic to cells when overexpressed and/or lead to protein aggregation.Citation29–Citation31
These and the other facts indicate that accumulation of the amyloid-like aggregates is a pathological process. This notion is further confirmed by the existence of mechanisms preventing amyloid-like protein aggregation, such as a specific chaperone preventing aggregation of excess α-globin chains.Citation32 As misfolded and potentially aggregating proteins are usually accumulated during aging, it is an intriguing possibility that aging could promote prion-like pathologies. Indeed, some aggregation-related diseases (e.g., Alzheimer's disease) in humans are frequently associated with advanced age.
Exact mechanism of cell death in amyloid and neural inclusion disorders remains unknown. At least in case of mammalian PrP, it is certainly not due to lack of the normal protein function, as deletion of the PrP-coding gene does not cause a disease in mice.Citation33 For Huntington's disease, it is proposed that aggregates sequester some essential cellular proteins.Citation34–Citation37 Poly-Q constructs introduced into Caenorhabditis elegans induce heat shock response at the stringency proportional to the length of the poly-Q stretch,Citation38 and disrupt the global quality control of protein folding, possibly by interfering with the disposal of misfolded proteins.Citation34 There is evidence that PrP and some other amyloidogenic proteins trigger cell death via apoptosis or autophagy.Citation39–Citation42
Pathological effects of amyloids in yeast.
Aggregated (prion) forms of the yeast proteins Sup35 and Ure2, called respectively [PSI+] and [URE3], are not found in the natural, industrial and clinical isolates of Saccharomyces yeast,Citation22,Citation43,Citation44 consistent with the possibility of their pathogenicity. However the prion form of Rnq1 protein, called [PIN+], was found in a few natural isolates.Citation22,Citation44 [URE3] decreases the growth rate of yeast.Citation22 While [PSI+] does not affect growth rates of exponential cells,Citation45 some [PSI+] strains exhibit facilitated cell death in the deep stationary phase, similar to apoptotic processes in higher eukaryotes (Y. Chernoff, J. Kumar, and G. Newnam, unpublished data). Activation of the apoptosis-like programmed cell death pathways in the starving yeast cells has been reported previously (refs. Citation46–Citation48). Some combinations of [PSI+] and [URE3] isolates exhibit the synthetic lethal or sublethal interactions.Citation49
Overproduced Sup35 protein or fragments containing the Sup35 prion domain (Sup35N) are toxic to the [PSI+] cells, or (at very high levels) to the [psi−] cells containing the [PIN+] prion, that facilitates de novo [PSI+] induction.Citation17,Citation50–Citation52 This toxicity is not simply due to accumulation of excess protein per se, as it is controlled by the same protein regions that are involved in prion formation, and is not seen in the [psi− pin−] background.Citation17,Citation52 It is shown that accumulation of aggregated Sup35 in the prion-containing cells is associated with cell death.Citation53,Citation54 This somewhat parallels mammalian prion diseases, where PrPSc-related pathology is usually detected only in neurons, cells known to produce mammalian prion protein (PrP) at high levels.Citation3
Some mammalian amyloidogenic proteins are also toxic to yeast. The poly-Q expanded fragment of human huntingtin, fused to the green fluorescent protein (GFP) generates aggregates and causes toxicity only in yeast cells containing the endogeneous QN-rich prions, [PIN+]Citation55 or [PSI+],Citation56 which manifest themselves as susceptibility factors for a poly-Q disorder. Prion-dependent poly-Q cytotoxicity in yeast is associated with a defect of endocytosis, apparently due to sequestration of some actin-assembly proteins, involved in formation of the endocytic vesicles, by poly-Q aggregates.Citation57 Sup35 aggregates also interact with some cytoskeletal proteins involved in the endocytic/vacuolar pathway, and cytotoxicity of overproduced Sup35 is increased in the strains with the cytoskeletal defects.Citation53,Citation58 Expression of mammalian α-synuclein in yeast leads to its aggregation and cytotoxicity with some characteristics of apoptosis.Citation59 Taken together, these data confirm that accumulation of prions and other amyloidogenic protein in yeast may lead to the pathological consequences, and establish yeast prions as appropriate models for studying the mechanisms of amyloid cytotoxicity.
Model of “Adaptive Prionization”
Evolutionary conservation of prion-forming properties.
One argument in favor of the adaptive role of prions is evolutionary conservation of prion-forming properties of some proteins. Prion properties of Sup35 are conserved in various species of Saccharomyces (see ref. Citation59a), as well as in Candida albicans and Pichia methanolica, budding yeast species that are distantly related to Saccharomyces cerevisiae.Citation43,Citation60–Citation63 Comparison of the Sup35 sequences among the different isolates of S. cerevisiae and between the sister species of S. cerevisiae and S. paradoxus demonstrates that while the prion domain (Sup35N) is evolving much faster than the C-proximal release factor domain (Sup35C), sequence of Sup35N still remains under the purifying selection pressure, confirming that this region of the protein is playing a certain positive biological role.Citation64 As the ability to form a prion is the only function of Sup35N known thus far, the simplest logical explanation would be that the ability to form a prion is adaptive under certain circumstances. Remarkably, the highest level of sequence conservation was observed within two subregions of Sup35N, the N-proximal QN-rich stretch (QN) and the region of oligopeptide repeats (ORs, see ), which are still clearly seen in the distantly related budding yeast species of Candida and Pichia, despite low overall conservation of the Sup35N aa sequence (reviewed in ref. Citation65). Both subregions play a major role in prion-related properties of Sup35N (reviewed in ref. Citation7). However, these observations can argue in both ways, as repetitive structure of OR region per se is not a requirement for prion propagation.Citation66 Then, conservation of OR region (and possibly of QN) could be related to some unknown function of this part of the protein that is distinct from its prion-propagating ability.
The Sup35N region of the distant relative of budding yeast, the fission yeast Schizosaccharomyces pombe, does not contain QN and ORs () and exhibits essentially no aa identity (only 18%) with the corresponding domain of S. cerevisiae, while Sup35C remains highly conserved (64% identity).Citation65 Likewise, neither sequence nor aa composition patterns of Sup35N are conserved between yeast and mammals, and the capability of Sup35 homologs (usually called eRF3) from species other than budding yeast to form prions is yet to be proven (). However, while aa composition of the Sup35N regions of higher eukaryotes is different from yeast Sup35N, it is still highly unusual. For example, N-terminal domain of the Sup35 homolog from mouse and human (GSPT1) contain a high percentage of P, S and G residues (10%, 15% and 20%, respectively). Instead of the QN and OR, mammalian eRF3 proteins contain poly-G and/or poly-S tracts. In mammals with two different eRF3-coding genes, all GSPT1 orthologs contain both poly-G and poly-S, while GSPT2 orthologs contain only poly-S. These homopeptide regions are usually coded almost exclusively by identical repeated trinucleotides, suggesting that they originate from trinucleotide expansions. Recent data confirm that the poly-G expansion can indeed occur in GSPT1 and is associated with susceptibility to gastric cancer.Citation67 Obviously eRF3 homologs of higher eukaryotes possess some unusual properties, although it remains to be seen whether these properties involve an ability to form amyloids.
At the current level of knowledge, it can not be ruled out that conservation of the Sup35N aa composition in budding yeast or unusual features of the aa composition of this region in other organisms are associated with its unknown function that is not directly related to prion formation. A variety of cellular proteins interact with Sup35N and/or Sup35M regions.Citation5 It is possible that Sup35N influences a function of the whole protein or targets it to a specific cell compartment. Indeed, the deletion of Sup35NM coding region leads to an alteration of the sexual cycle in Podospora,Citation68 implying that this region is not completely irrelevant to the cellular function of the protein.
Prion role in self/nonself recognition: Example of the [Het-s] prion in Podospora.
The first example of a prion having an adaptive biological function is [Het-s] of Podospora that controls vegetative incompatibility.Citation69 A cytoplasmic contact between the prion-containing and prion-free mycelia results in degeneration of the latter one. In this way, [Het-s] controls vegetative incompatibility, an adaptive trait in Podospora. Moreover, after meiotic division [Het-s] prion kills spores containing a het-S allele that is incapable of producing the prion state.Citation70 [Het-s] is abundant in natural Podospora populations. As adaptive function of [Het-s] is achieved via cytotoxic effect, [Het-s] combines features of both “prion pathology” and “adaptive prionization” models.
Role of [Het-s] in cytoplasmic incompatibility is related to one general characteristic feature of amyloids, that is, to a high level of sequence-specificity in amyloid propagation. While proteins of different sequences may possess amyloid properties, only molecules that contain the amyloid-forming domains of nearly identical sequences can join any given amyloid fiber. Recent data show that at least some amyloids are assembled together via parallel β-sheets, for which identity of aa sequences involved in β-sheet formation is extremely important.Citation11,Citation71 In terms of their stringency, sequence identity requirements for amyloid formation are not dissimilar from the rules that govern complementarity of DNA strands. These requirements may explain so-called “species barrier” in prion transmission, preventing transmission of the prion state between the divergent prion domains (reviewed in ref. Citation65). Sequence-specificity makes prions a useful tool for the self/nonself recognition systems, as demonstrated by the example of cytoplasmic incompatibility in Podospora.
Stress granules and protection against stresses.
In higher eukaryotes, the stress such as heat shock is followed by formation of the nuclear and/or cytoplasmic stress granules (SG).Citation72 Cytoplasmic SGs contain transcripts associated with 40S ribosomal subunits (48S complexes), unable to initiate translation in stress conditions. SG assembly is mediated by the RNA-binding protein TIA-1,Citation73 which contains the C-terminal RNA recognition motif and Q-rich domain () similar to prion domains of yeast prion proteins. Deletion of Q-rich domain blocks SG formation after arsenite-induced stress in the mammalian cell culture whereas substitution of TIA-1 “prion” domain for Sup35 prion domain (PrD) restores SG production. However in contrast to prion formation, TIA-1 aggregation and SG assembly are reversible after return to normal conditionsCitation72 (). Therefore, SGs provide an example of labile and economical post-trancriptional regulatory and protective mechanism contributing to the cellular function in stress conditions and based on prion-like properties.
There are several other examples of the protective mechanisms based on amyloid properties. Embryos of the fish Austrofundulus limnaeus are surrounded by an egg envelope composed of two proteins that together form a structure similar to amyloid fibrils.Citation74 Another fish protein, type I antifreeze protein that is normally a-helical, is converted into an amyloid upon freezing, that may possibly play a protective role by inhibiting ice formation.Citation75
As aggregation of the yeast prion proteins is increased in the stationary or non-dividing cells,Citation54,Citation76,Citation77 one attractive speculation is that reversible PrD-mediated aggregation is used to protect some important proteins (e.g., Sup35) during unfavorable conditions.
Other biological roles of amyloid-like structures.
Ability of prions to fix and “memorize” protein conformational changes make them ideal candidates for the role of memory molecules. Indeed, it has been hypothesized that a prion-like domain of the neuron-specific isoform of cytoplasmic polyadenylation element binding protein (CPEB) is connected to long-term memory in the shellfish Aplysia.Citation78
There is a number of other polymerized proteins that exhibit similarities to amyloid fibers, for example the spider silk protein, spidroin,Citation79 whose adaptive role in spiders is evident.Citation80 It has recently been shown that one of the mammalian proteins involved in melanin production adopts an amyloid structure, so that amyloid polymers likely serve as a scaffold for melanin polymerizationCitation81 (). This is a first clear evidence for the positive biological role of amyloids in mammals, although it is not known whether this specific kind of amyloid possesses prion properties.
There also are examples of a beneficial role of amyloid-like aggregates in bacteria, such as facilitation of biofilm formation in E. coli by the extracellular self-assembly of the major curli protein, CsgA, containing PrP-like oligopeptide repeats,Citation82 into typical amyloid fibrils.Citation83 Amyloid-forming proteins of Streptomyces coelicolor, called chaplins, are essential for aerial growth.Citation84 Moreover, it has been hypothesizedCitation85 that amyloid-like formations played an important role in the emergence of the primordial membranes and other structures at the early steps of the biological compartmentalization (reviewed in ref. Citation7).
Possible evolutionary consequences of Sup35 prionization.
Numerous attempts to identify an adaptive function of the prion state were made in case of Sup35 (eRF3), which is a translation termination factor. Formation of [PSI+] prion decreases supply of functional Sup35, leading to efficient read-through of the nonsense-mutations within ORFs. It remains unclear to which extent termination at the normal terminators, usually protected by nucleotide context,Citation86 is affected by [PSI+]. In some genotypic backgrounds, presence of [PSI+] induces heat shock responseCitation87 and increases resistance of yeast cells to some stresses.Citation88 Although “protective” in the artificially generated laboratory situations, such abnormalities in Hsp levels would not likely be adaptive in the long run in nature.
Systematic comparison of a variety of phenotypes (such as resistance to certain toxicants, etc.) between several isogenic pairs of [PSI+] and [psi−] strains has shown that the presence of [PSI+] was beneficial in some conditions for certain genotypes.Citation23 However, ancestors of these laboratory strains went through multiple rounds of mutagenesis and could therefore contain unidentified nonsense-alleles. While suppression of such alleles could be beneficial for these specific strains in the laboratory, the question remains whether or not this is directly applicable to natural conditions.
It was proposedCitation23 that the presence of [PSI+] could increase the “evolvability” of the yeast population and facilitate adaptation to environmental changes by generating new protein products from ORFs containing nonsense-mutations, weak terminators or frameshifting-prone sequences. Such a mechanism could in principle be applied to activation of the silent pseudogenes.Citation89,Citation90 As an extension of the modular principle in molecular evolution,Citation91 one could suggest that new genes can be created through recombination of inactivated (pseudogene) copies, which often have no introns and are “locked” by nonsense and frameshift mutations. As pseudogenes are not functional, they can easily accumulate new mutations potentially generating new functions.Citation92 Sporadic activation of pseudogenes through nonsense or frameshift suppression allows natural selection to choose combinations of mutations having beneficial effects. Analysis of whole genomes has revealed a number of cases, which can serve as examples of possible pseudogene resurrection.Citation93
If [PSI+] decreases termination efficiency and therefore allow pseudogene expression, such read-through events may take place at a frequency of at least one per every million years, as suggested by the quantitative model.Citation94 However, mutations in the genes coding for the components of translation machinery may have the same effect.Citation5 It is therefore not clear whether the proposed mechanism is specific to a prion. Although mutated translational components are likely to turn detrimental in natural environments, so is [PSI+], judging from analysis of the natural yeast isolates.Citation22,Citation43,Citation44
One potential advantage of [PSI+], not shared by most of the abovementioned gene mutants, could be that it is a dominant omnipotent suppressor affecting both termination and frameshifting. Another possibility that would give [PSI+] a preference at the population level over other mechanisms causing nonsense readthrough could be an easier transition between [psi−] and [PSI+] states. However, frequencies of spontaneous acquisition and loss of the typical “strong” [PSI+] variants are quite low, making them unlikely candidates for such a role. It is therefore possible that increased adaptability could be associated not with the “conventional” stable prion variants used in most laboratory experiments, but with the variants maintained only in certain conditions and eliminated after conditions are changed. Proof of the existence of such conditionally stable [PSI+] variants has been provided recently by identification of the [PSI+] isolate that can be maintained only at high levels of the chaperone Hsp104.Citation95 It still remains to be shown which (if any) conditions in nature could favor maintenance of such transient variants of [PSI+].
Conclusion
Prions, protein mutants and posttranslational feedback regulation.
While strong experimental data support “prion pathology” model, evidence in favor of the “adaptive prionization” model is of rather circumstantional nature. Most examples of the proven biologically positive effects of amyloid-like formations (melanin biosynthesis, stress granules, etc.) are so far dealing with the nonprion aggregates. The only prion that is clearly documented to play a biologically positive role in natural conditions, [Het-s] of Podospora, ironically does so by killing a nonprion partner.
However, one should remember that the majority of the known prions were identified by chance, due to extreme phenotypic effects caused by the corresponding proteins in the prion form, such as fatal transmissible disease in case of mammalian PrP or translation termination defect in case of yeast Sup35. It is possible that we are so far dealing only with a very top of the iceberg, and a large number of prion-like phenomena are still waiting for their discoverers. If prions are to be considered as “mutants” occurring at the protein level,Citation6 one should not expect that randomly chosen mutations would frequently turn beneficial for the organism. Rather, the majority of them would be expected to have either deleterious effect or no effect, as in case of DNA mutations. However, it does not exclude a possibility of some beneficial changes occurring by this mechanism that could be identified in the future.
Another possible dimension of this story is that beneficial effects could be associated with the transient prion variants, as hypothesized above in case of [PSI+], while the stably propagating and usually toxic prions might represent by-products of these processes. Normal cellular functions of Sup35 and Ure2 are decreased in the prion state, suggesting that transient formation of the prion-like multimers may serve as a mechanism of feedback regulation. This notion is supported by the existence of the shortened form of Ure2, generated by alternative translational initiation and lacking the prion domain.Citation96 Likewise, existence of the shortened transcript of the SUP35 gene in certain conditions has been reportedCitation97 but never studied carefully. Many proteins involved in DNA replication, repair and transcription contain PrD-like QN-rich domains.Citation98 In case of the yeast transcriptional repressor Gal11, existence of two alternative transcripts has been demonstrated, of which the shorter one is missing two QN-rich domains and codes for the protein that manifests itself as a transcriptional activator rather than repressor.Citation99 These data suggest that prion-like mechanisms of feedback regulation could be widespread, and this may explain evolutionary conservation of prion properties.
One should note that transient prion variants maintained only in certain conditions are hard to distinguish from both feedback regulatory circuits and so-called “heritable” modifications persisting for a few generations. Therefore, role of the transient prion variants in adaptive evolution, as hypothesized above, would be in agreement with the more general hypothesis of V. KirpichnikovCitation100 regarding the role of modifications in evolution. Moreover, prion model may provide a tool for even more direct relationship between phenotypic and “genotypic” (in traditional sense) inheritance. As prion state of a protein may influence probability of prionization of another protein,Citation17–Citation19 this opens a possibility for concerted modification (prionization) of several proteins at once. Such a prionization network, in turn, may potentially influence a DNA metabolism and rate of “classic” mutations, in case if some of the prionized proteins are involved in DNA replication/repair. This provides a mechanism for the possible effects of the heritable protein variations on the DNA material.
Figures and Tables
Figure 1 Structural organization of prion proteins. QN: the QN-rich stretch. OR: the region of oligopeptide repeats. PrD-prion domain. Numbers correspond to amino acid (aa) positions. Arrows indicate domain and subdomain boundaries. N, M and C-N-proximal, middle and C-proximal regions of Sup35, respectively. The N/M and M/C boundaries are arbitrarily assigned to the second (aa 124) and third (aa 254) methionine residues of the Sup35 protein. See text for details.
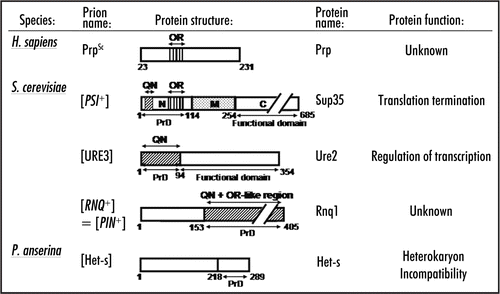
Figure 2 Evolutionary comparison of the N-terminal domains of Sup35 homologs. Sequences are from www.ncbi.nlm.nih.gov. Taxonomical relationships are from www.ncbi. nlm.nih.gov/Taxonomy. Scales do not correspond to evolutionary distances. For QN and OR designations, see . Numbers on the right correspond to the size of the N-terminal region (in aa) in each case. Sequence data were obtained from www.ncbi.nlm.nih.gov. ? -refers to the cases where search for prion activity in S. cerevisiae has been performed but have not yielded positive results (O. Zemlyanko, A. Petrova and G. Zhouravleva, unpublished; K. Gokhale and Y. Chernoff, unpublished). NT, not tested.
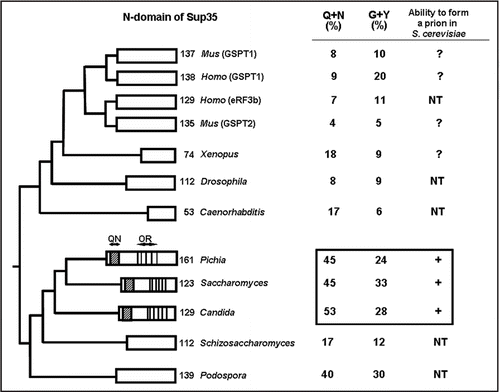
Figure 3 Formation of the stress granules. Schematic structure of TIA protein. (Q) the Q-rich stretch. Other designations are as in Model showing formation of stress granules. Ribosome subunits are shown as ovals and TIA as black asterisk. See text for more details.
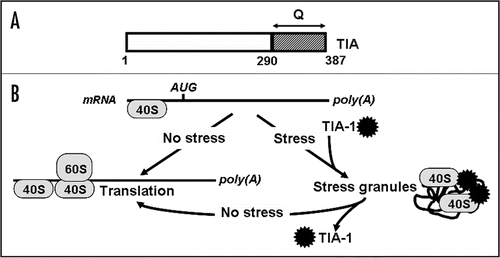
Figure 4 Role of amyloid in melanin polymerization. Glycoprotein Pmel17, that is a critical component of melanosome biogenesis, gives rise to two fragments, Mα and Mβ. Self-assembly of Mα leads to amyloid formation. Amyloid provides a scaffold for melanin polymerization.
Acknowledgements
We thank R.B. Wickner and G.P. Newnam for critical reading of the manuscript and helpful suggestions. This work was supported by grants ST-012 from CRDF, RAS Presidium Program “Biosphere origin and evolution” and (Lot 2006-12.2/001) from Federal Agency of Science and Innovations (to Sergey G. Inge-Vechtomov and Galina A. Zhouravleva), by grant 07-04-00605 from the Russian Foundation for Basic Research (to Galina A. Zhouravleva), and by grant R01GM58763 from NIH (to Yury O. Chernoff).
Note
This is a modified version of the previously published manuscript: Inge-Vechtomov SG, Zhouravleva GA, Chernoff YO. Biological Roles of Prion Domains. Protein-Based Inheritance Chernoff Y. Austin and New York Landes Bioscience and Kluwer Academic Press 2007; 93 - 105
References
- Crick FH. On protein synthesis. Symp Soc Exp Biol 1958; 12:138 - 163
- Koltsov NK. Inherent molecules. Cell Organisation 1936; Moscow-Leningrad State Publishing House of Biol and Med Lit 586 - 620
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93:337 - 348
- Wickner RB, Edskes HK, Ross ED, Pierce MM, Baxa U, Brachmann A, Shewmaker F. Prion genetics: New rules for a new kind of gene. Annu Rev Genet 2004; 38:681 - 707
- Inge-Vechtomov S, Zhouravleva G, Philippe M. Eukaryotic release factors (eRFs) history. Biol Cell 2003; 95:195 - 209
- Chernoff YO. Mutation processes at the protein level: Is Lamarck back?. Mutat Res 2001; 488:39 - 64
- Chernoff YO. Amyloidogenic domains, prions and structural inheritance: Rudiments of early life or recent acquisition?. Curr Opin Chem Biol 2004; 8:665 - 671
- Diaz-Avalos R, Long C, Fontano E, Balbirnie M, Grothe R, Eisenberg D, Caspar DL. Cross-beta order and diversity in nanocrystals of an amyloid-forming peptide. J Mol Biol 2003; 330:1165 - 1175
- Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 2005; 435:765 - 772
- Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997; 89:811 - 819
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005; 435:773 - 778
- Tanaka M, Chien P, Yonekura K, Weissman JS. Mechanism of cross-species prion transmission: An infectious conformation compatible with two highly divergent yeast prion proteins. Cell 2005; 121:49 - 62
- King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature 2004; 428:319 - 323
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature 2004; 428:323 - 328
- Jeffrey M, Gonzalez L. Pathology and pathogenesis of bovine spongiform encephalopathy and scrapie. Curr Top Microbiol Immunol 2004; 284:65 - 97
- Aguzzi A, Polymenidou M. Mammalian prion biology: One century of evolving concepts. Cell 2004; 116:313 - 327
- Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997; 147:507 - 519
- Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: The story of [PIN+]. Cell 2001; 106:171 - 182
- Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell 2001; 106:183 - 194
- Dobson CM. Protein folding and misfolding. Nature 2003; 426:884 - 890
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J Mol Med 2003; 81:678 - 699
- Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci USA 2005; 102:10575 - 10580
- True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000; 407:477 - 483
- Horwich AL, Weissman JS. Deadly conformations: Protein misfolding in prion disease. Cell 1997; 89:499 - 510
- Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci 1999; 24:329 - 332
- Pepys MB, Hawkins PN, Booth DR, Vigushin DM, Tennent GA, Soutar AK, Totty N, Nguyen O, Blake CC, Terry CJ, et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 1993; 362:553 - 557
- Faux NG, Bottomley SP, Lesk AM, Irving JA, Morrison JR, de la Banda MG, Whisstock JC. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res 2005; 15:537 - 551
- Orr HT. Beyond the Qs in the polyglutamine diseases. Genes Dev 2001; 15:925 - 932
- Dorsman JC, Pepers B, Langenberg D, Kerkdijk H, Ijszenga M, den Dunnen JT, Roos RA, van Ommen GJ. Strong aggregation and increased toxicity of polyleucine over polyglutamine stretches in mammalian cells. Hum Mol Genet 2002; 11:1487 - 1496
- Fandrich M, Dobson CM. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J 2002; 21:5682 - 5690
- Oma Y, Kino Y, Sasagawa N, Ishiura S. Intracellular localization of homopolymeric amino acid-containing proteins expressed in mammalian cells. J Biol Chem 2004; 279:21217 - 21222
- Kihm AJ, Kong Y, Hong W, Russell JE, Rouda S, Adachi K, Simon MC, Blobel GA, Weiss MJ. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature 2002; 417:758 - 763
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 1347
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 2006; 311:1471 - 1474
- Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for Huntington's disease pathology. Proc Natl Acad Sci USA 1999; 96:4604 - 4609
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 2000; 97:6763 - 6768
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001; 413:739 - 743
- Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci USA 2000; 97:5750 - 5755
- Leblanc AC. The role of apoptotic pathways in Alzheimer's disease neurodegeneration and cell death. Curr Alzheimer Res 2005; 2:389 - 402
- Liberski PP, Sikorska B, Bratosiewicz-Wasik J, Gajdusek DC, Brown P. Neuronal cell death in transmissible spongiform encephalopathies (prion diseases) revisited: From apoptosis to autophagy. Int J Biochem Cell Biol 2004; 36:2473 - 2490
- Rego AC, Oliveira CR. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: Implications for the pathogenesis of neurodegenerative diseases. Neurochem Res 2003; 28:1563 - 1574
- Roucou X, Leblanc AC. Cellular prion protein neuroprotective function: Implications in prion diseases. J Mol Med 2005; 83:3 - 11
- Chernoff YO, Galkin AP, Lewitin E, Chernova TA, Newnam GP, Belenkiy SM. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol Microbiol 2000; 35:865 - 876
- Resende CG, Outeiro TF, Sands L, Lindquist S, Tuite MF. Prion protein gene polymorphisms in Saccharomyces cerevisiae. Mol Microbiol 2003; 49:1005 - 1017
- Cox BS, Tuite MF, McLaughlin CS. The psi factor of yeast: A problem in inheritance. Yeast 1988; 4:159 - 178
- Fabrizio P, Battistella L, Vardavas R, Gattazzo C, Liou LL, Diaspro A, Dossen JW, Gralla EB, Longo VD. Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae. J Cell Biol 2004; 166:1055 - 1067
- Herker E, Jungwirth H, Lehmann KA, Maldener C, Fröhlich KU, Wissing S, Büttner S, Fehr M, Sigrist S, Madeo F. Chronological aging leads to apoptosis in yeast. J Cell Biol 2004; 164:501 - 507
- Madeo F, Herker E, Maldener C, Wissing S, Lachelt S, Herlan M, Fehr M, Lauber K, Sigrist SJ, Wesselborg S, Frohlich KU. A caspase-related protease regulates apoptosis in yeast. Mol Cell 2002; 9:911 - 917
- Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion “strains” in yeast. Proc Natl Acad Sci USA 2002; 99:16392 - 16399
- Chernoff YO, Inge-Vechtomov SG, Derkach IL, Ptyushkina MV, Tarunina OV, Dagkesamanskaya AR, Ter-Avanesyan MD. Dosage-dependent translational suppression in yeast Saccharomyces cerevisiae. Yeast 1992; 8:489 - 499
- Dagkesamanskaya AR, Ter Avanesyan MD. Interaction of the yeast omnipotent suppressors SUP1 (SUP45) and SUP2 (SUP35) with nonmendelian factors. Genetics 1991; 128:513 - 520
- Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996; 144:1375 - 1386
- Ganusova EE, Ozolins LN, Bhagat S, Newnam GP, Wegrzyn RD, Sherman MY, Chernoff YO. Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol Cell Biol 2006; 26:617 - 629
- Zhou P, Derkatch IL, Liebman SW. The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI+] and [PIN+]. Mol Microbiol 2001; 39:37 - 46
- Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. J Cell Biol 2002; 157:997 - 1004
- Gokhale KC, Newnam GP, Sherman MY, Chernoff YO. Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. J Biol Chem 2005; 280:22809 - 22818
- Meriin AB, Zhang X, Miliaras NB, Kazautsev A, Chernoff YO, McCaffery JM, Wendland B, Sherman MY. Aggregation of expanded polyglutamine domain in yeast leads to defects in endocytosis. Mol Cell Biol 2003; 23:7554 - 7565
- Bailleul PA, Newnam GP, Steenbergen JN, Chernoff YO. Genetic study of interactions between the cytoskeletal assembly protein Sla1 and prion-forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae. Genetics 1999; 153:81 - 94
- Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson's disease. J Mol Biol 2005; 351:1081 - 1100
- Chen B, Newnam GP, Chernoff YO. Prion species barrier between the closely related yeast proteins is detected despite coaggregation. Proc Natl Acad Sci USA 2007; 104:2791 - 2796
- Kushnirov VV, Kochneva-Pervukhova NV, Chechenova MB, Frolova NS, Ter-Avanesyan MD. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J 2000; 19:324 - 331
- Nakayashiki T, Ebihara K, Bannai H, Nakamura Y. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell 2001; 7:1121 - 1130
- Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell 2000; 100:277 - 288
- Zadorskii SP, Sopova I, Inge-Vechtomov SG. Prionization of the Pichia methanolica SUP35 gene product in the yeast Saccharomyces cerevisiae. Genetika 2000; 36:1322 - 1329
- Jensen MA, True HL, Chernoff YO, Lindquist S. Molecular population genetics and evolution of a prion-like protein in Saccharomyces cerevisiae. Genetics 2001; 159:527 - 535
- Zhouravleva G, Alenin V, Inge-Vechtomov S, et al. Pandalai SG. To stick or not to stick: Prion domains from yeast to mammals. Recent Res Devel Mol Cell Biol 2002; 3:185 - 218
- Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc Natl Acad Sci USA 2005; 102:12825 - 12830
- Brito M, Malta-Vacas J, Carmona B, Aires C, Costa P, Martius AP, Ramos S, Conde AR, Monteiro C. Polyglycine expansions in eRF3/GSPT1 are associated with gastric cancer susceptibility. Carcinogenesis 2005; 26:2046 - 2049
- Gagny B, Silar P. Identification of the genes encoding the cytosolic translation release factors from Podospora anserina and analysis of their role during the life cycle. Genetics 1998; 149:1763 - 1775
- Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA 1997; 94:9773 - 9778
- Dalstra HJ, Swart K, Debets AJ, Saupe SJ, Hoekstra RF. Sexual transmission of the [Het-s] prion leads to meiotic drive in Podospora anserina. Proc Natl Acad Sci USA 2003; 100:6616 - 6621
- Ross ED, Minton A, Wickner RB. Prion domains: Sequences, structures and interactions. Nat Cell Biol 2005; 7:1039 - 1044
- Anderson P, Kedersha N. Stressful initiations. J Cell Sci 2002; 115:3227 - 3234
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell 2004; 15:5383 - 5398
- Podrabsky JE, Carpenter JF, Hand SC. Survival of water stress in annual fish embryos: Dehydration avoidance and egg envelope amyloid fibers. Am J Physiol Regul Integr Comp Physiol 2001; 280:123 - 131
- Graether SP, Slupsky CM, Sykes BD. Freezing of a fish antifreeze protein results in amyloid fibril formation. Biophys J 2003; 84:552 - 557
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Aranesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J 1996; 15:3127 - 3134
- Bailleul-Winslett PA, Newnam GP, Wegrzyn RD, et al. An antiprion effect of the anticytoskeletal drug latrunculin A in yeast. Gene Expr 2000; 9:145 - 156
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003; 115:879 - 891
- Kenney JM, Knight D, Wise MJ, Vollrath F. Amyloidogenic nature of spider silk. Eur J Biochem 2002; 269:4159 - 4163
- Craig CL. Spider webs and silks: Tracing evolution from molecules to genes to phenotypes 2003; New York Oxford University Press
- Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol 2005; 4:e6
- Cherny I, Rockah L, Levy-Nissenbaum O, Gophna U, Ron EZ, Gazit E. The formation of Escherichia coli curli amyloid fibrils is mediated by prion-like peptide repeats. J Mol Biol 2005; 352:245 - 252
- Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002; 295:851 - 855
- Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, Boersma FG, Dijkhuizen L, Wosten HA. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev 2003; 17:1714 - 1726
- Zhang S, Holmes T, Lockshin C, Rich A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc Natl Acad Sci USA 1993; 90:3334 - 3338
- Bonetti B, Fu L, Moon J, Bedwell DM. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J Mol Biol 1995; 251:334 - 345
- Jung G, Jones G, Wegrzyn RD, Masison DC. A role for cytosolic hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress. Genetics 2000; 156:559 - 570
- Eaglestone SS, Cox BS, Tuite MF. Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism. EMBO J 1999; 18:1974 - 1981
- Inge-Vechtomov SG. A possible role of genetic translation ambiguity in evolution. Mol Biol (Mosk) 2002; 36:268 - 276
- Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet 2005; 6:435 - 450
- Gilbert W. Why genes in pieces?. Nature 1978; 271:501
- Koch AL. Enzyme evolution. I. The importance of untranslatable intermediates. Genetics 1972; 72:297 - 316
- Harrison P, Kumar A, Lan N, Echols N, Snyder M, Gerstein M. A small reservoir of disabled ORFs in the yeast genome and its implications for the dynamics of proteome evolution. J Mol Biol 2002; 316:409 - 419
- Masel J, Bergman A. The evolution of the evolvability properties of the yeast prion [PSI+]. Evolution Int J Org Evolution 2003; 57:1412 - 1498
- Borchsenius AS, Muller S, Newnam GP, Inge-Vechtomov SG, Chernoff YO. Prion variant maintained only at high levels of the Hsp104 disaggregase. Curr Genet 2006; 49:21 - 29
- Komar AA, Lesnik T, Cullin C, Merrick WC, Trachsel H, Altmann M. Internal initiation drives the synthesis of Ure2 protein lacking the prion domain and affects [URE3] propagation in yeast cells. EMBO J 2003; 22:1199 - 1209
- Surguchov AP, Telkov MV, Smirnov VN. Absence of structural homology between sup1 and sup2 genes of yeast Saccharomyces cerevisiae and identification of their transcripts. FEBS Lett 1986; 206:147 - 150
- Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: Implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci USA 2000; 97:11910 - 11915
- Ono B, Futase T, Honda W, Yoshida R, Nakano K, Yamamoto T, Nakajima E, Noskov VN, Negishi K, Chen B, Chernoff YO. The Saccharomyces cerevisiae ESU1 gene, which is responsible for enhancement of termination suppression, corresponds to the 3′-terminal half of GAL11. Yeast 2005; 22:895 - 906
- Kirpichnikov VS. Role of noninherent variability in the process of natural selection. Biological Journal 1935; 4:775 - 801