Abstract
Prion protein (PrP), the causative agent of Transmissible Spongiform Encephalopathies, is synthesized in the endoplasmic reticulum (ER) where it undergoes numerous covalent modifications. Here we investigate the interdependence and regulation of PrP oxidative folding, N-glycosylation, and GPI addition in diverse ER conditions. Our results show that formation of the single disulphide bond is a pivotal event, essential for PrP transport, and can occur post-translationally. Retarding its formation enhances N-glycosylation and GPI-anchoring. In contrast, lowering ER Ca2+ concentration inhibits N-glycosylation and GPI-anchoring. These data reveal tight interplays between the different ER covalent modifications, which collectively increase of PrP conformational diversity and may be important for its propagation.
Introduction
Upon translocation into the endoplasmic reticulum (ER), secretory and membrane proteins begin to fold under the assistance of resident chaperones and enzymes.Citation1 Numerous covalent modifications are executed in the ER, including the removal of the signal sequence,Citation2 formation of disulphide bonds,Citation3 N-glycosylationCitation4 and addition of GPI-anchors.Citation5 These modifications are important for the sequential folding of separate domains, but rather little is known on their order of execution, possible interplays and regulation. We addressed these questions using the prion protein (PrP) as a model system. PrP is the causative agent of transmissible spongiform encephalopathies (TSE).Citation6 One notable characteristic of PrP polypeptide chain is that it can fold in at least two alternative conformations called cellular PrP (PrPC) and PrP Scrapie (PrPSc). PrPSc represents the pathogenic conformer and catalyses the conversion of endogenous, non-toxic conformer PrPC thus allowing its own propagation. In addition, PrP polypeptide normally undergoes a variety of covalent modifications such as glycosylation, oxidation and GPI anchoring. Depending on the efficiency of these processes, multiple PrP isoforms arise (see ref. Citation7 and references therein). These modifications occur in a limited time frame, while or soon after PrP is translocated into the ER lumen. The two N-glycosylation sites of PrP are variably utilized resulting in a mixture of di, mono and unglycosylated forms.Citation7,Citation8 This variability has important implications, in that the N-glycan pattern of PrPSc is characteristic of the different TSE and could be used as diagnostic marker.Citation9 Furthermore, the signal sequence of PrP is inefficient in mediating translocation. This can lead to accumulation of cytosolic PrP, especially under ER stress conditions, and to alternative membrane orientation.Citation7,Citation10 The existence of many conformers, glycoforms and topological variants may suggest that PrP exploit posttranslational modifications to increase its diversity.
In this study, we explore the early folding of PrP in the ER and the impact of ER redox and ionic conditions on the maturation of PrP. We show that formation of the single disulfide bonds competes with N-glycosylation and GPI addition. The Ca2+ concentration in the ER ([Ca2+]ER) also affects glycosylation. The interplays between these modifications determine a dynamic heterogeneity in the early PrP biogenesis, increasing its structural diversity.
Materials and Methods
Cell cultures, transfection and plasmids.
HeLa cells (ATCC #CCL-2) were maintained in DMEM 5% FCS and transfected with 1 µg of plasmid and 4 µl of lipofectin (Invitrogen) per ml of transfection mix. Analysis was perfomed 36–48 h after transfection when cells were about 80–100% confluent. N2a cells (ATCC #CCL-131) were maintained in DMEM 10% FCS.
Plasmids encoding a murine PrP epitope-tagged for 3F4 antibody (MoPrP 3F4-tag) were described in references Citation11 and Citation12, Ero1-α and its C394A mutant in reference Citation13.
The C178-213S mutant was generated by mutagenesis with the Stratagene Quikchange Site-Direct Mutagenesis Kit from pCDNA MoPrP 3F4-tag. Cys178 and Cys213 were replaced with serines using the following primers:
C178S-fwd: CAA CTT CGT GCA CGA CAG CGT CAA TAT CAC CATC; C178S-rev: GAT GGT GAT ATT GAC GCT GTC GTG CAC GAA GTTG; C213S-fwd: GTG GTG GAG CAG ATG AGC GTC ACC CAG TAC; C213S-rev: GTA CTG GGT GAC GCT CAT CTG CTC CAC CAC.
PCR was performed in in Techne Progene Termal Cycler using Platinum Pfx DNA polymerase (Invitrogen).
Metabolic labelling, immunoprecipitation and SDS-PAGE.
Metabolic labelling, immunoprecipitation and SDS-PAGE were performed as described in reference Citation7. Unless differently indicated, cells were lysed in RIPA buffer (0.2% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 150 mM NaCl, 10 mM Tris-HCl, pH 7.6) containing protease inhibitors (Complete, Roche) and 10 mM N-ethylmaleimide.
Immunoblotting (WB).
Nitrocellulose membranes were immunodecorated with one of the following antibodies: 3F4 (mouse monoclonal anti-PrP, kind gift of R.J. Kascsak, reviewed in ref. Citation14), SPA-827 (α-KDEL, Stressgene), A5441 (α-βactin, Sigma), α-Thioredoxin (rabbit polyclonal, described in ref. Citation15) α-ERp57 (rabbit polyclonal, kind gift of N.J. Bulleid)Citation16 followed by incubation with the relative HRP-conjugated secondary antibody and revelation by ECL® (Amersham).
Assays for glycosylation, disulphide bonding and other cell treatments.
To remove N-linked glycans, lysates or immunoprecipitates were treated with Endoglycosydase H or PGNase F (New England Biolabs) according to the manufacturer's instructions. For detection of disulphide bonds immunoprecipitates were denatured for five min at 95°C in Sample Buffer (50 mM Tris-HCl pH 6.8, 1% SDS, 5% glycerol) supplemented with (R) or without (NR) 200 mM DTT. Both reduced and non-reduced samples were then incubated with 400 mM Iodoacetamide for ten min at RT before SDS-PAGE.
To inhibit N-glycosylation, cells were treated with 2.5 µg/ml of tunicamycin (Tm). To deplete ER Calcium stores, cells were treated with 2.5 µg/ml thapsigargin (Tg, Sigma-Aldrich) or 50 µM Cyclopiazonic Acid (CPA, Sigma-Aldrich). Xbp1 splicing assay on total mRNA was performed as described in reference Citation7.
Digitonin permeabilisation.
Hela cells were washed in PBS and then in KHM buffer (110 mM K-Acetate, 2 mM Mg-Acetate, 20 mM Hepes pH 7.2). 107 cells were resuspended in 5 ml of ice-cold KHM buffer and incubated with 50 µg/ml digitonin (Sigma) for 5 min on ice. Five ml of fresh KHM buffer were added to dilute digitonin and cells were promptly spun for 3 min at 1,200 rpm, 4°C. The supernatant was saved and pooled with two washes in Hepes buffer (50 mM K-Acetate, 90 mM Hepes pH 7.2) to obtain the cytosolic C-fraction. The pellet containing semi-permeabilised cells was lysed in 10 ml of Ripa buffer (E-fraction). In parallel, 107 cells were lysed in 10 ml of Ripa buffer to obtain total proteins (Tot-fraction). All the fractions were supplemented with protease inhibitors and centrifuged for 15 min at 13,000 rpm. For WB, 200 µg of protein from each fraction were precipitated with acetone and resuspended in an appropriate volume before SDS-PAGE. For IP, the amount of each sample was normalised by TCA counts. 1/3 volume of Ripa buffer 4x was added to the C-fraction before IP to mimic the conditions of the E- and Tot-fractions. The efficiency of the protocol was evaluated by trypan blue exclusion and by monitoring the staining patterns with ER-localised yellow fluorescent protein (ER-YFP) and a cytosolic red fluorescent protein (cyt-RFP). Most cells displayed an intact, yellow ER but had lost the cytosolic red fluorescence from the cytosol (not shown).
Results and Discussion
Oxidative folding is required for maturation and transport of PrP.
We first analyzed the role of oxidative folding in glycosylation and intracellular transport of PrP. We have previously shownCitation7 that seven isoforms of PrP can be distinguished after a short pulse with radioactive aminoacids. These forms have a MW comprised between 25 and 31 kDa.Citation6,Citation7,Citation17 Owing to inefficient translocation, one of them, SP-PrP, is localised in the cytosol and bears an uncleaved signal sequence. This form is less abundant in unstressed cells. The remaining six forms represent newly synthesised PrP molecules that localise at the ER and differ for their glycosylation and C-terminal processing. Doublet PrP bands can be easily resolved carrying two, one or no N-glycans (indicated as 2, 1, 0 in , lane 1).
As suggested by the mobility shift in reducing versus non-reducing conditions (, compare lanes 1–2), newly synthesised, ER-resident species rapidly acquire the disulfide bond linking cysteines 178 and 213. To confirm that the mobility shift was due to the presence of the disulphide, we pulsed cells in the presence of DTT, which inhibits oxidative folding (lanes 3–4). We also analyzed a mutant (C178-213S) lacking both cysteines and hence unable to form the bond (lanes 5–6). When oxidation was prevented, no mobility shifts were detected (, lanes 3–6). Remarkably, the failure to form the intra-chain disulfide bond correlated with increased glycosylation, and the mono- or un-glycosylated species (indicated as 1 and 0 in ) became almost undetectable.
As proteins travel through the Golgi apparatus, N-glycans are processed and acquire resistance to Endoglycosydase H (Endo H). As expected, Western blot analyses of cells expressing transport competent, wt PrP revealed abundant Endo H resistant species with apparent molecular weight of 40 kDa or more (, lanes 1–2). In contrast, the C178-213S mutant was mainly detected as a sharp, glycosidase sensitive band (lanes 3–4). Pulse-chase assays () confirmed that formation of the disulphide bond is important for Golgi transport. The C178-213S mutant (lanes 5–6) did not yield the characteristic smear of heterogeneous glycoforms seen in wt PrP (lane 2) after two hours of chase. Previous observations that a double cysteine mutant was transported to the cell surface, albeit weakly,Citation18 may reflect differences in the cell typesCitation10 or in the vectors used.
The finding that the presence of DTT during the chase did not prevent PrP processing seemed at first to contradict the notion that formation of the intra-chain disulfide is important for transport. However, we found that DTT did not block the formation of the disulphide, but rather retarded it. After digestion with PNGase F, which removes all N-glycans, virtually all the PrP present in cells chased in DTT was indeed oxidised (). To confirm that the formation of the disulphide precedes transport, we performed pulse-chase assays in the presence of reducing agents (). Already after five min of chase some PrP started to form the disulphide, even in the presence of 5 mM DTT (lanes 3–4). After ten min virtually all PrP was oxidised (lanes 5–6), and after 30 min Golgi-processed glycoforms were detectable (lanes 7–8).
Taken together, these findings indicated that oxidative folding is required for Golgi transport of PrP. Unlike other secretory proteins,Citation19 wild type PrP exited from the ER despite the presence of DTT.Citation10 However, virtually all transported PrP was oxidized, implying that disulphide formation can occur post-translationally, even in reducing conditions. Once formed, the bond likely becomes buried and DTT resistant. Such a collapse may reduce sequon accessibility to oligosaccharide transferase, explaining why inhibition of oxidation favored glycosylation of PrP ().
Disulphide bond formation impacts PrP N-glycosylation.
The above observation is in agreement with previous studiesCitation18 and suggests that oxidative folding might compete with N-glycosylation. Since oligosaccharide transferase is located in proximity of the translocon, such a competition likely occurs during or soon after translation/translocation, the rate of disulphide formation being a key factor. Therefore, we analyzed PrP processing in short pulses (1, 2 and 5 min, ). Similar amounts of radioactivity were loaded onto each lane to facilitate the comparison. Remarkably, mono- and un-glycosylated PrP were completely oxidized already after 1 min of synthesis. A small fraction of diglycosylated molecules lacking the disulphide bond were still detected at this time point (lane 2, see arrow). At later times, however, virtually all the detectable species were oxidised. To slow down oxidative folding, we pulsed cells in the presence of decreasing doses of DTT. Low doses of DTT () allowed PrP oxidation, but were sufficient to increase its glycosylation. To further confirm the redox-dependency of PrP glycosylation, we overexpressed Ero1α (), a rate-limiting factor in oxidative folding.Citation20 Likely owing to the rapidity with which PrP oxidizes, Ero1α did not have detectable effects in the absence of DTT. However, Ero1α clearly counteracted the DTT effects on PrP glycosylation ( and E). Expression of the inactive mutant C394A had no effects.
GPI-anchoring and covalent modifications of PrP.
Addition of GPI-anchors involves the removal of the C-terminal amino acids (231 to 254 in PrP, see ) before the addition of a lipid moiety. The process is catalysed by a transamidase complex, also localised in the proximity of the translocon.Citation21 To determine whether also GPI addition was influenced by oxidation, we used antibodies specific for the 231–254 peptide to quantify PrP intermediates lacking the GPI-anchor (αGP; ). About one fourth of PrP synthesised in a 10 min pulse was immunoprecipitated by αGP antibodies (, left). A similar figure was observed for endogenous PrP in N2a neuroblastoma cells (right), excluding that inefficient C-terminal processing was due to transfection or overexpression. Notably, αGP antibodies failed to decorate Golgi-processed PrP species, indicating that also GPI anchoring is important to leave the ER (). When disulphide bond formation was inhibited, less PrP was recognised by the αGP antibody, indicating a more efficient removal of the C-terminus peptide ( and E). Since only diglycosylated PrP is present in these conditions, it was possible that the processing of PrP C-terminus was favored by N-glycosylation. However, the N-glycosylation inhibitor tunicamycin (Tm), had little if any effect on the amount of PrP recognised by the αGP antibody (E and F), implying that N-glycan addition is not necessary for GPI-anchoring. In conclusion, and similarly to what described for N-glycosylation, the presence of the disulphide retards processing by the transamidase complex.
How does oxidative folding influence the extent of glycosylation and GPI processing? According to the available models,Citation22 when Cys213 enters the ER lumen and becomes available for oxidation, Asn196 should reach oligosaccharyl transferase active sites. Immediately thereafter, the GPI-transamidase complex can enter in action. The three covalent modifications are therefore temporally and spatially clustered. The velocity with which PrP traverses the channel seems a key determinant. Deleting the last 23 amino acids inhibits N-glycosylation.Citation23 In contrast, replacing them with a Class I transmembrane region does not impede N-glycosylation. Together, these findings suggest that engagement with the transamidase complex might prevent premature slipping form the membrane and allow oligosaccharyl transferase activity.
[Ca2+]ER and N-glycosylation of PrP.
Ca2+ levels undergo physiological fluctuations within cellular compartments and regulate the activity of many proteins, including ER resident enzymes and chaperones. Therefore, we wondered whether and how differences in [Ca2+]ER impact PrP folding and processing. Treatment with thapsigargin (Tg), an irreversible inhibitor of the Sarcoplasmatic-ER Ca2+ ATP-ase (SERCA) that severely reduces [Ca2+]ER, caused a significant depletion of diglycosylated PrP, matched by an increase in unglycosylated PrP ( and B). A short Tg treatment (10 min) had similar effects, arguing against the possibility that the observed effects were due to activation of a ER stress response, such as the Unfolded Protein Response (UPR). Nonetheless, Tg could affect translocation efficiency.Citation10 Therefore, we exposed cells to the reversible SERCA inhibitor cyclopiazonic acid (CPA) for 4 h. UPR induction was confirmed by monitoring Xbp1 splicing (). CPA was then removed allowing restoration of [Ca2+]ER (reviewed in ref. Citation24 and Andrea Orsi and Roberto Sitia, unpublished results). In the absence of SERCA inhibitors, PrP glycosylation returned to the basal levels (). On the contrary, the presence of CPA during the pulse resulted in a reduction on PrP glycosylation. Ca2+ depletion might affect oligosaccharyl transferase activity. However, when cells were exposed to both Tg and DTT, di-glycosylated PrP became preponderant (). In addition, Tg treatment did not alter N-glycosylation of Ero1α (). We conclude that lowering [Ca2+]ER specifically inhibits PrP glycosylation, independently from an ongoing UPR.
Lowering [Ca2+]ER impacted also GPI-anchoring: more PrP retained the C-terminal GP peptide in cells exposed to Tg (). In low [Ca2+]ER therefore, both N-glycosylation and GPI-anchoring are retarded. A possible underlying mechanism would be impaired cotranslational translocation of PrP.Citation10 However, the observations that DTT restored glycosylation and that the majority of PrP species are oxidised (, lanes 1–2), argue against this hypothesis.
The above results did not exclude that PrP transiently entered the ER, underwent glycosylation and oxidation but then slipped back in the cytosol before C-terminal processing. Therefore, we analyzed the subcellular distribution of the different PrP isoforms. Grp94, BiP and ERp57, but not actin and thioredoxin (TRX), two abundant cytosolic proteins, were enriched in the ER fraction (). In the absence of prolonged ER stress, none of the forms recognised by αGP were detectable in the ER fraction (, upper, lanes 3 and 6). As expected,Citation7 SP-PrP were detectable with αGP in the cytosolic fraction of Tg-treated cells (lower, lanes 3 and 6). However, most of the other αGP-reactive isoforms accumulated in the ER.
Therefore, depletion of Ca2+ levels in the ER inhibited PrP N-glycosylation and GPI addition, and only in part was this due to mistargeting of PrP to the cytosol or back-slipping after SP removal.Citation10 Most of the molecules retaining the C-terminal peptide displayed intra-chain disulfide bonds and accumulated in the ER lumen.
DTT and Tg displayed antagonistic effects on N-glycosylation and GPI addition (). The mechanisms by which [Ca2+]ER depletion inhibits N-glycosylation and GPI-anchoring of PrP are not clear. Ca2+ could be required for the activity of chaperones that help PrP to remain in a status competent to undergo posttranslational modifications ().Citation18
Concluding remarks.
Taken together with the results of Kang et al.,Citation10 our observations suggest that cells can differently handle difficult-to-fold proteins, precisely coordinating their post-translational modifications. This might be particular important under ER stress conditions, in order to limit the potential toxicity of these proteins. From the PrP biology viewpoint, it is tempting to speculate that the unconventional processing represents an evolutionary PrP feature, which increases its conformational diversity and allows exploring different subcellular compartments.
In view of the importance that glycosylation and topology have in the pathogenesis of prion diseases,Citation9,Citation25 the observations that different redox or ionic conditions alter PrP conformation may be relevant in the context of PrP variability and susceptibility to infection.Citation26,Citation27
Abbreviations
| DTT | = | dithiothreitol |
| Endo H | = | endoglycosydase H |
| ER | = | endoplasmic reticulum |
| GPI | = | glycosylphosphatidyl inositol |
| IP | = | immunoprecipitation |
| OST | = | oligosaccharyl transferase |
| PrP | = | prion protein |
| SP | = | signal peptide |
| Tg | = | thapsigargin |
| Tm | = | tunicamycin |
Figures and Tables
Figure 1 Oxidative folding is required for PrP transport and competes with N-glycosylation. (A) Schematic representation of the murine PrP used in this study (not to scale). SP, signal peptide; GP, C-terminal propeptide for GPI-anchor addition. N-glycans and the single intra-chain disulfide bond formed by cysteines 178 and 213 are represented above and below the central region respectively. Numbers indicate amino acid residues. (B) HeLa cells expressing wt PrP or C178-213S mutant lacking both cysteines, were pulse labelled for 10 min, with (lanes 3–4) or without 5 mM DTT (lanes 1–2, 5–6). PrP was recovered by IP with 3F4 and resolved by SDS-PAGE under reducing (R) or non-reducing (NR) conditions. The numbers on the left margin indicate the number of PrP N-glycans. Note that after 10 min pulse, PrP is still in the ER and only immature glycans (imm) are present.Citation7 Migration of a 30 kDa marker is indicated on the right. (C) Lysates of cells expressing wt PrP (lane 1–2) or C178-213S mutant (lanes 3–4) were treated with or without Endo H as indicated and then analysed by SDS-PAGE and immunoblot with 3F4 antibody. Mat; mature glycosylated species, mainly consisting of Endo-H resistant species, Imm; Endo-H sensitive, immature glycoforms. 0 points to the migration of unglycosylated molecules. Migration of MW markers is shown on the right hand margin. (D) After a 10 min pulse, cells expressing wt PrP or C178-213S were chased for 0 or 2 hours. For the samples in lanes 3–4, 5 mM DTT was present throughout the pulse and the chase. PrP was immunoprecipitated with 3F4 and resolved by SDS-PAGE under reducing conditions. (E) Aliquots of immunoprecipitates in lanes 2 and 4 from (D) were digested with PNGase F (PF) and analyzed under reducing (R) or non-reducing (NR) conditions. Only the relevant area of the gel is shown. (F) Cells expressing PrP were pulsed for 5 min and chased for 5, 10 or 30 min in the constant presence of 5 mM DTT. After IP with 3F4, proteins were analyzed under reducing (R) or non-reducing (NR) conditions. Mat; mature glycoforms, Imm; immature ER-resident glycoforms.
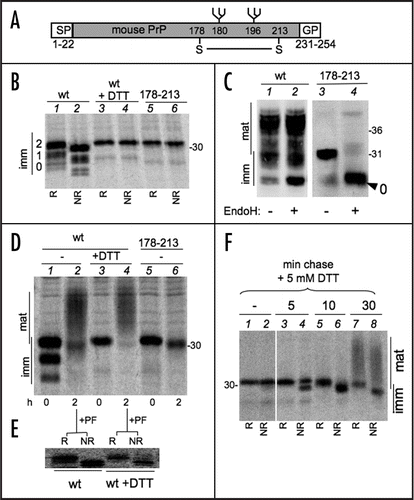
Figure 2 Redox dependency of PrP N-Glycosylation. (A) Cells expressing PrP were pulse-labelled for 1, 2 or 5 min. Lysates were immunoprecipitated with 3F4 and analyzed under reducing (R) or non-reducing (NR) conditions. Note that reduced, diglycosylated species are detectable only after a 1 min pulse (see lane 2, arrow). In (A, B and D), numbers on the left indicates the number of PrP immature N-glycans. (B) Same as in (A) but cells were pulsed for 5 min in the presence of the indicated concentration of DTT. (C) HeLa cells were transfected to express PrP alone (-) or together with exogenous wt Ero1α or its inactive C394A mutant. The Ero1α levels were determined by WB with Ero1α-specific antibodies. (D) HeLa cells transfected as in (C) were pulsed for 5 min in the presence of the indicated concentrations of DTT. After IP with 3F4, proteins were analysed under reducing (R) or non-reducing (NR) conditions. (E) The percentage of diglycosylated PrP relative to total PrP was determined by densitometric analyses from three independent experiments, as in (C). Error bars (SEM) are shown for PrP and PrP + wt Ero1α transfectants.
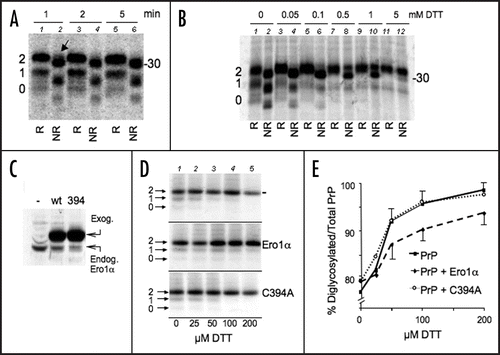
Figure 3 PrP GPI is slow and depends on disulphide formation. (A) Cells expressing PrP were pulsed for 10 min. Lysates were immunoprecipitated with αGP (lane 2) or 3F4 (lane 3) antibodies. Leftovers from these IPs were then immunoprecipitated with 3F4 (lane 1) and αGP (lane 4) respectively. (B) HeLa transfectants expressing exogenous PrP (left) or N2a cells expressing endogenous PrP (right) were pulsed for 10 min. PrP from cell lysates was immunoprecipitated with 3F4, αGP or P45–66 (α-PrP) antibodies. The percentage of signal recognised by αGP relative to 3F4 or α-PrP, calculated by densitometry, is indicated below the gels. (C) Lysates from HeLa cells expressing PrP were treated with PNGase F (PF), Endo-H (EH) or left untreated (-) before SDS-PAGE and WB with αGP (lanes 2–4). Note that PrP molecules with uncleaved C-terminal peptides do not acquire Endo-H resistance. The gel was stripped and reprobed with 3F4. Lane 1 containing untreated sample is shown to illustrate the normal pattern of PrP maturation. Mat and Imm indicate mature and immature glycoforms, respectively. (D) HeLa cells expressing wt PrP (lanes 1–2, 5–6) or C178–213S (lanes 3–4) were pulsed for 10 with 5 mM DTT or 2.5 µg/ml Tunicamycin (Tm). Lysates were immunoprecipitated with 3F4 or αGP as indicated. (E) Percent of signal recovered by IP with αGP relative to 3F4. Average from three (DTT and Tm) or two (178–231) independent experiments as in (D) is shown. SEM bar are present when applicable. The value in the first column (untreated) is taken from (B) for comparison. Note that the higher this value, the less is the GPI-anchoring.
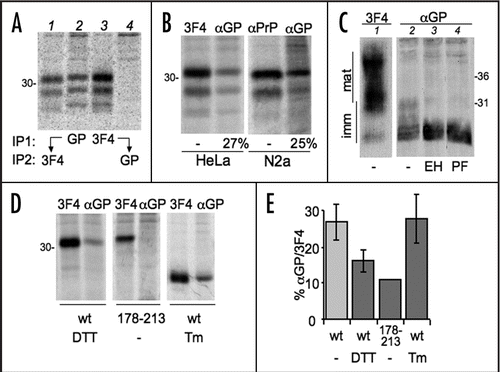
Figure 4 Tg treatment reduces PrP glycosylation. (A) HeLa cells expressing PrP were treated with 2.5 µg/ml thapsigargin for 0 (lane 1–2) or 230 min and then pulsed for 10 min in the absence (lane 1) or presence of the same concentration of thapsigargin (lanes 2–3). Lysates were immunoprecipitated with 3F4. (B) The percentages of the three PrP glycoforms were determined by densitometry from the experiment shown in (A) and two additional independent ones. Dark gray, diglycosylated; light gray, monoglycosylated; white, unglycosylated PrP. SEM bars are shown.
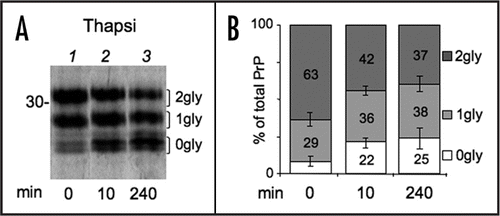
Figure 5 Ca2+ depletion reduces N-glycosylation of PrP independently from ER stress. (A) Cells expressing PrP were left untreated or incubated with cyclopiazonic acid (CPA) for 4 h. Cells were then starved for 20 min and pulsed for 10 min with (wash out = 0 min) or without CPA (wash out 20 + 10 = 30 min). Lysates were immunoprecipitated with 3F4 antibody (not shown). Percent of diglycosylated (dark gray), monoglycosylated (light gray) and unglycosylated PrP (white bars) were calculated by densitometry from at least two independent experiments. SEM bar are present for n = 3. (B) As an indicator of ER stress, XBP1 splicing was evaluated by PCR and electrophoresis on agarose gel in parallel samples treated with Tg or CPA for 4 h. (C) Cells were left untreated or incubated with Tg for 4 h and then pulsed for 10 min in the presence or absence of 5 mM DTT as indicated. After IP with 3F4, proteins were analysed under reducing conditions. Note that virtually all PrP is diglycosylated when cells are pulsed in the simultaneous presence of Tg and DTT, excluding substantial alterations of the OST complex at lower [Ca++]ER. (D) HeLa cells expressing myc-tagged Ero1α were incubated for 3.5 h with Tg or Tunicamycin (Tm). Glycosylated and unglycosylated Ero1α are indicated on the right. The percentage of glycosylated relative to total Ero1a in each condition is reported below each lane.
![Figure 5 Ca2+ depletion reduces N-glycosylation of PrP independently from ER stress. (A) Cells expressing PrP were left untreated or incubated with cyclopiazonic acid (CPA) for 4 h. Cells were then starved for 20 min and pulsed for 10 min with (wash out = 0 min) or without CPA (wash out 20 + 10 = 30 min). Lysates were immunoprecipitated with 3F4 antibody (not shown). Percent of diglycosylated (dark gray), monoglycosylated (light gray) and unglycosylated PrP (white bars) were calculated by densitometry from at least two independent experiments. SEM bar are present for n = 3. (B) As an indicator of ER stress, XBP1 splicing was evaluated by PCR and electrophoresis on agarose gel in parallel samples treated with Tg or CPA for 4 h. (C) Cells were left untreated or incubated with Tg for 4 h and then pulsed for 10 min in the presence or absence of 5 mM DTT as indicated. After IP with 3F4, proteins were analysed under reducing conditions. Note that virtually all PrP is diglycosylated when cells are pulsed in the simultaneous presence of Tg and DTT, excluding substantial alterations of the OST complex at lower [Ca++]ER. (D) HeLa cells expressing myc-tagged Ero1α were incubated for 3.5 h with Tg or Tunicamycin (Tm). Glycosylated and unglycosylated Ero1α are indicated on the right. The percentage of glycosylated relative to total Ero1a in each condition is reported below each lane.](/cms/asset/4509afb3-2e35-4a38-8b49-704e8e5c162a/kprn_a_10905727_f0005.gif)
Figure 6 [Ca++]ER affects GPI-anchoring of PrP. (A) Cells expressing PrP were incubated with thapsigargin for 0 (lanes 1–2) or 230 min (lanes 3–4) and then pulsed for 10 min in the presence of thapsigargin. Lysates were immunoprecipitated with 3F4 or αGP antibodies. The percentage of signal recognised by αGP relative to 3F4, calculated by densitometry, is indicated below the gels. (B) Cells expressing PrP were pulsed for 10 min. After IP with αGP the samples were analysed under reducing (R; lanes 2–3) and non-reducing conditions (NR, lane 1) or after treatment with Endo H (EH, lane 3).
![Figure 6 [Ca++]ER affects GPI-anchoring of PrP. (A) Cells expressing PrP were incubated with thapsigargin for 0 (lanes 1–2) or 230 min (lanes 3–4) and then pulsed for 10 min in the presence of thapsigargin. Lysates were immunoprecipitated with 3F4 or αGP antibodies. The percentage of signal recognised by αGP relative to 3F4, calculated by densitometry, is indicated below the gels. (B) Cells expressing PrP were pulsed for 10 min. After IP with αGP the samples were analysed under reducing (R; lanes 2–3) and non-reducing conditions (NR, lane 1) or after treatment with Endo H (EH, lane 3).](/cms/asset/35246def-76f0-42a2-8c2b-51116fbe6c5a/kprn_a_10905727_f0006.gif)
Figure 7 PrP molecules bearing an unprocessed C-terminus are mostly associated with the ER. (A) HeLa cells were lysed in SDS containing buffer (Tot) or permeabilised with 50 µg/ml digitonin to extract cytosolic (C) molecules. The digitonin insoluble fraction includes ER resident proteins (E). 50 µg of protein from each fraction were resolved and the blot was decorated with antibodies against cytosolic (Actin, TRX = thioredoxin) or luminal ER proteins (Grp94, BiP and ERp57). (B) Hela transfectants expressing PrP were left untreated (upper) or pre-incubated with Tg for 3.5 h (lower) and then pulsed for 10 min. 10 µM MG132 was added during the pulse to inhibit proteasomal degradation of cytosolic proteins. Cells were treated as in (A) and fractions immunoprecipitated with 3F4 or αGP antibodies. To facilitate the detection of PrP cytosolic species, twice as much of the cytosolic fraction was loaded.
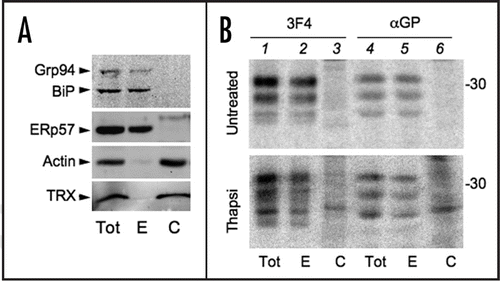
Figure 8 Increased PrP variability from competing posttranslational modifications. (A and B) Formation of the intra-chain disulphide bond might hamper substrate accessibility to the oligosaccharyl transferase (dark grey) and transamidase (black) complexes. The competition occurs in a narrow time frame, after PrP has left the ribosome and its C-terminus is moving through the translocon. (C) In the absence of Ca2+, PrP may adopt a structure that further inhibits processing by oligosaccharyl transferase and transamidase. Alternatively, Ca2+ could be important for chaperones or other folding assistants (chap). (D) Summary of PrP covalent modifications and their interplays. Inhibiting disulphide bond formation and lowering [Ca2+]ER similarly induce ER stress and weaken PrP ER targeting and translocation.Citation7,Citation10 In contrast, Tg and DTT have antagonistic effects on N-glycosylation and GPI-anchoring. N-glycosylation affects neither disulphide bond formation, nor GPI-anchoring.
![Figure 8 Increased PrP variability from competing posttranslational modifications. (A and B) Formation of the intra-chain disulphide bond might hamper substrate accessibility to the oligosaccharyl transferase (dark grey) and transamidase (black) complexes. The competition occurs in a narrow time frame, after PrP has left the ribosome and its C-terminus is moving through the translocon. (C) In the absence of Ca2+, PrP may adopt a structure that further inhibits processing by oligosaccharyl transferase and transamidase. Alternatively, Ca2+ could be important for chaperones or other folding assistants (chap). (D) Summary of PrP covalent modifications and their interplays. Inhibiting disulphide bond formation and lowering [Ca2+]ER similarly induce ER stress and weaken PrP ER targeting and translocation.Citation7,Citation10 In contrast, Tg and DTT have antagonistic effects on N-glycosylation and GPI-anchoring. N-glycosylation affects neither disulphide bond formation, nor GPI-anchoring.](/cms/asset/b8ace661-aef9-478c-ac9f-1caa06830afb/kprn_a_10905727_f0008.gif)
Acknowledgements
We thank Drs. N.J. Bulleid, S. Cenci, R. Chiesa, M. Nuvolone, M. Otsu, L. Rampoldi and S. Tooze for stimulating discussions and advice, D. Harris, N. Hooper and R.J. Kascsak, for providing excellent reagents, Claudio Fagioli and Elena Pasqualetto for technical help, and Ana Fella and Raffaella Brambati for expert secretarial assistance. The financial support of Associazione Italiana per la Ricerca sul Cancro (AIRC), Cariplo (Project NOBEL), Ministero Universita' e Ricerca (MIUR CoFin) and Telethon-Italy (Grant no. GGP06155) is gratefully acknowledged.
References
- Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature 2003; 426:891 - 894
- Johnson AE, van Waes MA. The translocon: a dynamic gateway at the ER membrane. Annu Rev Cell Dev Biol 1999; 15:799 - 842
- Frand AR, Cuozzo JW, Kaiser CA. Pathways for protein disulphide bond formation. Trends Cell Biol 2000; 10:203 - 210
- Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science 2001; 291:2364 - 2369
- Udenfriend S, Kodukula K. How glycosylphosphatidylinositol-anchored membrane proteins are made. Annu Rev Biochem 1995; 64:563 - 591
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363 - 13383
- Orsi A, Fioriti L, Chiesa R, Sitia R. Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J Biol Chem 2006; 281:30431 - 30438
- Walmsley AR, Zeng F, Hooper NM. Membrane topology influences N-glycosylation of the prion protein. Embo J 2001; 20:703 - 712
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996; 39:767 - 778
- Kang SW, Rane NS, Kim SJ, Garrison JL, Taunton J, Hegde RS. Substrate-Specific Translocational Attenuation during ER Stress Defines a Pre-Emptive Quality Control Pathway. Cell 2006; 127:999 - 1013
- Fioriti L, Dossena S, Stewart LR, Stewart RS, Harris DA, Forloni G, Chiesa R. Cytosolic prion protein (PrP) is not toxic in N2a cells and primary neurons expressing pathogenic PrP mutations. J Biol Chem 2005; 280:11320 - 11328
- Lehmann S, Harris DA. A mutant prion protein displays an aberrant membrane association when expressed in cultured cells. J Biol Chem 1995; 270:24589 - 24597
- Bertoli G, Simmen T, Anelli T, Molteni SN, Fesce R, Sitia R. Two conserved cysteine triads in human Ero1alpha cooperate for efficient disulfide bond formation in the endoplasmic reticulum. J Biol Chem 2004; 279:30047 - 30052
- Kascsak RJ, Rubenstein R, Merz PA, Tonna DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 1987; 61:3688 - 3693
- Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, Clarke F, Sitia R, Rubartelli A. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci USA 2002; 99:1491 - 1496
- Jessop CE, Bulleid NJ. Glutathione directly reduces an oxidoreductase in the endoplasmic reticulum of mammalian cells. J Biol Chem 2004; 279:55341 - 55347
- Turk E, Teplow DB, Hood LE, Prusiner SB. Purification and properties of the cellular and scrapie hamster prion proteins. Eur J Biochem 1988; 176:21 - 30
- Capellari S, Zaidi SI, Urig CB, Perry G, Smith MA, Petersen RB. Prion protein glycosylation is sensitive to redox change. J Biol Chem 1999; 274:34846 - 34850
- Valetti C, Sitia R. The differential effects of dithiothreitol and 2-mercaptoethanol on the secretion of partially and completely assembled immunoglobulins suggest that thiol-mediated retention does not take place in or beyond the Golgi. Mol Biol Cell 1994; 5:1311 - 1324
- Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J 2001; 20:6288 - 6296
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987; 51:229 - 240
- Whitley P, Nilsson IM, von Heijne G. A nascent secretory protein may traverse the ribosome/endoplasmic reticulum translocase complex as an extended chain. J Biol Chem 1996; 271:6241 - 6244
- Walmsley AR, Hooper NM. Distance of sequons to the C-terminus influences the cellular N-glycosylation of the prion protein. Biochem J 2003; 370:351 - 355
- Mattioli L, Anelli T, Fagioli C, Tacchetti C, Sitia R, Valetti C. ER storage diseases: a role for ERGIC-53 in controlling the formation and shape of Russell bodies. J Cell Sci 2006; 119:2532 - 2541
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996; 383:685 - 690
- Lehmann S, Harris DA. Blockade of glycosylation promotes acquisition of scrapie-like properties by the prion protein in cultured cells. J Biol Chem 1997; 272:21479 - 21487
- Taraboulos A, Rogers M, Borchelt DR, McKinley MP, Scott M, Serban D, Prusiner SB. Acquisition of protease resistance by prion proteins in scrapie-infected cells does not require asparagine-linked glycosylation. Proc Natl Acad Sci USA 1990; 87:8262 - 8266