Abstract
Prion disease is a neurodegenerative disorder, in which the normal prion protein (PrP) changes structurally into an abnormal form and accumulates in the brain. There is a great demand for the development of a viable approach to diagnosis and therapy. Not only has the ligand against PrP been used for diagnosis, but it has also become a promising tool for therapy, as an antibody. Aptamers are a novel type of ligand composed of nucleic acids. DNA aptamers in particular have many advantages over antibodies. Therefore, we tried to isolate the DNA aptamer for mouse PrP. We developed a competitive selection method and tried to screen the DNA aptamer with it. In the fourth round of selection, several clones of the aptamer with an affinity to PrP were enriched, and clone 4-9 showed the highest affinity of all. The investigation by aptamer blotting and Western blotting showed that clone 4-9 was specifically able to recognize both α-PrP and β-PrP. Moreover, it was indicated that clone 4-9 could recognize the flexible region of the N-terminal domain of PrP. These characteristics suggest that clone 4-9 might be a useful tool in prion-disease diagnosis and research.
Introduction
Prion protein (PrP), which is mainly located in the neuronal cells of the central nervous system in mammals, is thought to be the disease agent of transmissible spongiform encephalopathy, otherwise known as prion disease. This disease has many forms: scrapie in sheep, bovine spongiform encephalopathy (BSE) in cattle and Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker syndrome in humans. The cause of these diseases is thought to be the conformational change of normal cellular PrP (PrPC), which is an α-helix-rich isoform sensitive to protease digestion, into abnormal PrP (PrPSc), which is a β-sheet-rich isoform resistant to protease digestion.Citation1 Additionally, the conformational conversion is considered to induce the aggregation of PrPSc into amyloid fibrils.
PrPSc is the marker of prion disease. In current diagnosis kits, anti-PrP antibody is generally used. In addition, it has been recently reported that the formation of PrPSc was prevented by an anti-PrP antibody in animal experiments.Citation2 Therefore, the use of antibodies is expected to lead to the development of a therapy. For these reasons, a ligand against PrP would be the key to the development of not only a diagnostic technique but also a treatment approach for prion disease.
Aptamers are a type of single-stranded DNA or RNA ligand that bind to the target molecule with high affinity and specificity, sometimes equivalent to those of an antibody. Aptamers with dissociation constants (Kd) in the nanomolar-to-picomolar range have been reported, and the specificity is good enough to enable them to discriminate between similar proteins that share common structural domains.Citation3,Citation4
DNA aptamers, in particular, have certain advantages over antibodies and/or RNA aptamers as ligands. For instance, DNA aptamers can be easily and inexpensively prepared by chemical synthesis and are biochemically stable. In addition, their utilization allows the construction of high-through-put microarray detection systems because a considerable number of studies have already been conducted on DNA array technology. Due to these attributes, DNA aptamers can be applied in a wide range of technologies, such as target validation, drug discovery, affinity chromatography, in vivo imaging and therapy. There is no doubt that DNA aptamers would be a powerful type of ligand in many fields, since they have an affinity and specificity equivalent to those of antibodies. In particular, DNA aptamers against PrP would be useful in several fields of study related to prion disease.
Several RNA aptamers and a DNA aptamer against PrP have been reported already.Citation5–Citation12 These RNA aptamers were reported to have high affinity, with Kd values in the range of 1 × 10−8∼10−9 M, and some were found to have the ability to inhibit the formation of PrPSc.Citation5,Citation6 These aptamers are therefore promising tools in several fields related to prion disease. However, there is to date only a few reports of DNA aptamer screening against PrP, and the aptamers isolated in those studies could recognize PrPC but not PrPSc.Citation9,Citation10
In our present study, we therefore tried to screen DNA aptamers against PrP and isolate that DNA aptamer which can be applied to the diagnosis and/or therapy of prion disease. Aptamers are screened from a chemically synthesized nucleic-acid combinatorial library by an in vitro selection method called “systematic evolution of ligands by exponential enrichment (SELEX).”Citation13 In the screening of DNA aptamers against PrP, it might be difficult to enrich aptamers with high specificity to PrP, because PrP has the propensity to nonspecifically interact with nucleic acids due to the electrostatic interactions promoted by its inherent positive charge. In fact, it is reported that PrP can interact and form complexes with nucleic acids, just as nucleic-acid-binding proteins do.Citation14–Citation16 Due to this trait, those aptamers that are specifically able to bind to PrP cannot be concentrated from the DNA library. Therefore, we developed a novel competitive selection method in SELEX. We expected that this method would allow us to screen DNA aptamers against PrP efficiently. shows a schematic diagram of the operations of our novel competitive selection method. Thus, our aim was to screen, by means of this competitive selection method, those DNA aptamers which bind to PrP, and evaluate the isolated DNA aptamer and its potential for application to the diagnosis and research of prion disease.
Material and Methods
Reagents and DNAs.
All the reagents used in all the experiments were analytical-grade. AmpliTaq Gold (Applied Biosystems) was used for PCR. The sequence of the DNA libraries used in the competitive selection was 5′-ATACCAGTCTATTCAATT-30N-AGATAGTATGTGCAATCA-3′ (30N is the 30-mer randomized region), and these DNAs were labeled with fluorescein isothiocyanate (FITC) at their 5′-ends. The sequences of the primers used for PCR amplification were 5′-ATACCAGTCTATTCAATT-3′ (FITC-labeled at the 5′-end) and 5′-TGATTGCACATACTATCT-3′ (biotin-labeled at the 5′-end). Clone 4–9, isolated through the selection, was also labeled with FITC at its 5′-end. All the DNA oligonucleotides used were synthesized by Invitrogen.
Preparation of proteins.
Mouse-derived full-length recombinant PrP (mrPrP), corresponding to residues 23–230, was cloned, expressed and purified as reported previously,Citation17,Citation18 with the following modification. PrP corresponding to residues 23–231 without any tag was obtained by PCR amplification of cDNA and inserted as Nde I and BamH I restriction fragments into the expression plasmid pET22b(+) and amplified in Escherichia coli Rosetta (DE3) pLysS. Culture growth and expression of mrPrP were conducted in accordance with the instructions in the pET system manual. The cultured cells were harvested by centrifugation at 5,000 × g for 10 min. The pellets were resuspended and lysed in ice-cold lysis buffer (150 mM NaCl, 5 mM EDTA, 50 mM Tris/HCl, pH 8.0) using a French press. The lysate was centrifuged at 20,000 × g for 30 min at 4°C. The proteins in each pellet were dissolved in solubilization buffer (8 M urea, 20 mM HEPES, pH 7.5) and reduced by the addition of 100 mM dithiothreitol. Cell debris and insoluble materials were removed by centrifugation at 20,000 × g for 30 min. The supernatant was loaded onto a cation exchange chromatography column (Resource S column, GE Healthcare) pre-equilibrated with the solubilization buffer. After washing the column with the same buffer, the bound proteins were eluted with the solubilization buffer over a gradient of 0-600 mM NaCl. The eluted fractions of mrPrP were pooled, and the concentration of PrP was adjusted in 0.1 mg/mL. The disulfide bonds of PrP were oxidized by stirring overnight in a 2:1 molar excess of CuSO4. A three-fold volume of 0.1% (v/v) trifluoroacetic acid (TFA) in distilled water (DW) was added to the oxidized protein solution, and the solution was loaded onto a reverse-phase chromatography column (Resource RPC column, GE Healthcare) pre-equilibrated with 0.1% TFA in DW for solubilization. After washing the column with the same buffer, the bound proteins were eluted with 0.1% TFA in acetonitrile. The eluted fractions of mrPrP were pooled and lyophilized. For the preparation of α-PrP, lyophilized mrPrP was solubilized in 9 M urea. This solution was adjusted to 0.3 mg/mL mrPrP in buffer containing 3 M urea and 20 mM sodium acetate at pH 5.5, followed by dialysis against the same buffer with three changes of buffer. The purification of α-PrP was analyzed by SDS-PAGE and circular dichroic analysis.
In the preparation of β-PrP, cell-free conversionCitation18 was used, as previously reported. Lyophilized mrPrP was solubilized in 6 M guanidine hydrochloride (GdnHCl). This solution was adjusted to 0.5 mg/mL mrPrP in buffer containing 1 M GdnHCl, 3 M urea, 150 mM NaCl and 20 mM HEPES at pH 6.7, and incubated at 37 °C under agitation at 900 rpm by a Thermomixer Comfort (Eppendorf). The formation of β-PrP was confirmed by the resistance to protease K (PK) digestion and thioflavine T binding assay. β-PrP was partially digested by protease, and by Western blot analysis we were able to observe its band, which shifted to the lower-molecular-weight side relative to that of full-length PrP. This pattern was in agreement with the report by Bocharova et al.Citation18 PQQGDH and GST-Zif268 were prepared as described previously.Citation19,Citation20
Competitive selection.
The DNA library was denatured at 90°C and then refolded by gradual cooling to room temperature for 30 min in PBS buffer prior to each selection cycle. α-PrP was used as the objective target, and GST-Zif268 and PQQGDH as the competitors in SELEX. These target proteins were spotted onto a nitrocellulose membrane and then air-dried. This protein-spotted membrane was incubated with 4% skim milk in PBS containing 0.5% Tween 20 (PBS-T) buffer for 1 h with gentle shaking, followed by incubation with the DNA library in PBS-T buffer with gentle shaking. At this step, various concentrations of transfer RNA were added to the solution containing the DNA library in order to reduce the nonspecific binding of DNA to α-PrP. The unbound DNAs were removed by intensive washing with PBS-T buffer. The α-PrP-spotted region of the membrane was cut out, and the bound DNA was eluted by incubation in a 7-M urea solution. The eluted DNAs were purified by phenol extraction and concentrated by ethanol precipitation. The purified DNAs were amplified by PCR. For the preparation of the single-stranded DNA, the amplified DNAs were mixed and incubated with avidin-immobilized beads. Then, only the FITC-labeled DNA was eluted by the addition of 0.15 M NaOH, and this DNA pool was used in the next selection round. This cycle was repeated several times. When an enriched DNA pool with good affinity to α-PrP was obtained, it was cloned. This selection process is schematized in . The concentration condition of DNA, proteins and tRNA in each round of SELEX is shown in . The sequences of each clone were analyzed using an ABI PRISM 310 Genetic Analyzer (GE Healthcare).
Binding assay by aptamer blotting and Western blotting.
A binding assay of the enriched DNA pool was carried out at each round by visualizing the interaction of the DNAs with their targets on a membrane, as previously described.Citation21 The incubation of the DNAs with their targets on the membrane was carried out by the competitive selection method mentioned above. Then, the membrane was incubated with horseradish peroxidase (HRP)-labeled anti-FITC antibody and washed with PBS-T buffer for 1 h with gentle shaking, followed by intensive washing with PBS-T buffer. The membrane was incubated with ECL Plus (GE Healthcare) in accordance with the recommended protocol, and a chemiluminescence image was obtained using a Typhoon 8600 (GE Healthcare). The obtained chemiluminescence image was analyzed with IMAGE QUANT software (GE Healthcare).
Western blotting was carried out by SDS-PAGE using 15% gels and blotting of the membrane. The subsequent incubation of the DNA and chemiluminescence detection was carried out through aptamer blotting. A positive control experiment was conducted using the anti-mouse PrP antibody 7D9 (AbD Serotec) as the primary antibody and anti-mouse IgG antibody-conjugated HRP and STAR105P (AbD Serotec) as the secondary antibody. β-PrP was treated with PK at various concentrations for 1 h or not treated at all. The reaction was stopped by adding a stop solution (9 M urea, 50 mM Hepes-NaOH pH 7.0 containing 1 mg/mL Pefabloc SC; Merck) at twice the volume of the reaction mixture.
Binding assay by SPR.
A binding assay of the isolated DNA clone was carried out by SPR. α-PrP or β-PrP was immobilized on a CM5 sensor chip (Biacore) via covalent bonds, and the DNA clone was applied to each. SPR signals were measured with BIACORE X (Biacore) at a PBS-buffer flow rate of 5–20 µL/min. The equilibrium signals were used for the construction of a Scatchard plot.
Detection of PrP by fluorescence depolarization.
The changes in the fluorescence depolarization of the FITC-modified DNA were measured. The DNA was folded, as described above. DNA at a concentration of 1 µM and α-PrP at various concentrations were mixed into PBS buffer. The mixture was incubated at room temperature for more than 10 min, and its fluorescence depolarization was measured. As a control, bovine serum albumin (BSA) was used.
Results
Competitive selection.
The screening of aptamers against α-PrP, which represents the isoform of PrPC prepared in vitro, was carried out using the competitive selection method illustrated in . Although the DNA library is usually incubated with a single target in normal SELEX, in this competitive selection, the DNA library was simultaneously incubated with multiple targets, which included the objective target and other targets as competitors. In this method, competitors with properties similar to those of the objective target can induce the exclusion of the aptamer pool which binds to both the competitors and the objective target, and consequently the aptamer which is able to bind only to the objective target is enriched.
In this experiment, GST-fused Zif268 (pI = 8.86; MW = 38,272)Citation20,Citation22 and pyrroloquinoline quinone glucose dehydrogenase (PQQGDH) (pI = 8.75; MW = 50,237)Citation19 were used as competitors with similar properties, among them the propensity for nonspecifically interacting with DNA, as PrP does (pI = 9.58; MW = 22,932). Zif268 is a DNA-binding protein with a zinc finger motif, and PQQGDH has a high positive charge. Using this method, we tried to screen the DNA aptamer specific to α-PrP.
At every selection round, the binding ability of the enriched DNA pool was analyzed by binding assay with aptamer blotting (data not shown).Citation21 Chemiluminescent spots were observed where α-PrP and GST-Zif268 were spotted onto the membrane, but not when PQQGDH was spotted, although it was confirmed by Ponceau-S staining of the membrane that all blotted proteins remained on the membrane. Therefore, this result suggests that α-PrP and Zif268 have the propensity to interact with DNA, and that Zif268 is appropriate as a competitor. For a more exact evaluation of the binding ability of the DNA pool in each round, a binding assay was carried out using the membrane, as shown in . In this assay, PQQGDH was not used because it did not show a high affinity for DNA under these experimental conditions. The results are shown in . In the fourth round, the enriched DNA pool showed a larger increase in the binding to α-PrP than in that to Zif268.
Some DNA clones were isolated from the DNA pool in the fourth round. Thereafter, their binding abilities were evaluated by surface plasmon resonance (SPR) measurement. The interaction of each clone with the α-PrP immobilized on the chip was measured. Twenty-one DNA sequences were analyzed, and two pairs consisting of the same sequences were identified; that is, nineteen different sequences were identified. Their sequences are shown in Table S1 in Supplementary Data. Subsequently, ten clones predicted to have a stable structure by the Mfold web serverCitation23 were picked out based on the fact that an aptamer recognizes its target molecule by folding into a distinctive structure, and then their affinities to α-PrP were measured by SPR. The SPR sensorgrams are shown in . About half of these clones had a higher ability to bind to α-PrP than to the initial library. We can say that the objective aptamer was effectively enriched by this method, although no obvious convergence of sequences was observed at the fourth round (data not shown). The clone with the highest binding ability was clone 4–9 (5′ATACCAGTCTATTCAATTGGGCCCTTTTCTGTGTCTTTCTGTGCCCTTAGATAGTATGTGCAATCA3′).
Characterization of the specificity of clone 4–9.
The specificity of clone 4–9 was evaluated by binding assay with aptamer blotting (). We investigated the binding of clone 4–9 to α-PrP, Zif268, BSA and β-PrP, whose amyloid fibril was prepared from PrP in vitro and is thought to be the isoform of PrPSc.Citation18 The preparation of β-PrP was confirmed by TfT assay and the resistance to protease digestion (data not shown). If an aptamer is able to bind to β-PrP, it will be useful for applications such as therapy and diagnosis. In addition, Zif268 has been investigated particularly in order to evaluate the possibility of its application to diagnosis. If a DNA aptamer is to be applied to the diagnosis of prion disease, we must investigate whether or not the DNA aptamer interacts with DNA-binding proteins and positively charged proteins such as Zif268, because the samples used for the diagnosis of prion disease are homogenates containing such proteins, which may lead to false-positive detection signals.
No chemiluminescent spots were observed on the membranes prepared with Zif268 and BSA. However, increases in chemiluminescence intensity as a function of the concentration of α-PrP and β-PrP were observed. This means that clone 4–9 bound to α-PrP and β-PrP, but not to Zif268 and BSA, which indicates that clone 4–9 could recognize α-PrP and β-PrP with high specificity.
Moreover, its applicability to Western blot analysis was evaluated. Interestingly, bands derived from α-PrP and β-PrP were observed (data not shown), although in this method PrP is denatured and the conformation of both the α-form and the β-form is disrupted.
Next, we tried to detect PK-digested β-PrP by Western blot analysis using clone 4–9 (). In the positive control experiment using anti-PrP antibody, both the band of PK-digested β-PrP and that of non-digested β-PrP were observed. Meanwhile, in the experiment using clone 4–9 a band of full-length β-PrP was detected, but no band of PK-digested β-PrP. This means that clone 4–9 can recognize the region digested by PK, which may be a flexible region in the N-terminus of PrP.
Characterization of the binding ability of clone 4–9.
The binding ability of clone 4–9 to α-PrP was characterized using SPR. The interaction with α-PrP immobilized on a chip was measured at various concentrations of clone 4–9. The signal was observed to increase with increasing concentration of clone 4–9 (Fig. S1). The Scatchard plot of the signal shows that the Kd value was 1.13 × 10−7 M.
The binding ability of clone 4–9 to β-PrP was characterized as well. The signal was observed to increase with increasing concentration of clone 4–9 (Fig. S1). The Kd value was 1.00 × 10−7 M. The similarity in the binding ability of clone 4–9 to α-PrP and β-PrP is in agreement with the results of aptamer blotting () and Western blotting (data not shown) analyses.
The binding region in clone 4–9 was identified using SPR. The folding of clone 4–9 was predicted by the Mfold web server, and showed two stem-loop structures (). At first, the binding ability of the deletion mutant clone 4–9 (19–66) to α-PrP was evaluated. This mutant is a partial sequence lacking the 18-mer primer sequence at the 5′-end (Fig. S2). The signal was observed to increase with increasing concentration of clone 4–9 (19–66) (). The Scatchard analysis of these data gave almost the same Kd value, 1.32 × 10−7 M. Next, another deletion mutant, clone 4–9 (19–46), to α-PrP was evaluated. This mutant is a partial sequence lacking the 18-mer primer sequence from the 5′-end and the small stem-loop at the 3′-end (Fig. S2 in Supplementary Data). The signal was observed to increase with increasing concentration of clone 4–9 (19–46) (Fig. S2 in Supplementary Data). However, the signal did not reach a plateau at high concentrations of DNA. The Scatchard analysis of these data gave a higher Kd value, 1.04 × 10−6 M. In addition, it was also found that the two oligonucleotides at the primer regions had no affinity to α-PrP (data not shown).
Application of clone 4–9 to a detection system with fluorescence depolarization.
The results mentioned above indicate that clone 4–9 can be applied to immunochromatographic assay, ELISA and Western blot analysis. For further investigation of the possibility of its application to diagnosis, the detection of α-PrP by clone 4–9 by fluorescence depolarization measurement was carried out. It was observed that the changes in polarization depended on increases in PrP concentration, but not on increases in BSA concentration (). This result suggests that clone 4–9 can indeed be applied to PrP detection.
Discussion
We screened DNA aptamers against α-PrP in SELEX using our competitive selection method illustrated in . The evaluation of the binding ability of the DNA pool in each round ( and ) showed that the enriched DNA pool in the fourth round contained an aptamer with higher affinity to α-PrP. In the current SELEX, 10 to 20 rounds usually need to be carried out.Citation24,Citation25 This means that the DNA aptamers seemed to have been efficiently enriched by our competitive selection method. Numerous attempts have been made by researchers to develop a method of enriching aptamers with high affinity to the target in SELEX. For example, the use of techniques for measuring biomolecular interaction has been reported, such as capillary electrophoresisCitation26 and SPR,Citation27 in the process of separation of the target-bound nucleic acids from the unbound ones. However, only a few attempts have been made so far to develop a selection method focused on the specificity of aptamers and not on negative selection.Citation12,Citation28 Therefore, the competitive selection method developed in this study might serve as a novel method of determining specificity, although its precise efficiency has not yet been demonstrated. In addition, the utilization of this method enables us to simultaneously screen aptamers against multiple targets. In that case, the more specific the aptamer, the more it can be enriched by competition with multiple targets.
The DNA aptamer clone 4–9, isolated from the DNA pool in the fourth round, specifically detects α-PrP and β-PrP, but not Zif268 and BSA in aptamer blotting (). The SPR analysis showed that it has a Kd value of 1.13 × 10−7 M for α-PrP and 1.00 × 10−7 M for β-PrP. In addition, the affinity analysis of the two deletion mutants showed that clone 4–9 (19–66) retained the binding ability of clone 4–9 to α-PrP but that clone 4–9 (19–46) did not (). This indicates that the region 47–66 residues away from the 5′-end of clone 4–9 is related to the interaction between clone 4–9 and PrP, even though it is the primer region. We also confirmed that only the primer region had no affinity to α-PrP. Therefore, this primer region may not have any ability to bind to α-PrP and is related to the stability of another stem-loop located in the randomized region of clone 4–9. Moreover, clone 4–9 (19–66) is a minimized sequence which includes secondary structures, i.e., two stem-loop structures. The binding ability of an aptamer is usually correlated to that of the secondary structure, so these results also indicate that secondary structure may have been correctly predicted.
Clone 4–9 was successfully applied to the detection of PrP with aptamer blotting, Western blotting and fluorescence depolarization (, , and ). This indicates that clone 4–9 might be applicable to prion diagnosis, although it might not to be applicable to the current method of detection which is based on PK treatment. DNA aptamers which can bind to PrPC have already been reported.Citation9,Citation10 Some DNA aptamers have a high affinity to PrP, with Kd values in the range of 1.0 × 10−9∼10−10.Citation10 But these aptamers cannot recognize PrPSc. This is the first report of a DNA aptamer which can bind to β-PrP.
Moreover, clone 4–9 can bind to PrP that has undergone denaturation by sodium dodecyl sulfate (). This ability to bind PK-digested β-PrP () suggests that clone 4–9 may recognize the unfolded region in the N-terminal domain of PrP. These two properties are very particular to clone 4–9 and set it apart from the anti-prion DNA aptamers reported previously.Citation9,Citation10
Several RNA aptamers against PrP have already been reported.Citation5,Citation6,Citation8,Citation11,Citation12 Many RNA aptamers can recognize α-PrP but not β-PrP.Citation5,Citation11,Citation12 An RNA aptamer that can bind specifically to β-PrP has been found by Rhie et al.Citation6 In addition, this RNA aptamer could inhibit the conversion of α-PrP to β-PrP in an in vitro assay. Another RNA aptamer has been reported by Proske et al. to inhibit the conversion of α-PrP to β-PrP in an assay using prion-infected cells.Citation5 According to these researchers, this aptamer prevents prion conversion by binding to α-PrP, which inhibits the association between α-PrP and β-PrP. These aptamers may constitute a valuable tool for drug discovery and mechanism analysis in prion disease. In terms of prion-disease diagnosis, the RNA aptamer reported by Rhie et al.Citation6 has a suitable characteristic: it can bind specifically to β-PrP. However, the RNA aptamer must be modified to be resistant to ribonuclease digestion, because RNA can be easily digested by the many ribonucleases present in the in vivo environment. Therefore, clone 4–9 has an advantage as a diagnosis tool, namely that it consists of DNA (although RNA aptamers have other merits).
As mentioned above, clone 4–9 cannot be applied to current prion diagnosis because it binds to the region outside the PK-resistant core. PrPC and PrPSc have different characteristics vis-à-vis protease digestion. PrPSc has partial resistance to PK. In contrast, PrPC is sensitive to PK and can be completely digested by it. Since many antibodies and aptamers bind to both PrPC and PrPSc, pre-treatment by PK digestion is required, which results in samples containing no PrPC. In this case, because the N-terminus of PrPSc is digested and only the PK-resistant core remains, the antibody or aptamer must recognize the PK-resistant core. Recently, some aptamers have been reported to bind specifically to PrPC.Citation9,Citation10 We think that if PrPC can be removed by those aptamers, clone 4–9 can be applied to prion-disease diagnosis. In addition, aptamers, especially DNA aptamers, have characteristics which make them useful for PrP detection: they are easily and inexpensively prepared, are regenerable and are biochemically stable. Thus, we believe that clone 4–9 would be useful in prion-disease diagnosis.
In the field of prion-disease diagnosis, the development of a rapid detection system using a ligand which recognizes PrP is crucial. In particular, such a system is necessary for the detection of BSE. Therefore, a detection system using clone 4–9 which can recognize bovine PrP is essential. The amino-acid sequences of PrPs have high homology and identity across different species; for example, mouse PrP has a 92% identity to bovine PrP and a 93% identity to human PrP. Moreover, almost all mammalian PrPs have an unfolded N-terminal domain. Therefore, clone 4–9 might recognize PrPs derived from certain species. This remains to be investigated in future studies.
Abbreviations
| PrP | = | prion protein |
| BSE | = | bovine spongiform encephalopathy |
| PrPC | = | normal cellular prion protein |
| PrPSc | = | scrapie-associated prion protein |
| SELEX | = | systematic evolution of ligands by exponential enrichment |
| α-PrP | = | isoform of PrPC prepared in vitro |
| β-PrP | = | isoform of PrPSc prepared in vitro |
Figures and Tables
Figure 1 Sequence of operations in the competitive selection method. The protein-blotted membrane was incubated with the DNA library. After the removal of the unbound DNAs, the portion of the membrane on which the target was immobilized was cut out. The target-bound DNAs were eluted and then amplified by PCR to obtain a new library. This cycle was repeated.
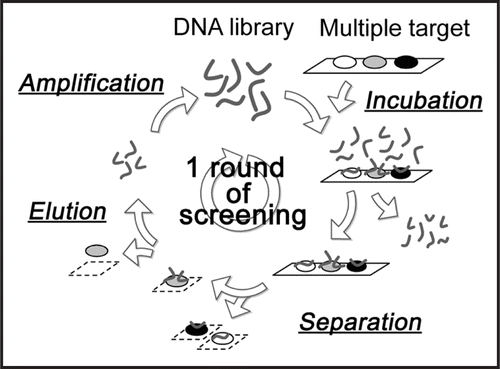
Figure 2 Binding assay of DNA pools to target proteins by aptamer blotting. At the top left, the immobilized proteins on the membrane are shown. α-PrP and Zif268 were spotted onto the membrane at 0.9 pmol and 79 pmol, respectively. The other figure shows a chemiluminescent image of the aptamer blotting. DNA at a concentration of 1 µM was incubated with the membrane. The black spots indicate chemiluminescence, which reflects the amount of target-bound DNA. The number of rounds is shown on the left side of the image.

Figure 3 Binding assay of DNA clones by SPR. The constantly appearing DNA clones were injected on a chip, onto which α-PrP was already immobilized. The names of the clones are shown on the right. The clones are ordered according to the strength of the signal.
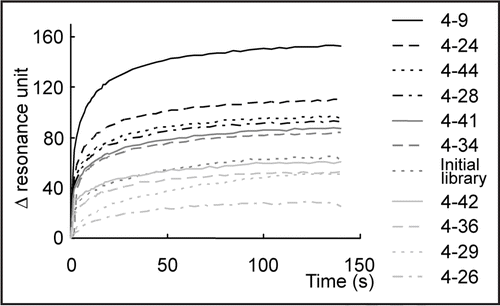
Figure 4 Evaluation of the specificity of clone 4–9 by aptamer blotting. Images of chemiluminescence detection are shown. On the left, the immobilized proteins are indicated. The upper number shows the quantitative ratio of proteins immobilized on the membrane. “1” means 1 pmol proteins in the case of α-PrP, Zif268 and BSA, whereas “1” means 4 pmol proteins in the case of β-PrP.
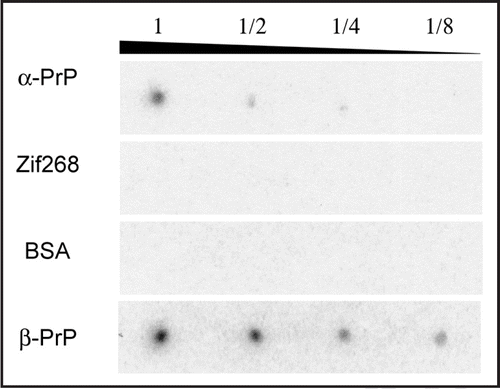
Figure 5 Detection of PK-digested β-PrP by Western blotting. Images of chemiluminescence detection using clone 4–9 and the anti-PrP antibody 7D9 are shown. β-PrP at a concentration of 0.2 mg/mL was treated with PK at various concentrations. The numbers show the concentration of PK: (1) no PK, (2) 4 ng/mL, (3) 8 ng/mL, (4) 40 ng/mL, (5) 80 ng/mL.
Figure 6 Predicted folding of clone 4–9. The folding of clone 4–9 was predicted using the Mfold web server (Zuker, 2003).
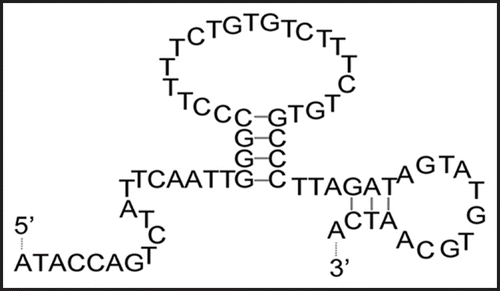
Figure 7 Application of clone 4–9 to a detection system with fluorescence depolarization. FITC-labeled DNA at a concentration of 1 µM and α-PrP at various concentrations were mixed in PBS buffer. The mixture was incubated at room temperature for more than ten min, and then its fluorescence depolarization was measured. BSA was used as a control.
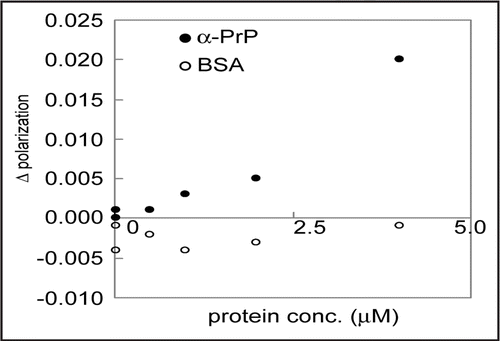
Table 1 The concentrations of DNA, proteins and tRNA at each round of SELEX
Acknowledgements
This work is partly supported by the Grant-in-Aid for the 21st Century COE “Future Nano- Materials” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and also by Industrial Technology Research Grant Project in 2006 from New Energy and Industrial Technology Development Organization (NEDO) of JAPAN.
Note
Supplemental information can be found at: www.landesbioscience.com/supplement/Ogasawara PRION1-4-Suppl.pdf
References
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 13383
- White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003; 422:80 - 83
- Doudna JA, Cech TR, Sullenger BA. Selection of an RNA molecule that mimics a major autoantigenic epitope of human insulin receptor. Proc Natl Acad Sci U S A 1995; 92:2355 - 2359
- Rusconi CP, Scardino E, Layzer J, Pitoc GA, Ortel TL, Monroe D, Sullenger BA. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 2002; 419:90 - 94
- Proske D, Gilch S, Wopfner F, Schatzl HM, Winnacker EL, Famulok M. Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem 2002; 3:717 - 725
- Rhie A, Kirby L, Sayer N, Wellesley R, Disterer P, Sylvester I, Gill A, Hope J, James W, Tahiri-Alaoui A. Characterization of 2′-fluoro-RNA aptamers that bind preferentially to disease-associated conformations of prion protein and inhibit conversion. J Biol Chem 2003; 278:39697 - 39705
- Davidowitz E, Eljuga L, Dover K, Tian J, Grossman A. Concentration of prion protein from biological samples to increase the limits of detection by immunoassay. Biotechnol Appl Biochem 2005; 41:247 - 253
- Sekiya S, Noda K, Nishikawa F, Yokoyama T, Kumar PK, Nishikawa S. Characterization and application of a novel RNA aptamer against the mouse prion protein. J Biochem (Tokyo) 2006; 139:383 - 390
- Takemura K, Wang P, Vorberg I, Surewicz W, Priola SA, Kanthasamy A, Pottathil R, Chen SG, Sreevatsan S. DNA aptamers that bind to PrP(C) and not PrP(Sc) show sequence and structure specificity. Exp Biol Med (Maywood) 2006; 231:204 - 214
- King DJ, Safar JG, Legname G, Prusiner SB. Thioaptamer interactions with prion proteins: sequence-specific and non-specific binding sites. J Mol Biol 2007; 369:1001 - 1014
- Mercey R, Lantier I, Maurel MC, Grosclaude J, Lantier F, Marc D. Fast, reversible interaction of prion protein with RNA aptamers containing specific sequence patterns. Arch Virol 2006; 151:2197 - 2214
- Weiss S, Proske D, Neumann M, Groschup MH, Kretzschmar HA, Famulok M, Winnacker EL. RNA aptamers specifically interact with the prion protein PrP. J Virol 1997; 71:8790 - 8797
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990; 249:505 - 510
- Gabus C, Auxilien S, Pechoux C, Dormont D, Swietnicki W, Morillas M, Surewicz W, Nandi P, Darlix JL. The prion protein has DNA strand transfer properties similar to retroviral nucleocapsid protein. J Mol Biol 2001; 307:1011 - 1021
- Gabus C, Derrington E, Leblanc P, Chnaiderman J, Dormont D, Swietnicki W, Morillas M, Surewicz WK, Marc D, Nandi P, Darlix JL. The prion protein has RNA binding and chaperoning properties characteristic of nucleocapsid protein NCP7 of HIV-1. J Biol Chem 2001; 276:19301 - 19309
- Lima LM, Cordeiro Y, Tinoco LW, Marques AF, Oliveira CL, Sampath S, Kodali R, Choi G, Foguel D, Torriani I, Caughey B, Silva JL. Structural insights into the interaction between prion protein and nucleic acid. Biochemistry 2006; 45:9180 - 9187
- Hornemann S, Korth C, Oesch B, Riek R, Wider G, Wuthrich K, Glockshuber R. Recombinant full-length murine prion protein, mPrP(23–231): purification and spectroscopic characterization. FEBS Lett 1997; 413:277 - 281
- Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J Mol Biol 2005; 346:645 - 659
- Igarashi S, Ohtera T, Yoshida H, Witarto AB, Sode K. Construction and characterization of mutant water-soluble PQQ glucose dehydrogenases with altered K(m) values—site-directed mutagenesis studies on the putative active site. Biochem Biophys Res Commun 1999; 264:820 - 824
- Green A, Sarkar B. Alteration of zif268 zinc-finger motifs gives rise to non-native zinc-co-ordination sites but preserves wild-type DNA recognition. Biochem J 1998; 333:85 - 90
- Noma T, Ikebukuro K, Sode K, Ohkubo T, Sakasegawa Y, Hachiya N, Kaneko K. A screening method for DNA aptamers that bind to a specific, unidentified protein in tissue samples. Biotechnol Lett 2006; 28:1377 - 1381
- Cao XM, Koski RA, Gashler A, McKiernan M, Morris CF, Gaffney R, Hay RV, Sukhatme VP. Identification and characterization of the Egr-1 gene product, a DNA-binding zinc finger protein induced by differentiation and growth signals. Mol Cell Biol 1990; 10:1931 - 1939
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31:3406 - 3415
- Green LS, Jellinek D, Jenison R, Ostman A, Heldin CH, Janjic N. Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry 1996; 35:14413 - 14424
- Hicke BJ, Watson SR, Koenig A, Lynott CK, Bargatze RF, Chang YF, Ringquist S, Moon-McDermott L, Jennings S, Fitzwater T, Han HL, Varki N, Albinana I, Willis MC, Varki A, Parma D. DNA aptamers block L-selectin function in vivo. Inhibition of human lymphocyte trafficking in SCID mice. J Clin Invest 1996; 98:2688 - 2692
- Mendonsa SD, Bowser MT. In vitro selection of high-affinity DNA ligands for human IgE using capillary electrophoresis. Anal Chem 2004; 76:5387 - 5392
- Misono TS, Kumar PK. Selection of RNA aptamers against human influenza virus hemagglutinin using surface plasmon resonance. Anal Biochem 2005; 342:312 - 317
- Plummer KA, Carothers JM, Yoshimura M, Szostak JW, Verdine GL. In vitro selection of RNA aptamers against a composite small molecule-protein surface. Nucleic Acids Res 2005; 33:5602 - 5610