Abstract
The yeast, fungal and mammalian prions determine heritable and infectious traits that are encoded in alternative conformations of proteins. They cause lethal sporadic, familial and infectious neurodegenerative conditions in man, including Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), kuru, sporadic fatal insomnia (SFI) and likely variable protease-sensitive prionopathy (VPSPr). The most prevalent of human prion diseases is sporadic (s)CJD. Recent advances in amplification and detection of prions led to considerable optimism that early and possibly preclinical diagnosis and therapy might become a reality. Although several drugs have already been tested in small numbers of sCJD patients, there is no clear evidence of any agent’s efficacy. Therefore, it remains crucial to determine the full spectrum of sCJD prion strains and the conformational features in the pathogenic human prion protein governing replication of sCJD prions. Research in this direction is essential for the rational development of diagnostic as well as therapeutic strategies. Moreover, there is growing recognition that fundamental processes involved in human prion propagation – intercellular induction of protein misfolding and seeded aggregation of misfolded host proteins – are of far wider significance. This insight leads to new avenues of research in the ever-widening spectrum of age-related human neurodegenerative diseases that are caused by protein misfolding and that pose a major challenge for healthcare.
Introduction
Prion diseases,Citation1 originally called transmissible spongiform encephalopathies,Citation2 are invariably fatal neurodegenerative diseases that affect humans and animals. The key characteristics of human spongiform encephalopathies are (1) heterogeneity of the clinical and pathologic phenotype,Citation3-Citation5 (2) a single pathologic process, which may present as a sporadic, genetic, or infectious illness,Citation6 and (3) the age dependence of genetic as well as sporadic forms: the annual peak incidence is 3–6 cases per million people between 65 and 79 years of age.Citation6,Citation7 Despite their rarity, human prion diseases have gained considerable importance because their unique etiology and pathogenesis challenged basic principles of biology. Furthermore, prion diseases can be transmitted between humans as well as from animals to humans by an agent that is highly resistant to inactivation and which thus poses novel problems to disease control and public health. Finally, because of the marked heterogeneity of their clinical phenotype, prion diseases are difficult to differentiate from other age-related brain neurodegenerations, a feature that has prompted the establishment of specialized prion disease surveillance centers worldwide. Human prion diseases also include inherited forms as well as forms acquired by infection (). However, this review focuses on the molecular aspects of the pathogenesis of sporadic forms.
Table 1. Etiologic classification of human prion diseases
From slow virus to the prion concept
Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common form of human spongiform encephalopathy, accounting for ~85% of all human prion disease.Citation8 The term “Creutzfeldt-Jakob disease” (CJD) was introduced by Alfons Maria Jakob in 1921, who referred to a previous case described by Hans Gerhard Creutzfeldt in 1920.Citation9,Citation10 These original cases were clinically heterogeneous, and neuropathologic review of the historical material with modern tools provided confirmation of the diagnosis of CJD in only two of the original five cases.Citation11 Over the next two decades, the clinicopathological classification of CJD remained uncertain and for example Wilson concluded that CJD is a “dumping ground for several rare cases of presenile dementia”Citation12. Although monograph by KirschbaumCitation13 listed the clinical and pathologic characteristics of 150 cases diagnosed before 1965, it included also cases such as Creutzfeldt’s original case, which would not fulfill criteria for the diagnosis of CJD today. Importantly, in the 1960s, Nevin and Jones described the typical clinical symptoms, electroencephalogram (EEG), and neuropathologic changes, including spongiform change, which together are now recognized as the paradigm features of sporadic CJD.Citation14 Subsequently, new sporadic forms of the spongiform encephalopathies with unique phenotypic features were described; specifically, sporadic fatal insomnia (SFI) in 1997,Citation15,Citation16 and the latest likely new candidate, variable protease-sensitive prionopathy (VPSPr) in 2008.Citation17
Controversy concerning the cause of human spongiform encephalopathies has polarized the scientific community for decades. The 1950s saw considerable interest in an epidemic of a neurodegenerative disease, kuru, characterized principally by a progressive cerebellar ataxia, among the Fore people of the Eastern Highlands of Papua New Guinea.Citation18,Citation19 Fieldwork by Carleton Gajdusek and Vincent Zigas suggested that kuru was transmitted during cannibalistic feasts.Citation20 Importantly, in 1959 Hadlow pointed out the similarities between kuru and scrapie of sheep at the neuropathologic and clinical levels and suggested that human diseases might also be transmissible.Citation21 Subsequent transmission of kuru (in 1966) and then CJD (in 1968) by intracerebral inoculation of brain homogenates into chimpanzees, work which was conducted by Gajdusek, Gibbs, and colleagues, was a landmark discovery which led to the concept of the “transmissible spongiform encephalopathies”Citation22,Citation23. The transmission of Gerstmann-Sträussler-Scheinker disease (GSS) followed in 1981Citation24 and fatal familial insomnia (FFI) in 1995.Citation25 Interestingly, Jakob, suspecting in his original observations that the condition might be transmissible, inoculated rabbits experimentally, in an attempt to demonstrate this in the 1920s.Citation26 However, thanks to the important role of serendipity in science, his experiment was unsuccessful and we know now that rabbits are uniquely resistant to prion infections.Citation27
The transmission of CJD and of kuru allowed refinement of the diagnostic pathologic criteria for human spongiform encephalopathy and led to the conclusion that all the human conditions share common histopathologic features: spongiform vacuolation (affecting any part of the cerebral gray matter), neuronal loss, and astrocytic proliferation that may be accompanied by amyloid plaques.Citation28-Citation31 On analogy with scrapie in sheep, it was assumed that the causative agent must be some type of atypical “slow virus,” the term Sigurdsson coined in 1954 for scrapie infection.Citation2,Citation32 Regrettably, despite extensive efforts in Europe and the US, no non-host DNA or RNA could be found, and a growing body of data pointed to a causative agent having unique characteristics.Citation33,Citation34 Most researchers today accept the model according to which the infectious pathogen responsible for human prion diseases is an abnormal protein, designated PrPSc 1.
The discovery that proteins could be infectious represented a new paradigm of molecular biology and medicine. Although originally deemed heretical, this protein-only model is now supported by a wealth of biochemical, genetic and animal studies.Citation6,Citation35-Citation38 Moreover, the concept of prion diseases has important implications for other neurodegenerative disorders. Recent studies with Amyloid β, tau, α-synuclein, huntingtin, and superoxide dismutase 1 suggest that molecular and cellular mechanims that were first discovered in studies of prions are involved in the pathogenesis of other neurodegenerative disorders associated with the accumulation of misfolded proteins, including Parkinsonism, Huntington disease, amyotrophic lateral sclerosis (ALS), and Alzheimer disease.Citation39-Citation43
Pathogenetic mechanisms of human sporadic prion diseases
The basic event shared by all three forms of prion diseases – sporadic, inherited and acquired by infection – is a change in conformation of the normal or cellular PrP (PrPC) which is converted into a pathogenic PrP isoform commonly identified as PrPSc for prototypic scrapie (Sc) PrP.Citation1 Human PrPC is encoded by the PrP gene (PRNP) on chromosome 20 and expressed at different levels in most mammalian cells. It comprises 209 amino acids (23–231), two sites of N-linked glycosylation, a disulfide bond and a glycolipid anchor.Citation44-Citation49 The variable degree of glycosylation is responsible for the presence of di-, mono- and un-glycosylated forms of PrPC. Because of the glycolipid anchor, most of the PrPC is extracellularly linked in specialized cholesterol-rich domains (caveoli) of the cell plasma membrane.Citation48,Citation50 In normal conformation, human PrPC comprises a C-terminal globular domain (involving residues 127–231), which consists of three α-helices and two short β-sheets.Citation51 In contrast, the N-terminus is unstructured.Citation51
The PrPSc, in contrast, is pathogenic, infectious and displays different biophysical features, forming insoluble aggregates of different sizes with predominantly β-sheet secondary structure; the C-terminal region is relatively resistant to proteolytic degradation.Citation52-Citation54 The conformational transition that underlies these pronounced changes is believed to involve refolding of the C-terminal region whereby the α-helical structures of PrPC are replaced by β-sheet to different extents and with variable patterns.Citation55 Although insolubility and protease resistance were the original defining features of PrPSc, the finding of protease-sensitive small oligomers of pathogenic PrPSc 56, 57 significantly broadened the spectrum of pathogenic conformers.
Although some authors believe that the toxicity in prion disease can be explained as a loss of function of PrPC due to the conformational transformation,Citation58 others argue that a gain of toxic function is more likely.Citation59,Citation60 Nevertheless, there is general agreement that PrPC serves as a substrate for conversion to PrPSc and as a major receptor for toxic effects of PrPSc. Thus the neuroprotective function physiologically provided by PrPC could be lost following its conversion to PrPSc. The prevailing presence of PrPSc on neuronal plasma membrane also at the synapse might explain the widespread spongiform degeneration, which can be viewed as intracellular edema. All these changes are pathognomonic of sCJD. Apoptosis and oxidative stress are reported to occur when PrPSc aggregates form at the cell surface and may well contribute to the neuronal cell loss that is prominent in prion diseases of long duration and in prion diseases like SFI, which manifest no or minimal spongiosis as well as severe neuronal loss.Citation61 Astrogliosis, a common reaction to injury, is considered a secondary response.
The origin and phenotypic heterogeneity of sCJD
Several explanations have been proposed for the etiology of sporadic prion diseases. These include spontaneous somatic mutations in the PRNP gene or rare stochastic conformational changes in the structure of PrPC 62. These explanations presuppose that the mutant PrPSc would have to be capable of recruiting wild-type PrPC; however, this process which might occur with some mutations or conformations but is unlikely with others.Citation63 According to a second explanation, low amounts of PrPSc-like isoforms are normally present in brain, and possibly bound to other proteins such as heat shock proteins, however, this protective mechanism fails with aging.Citation64,Citation65 Finally, it has been suggested that at least some cases of apparent sCJD result from covert, low-level exposure to a “common external factor”Citation66.
With more cases investigated after the original reports by Jakob, it became clear that the clinical as well as histopathologic features of sCJD are remarkably diverse, perhaps making the human prion diseases the most heterogeneous of all neurodegenerative disease. On the basis of predominant clinical and pathologic features, the following phenotypic subtypes of sCJD were proposed in the 1980s: 1) myoclonic or cortico-striatal-spinal, 2) amaurotic or Heidenhain, 3) classical or diffuse, 4) thalamic, 5) ataxic or cerebellar, and 6) amyotrophic.Citation67-Citation69 Researchers today generally agree that the genotype at codon 129 of the chromosomal gene PRNP, and to some degree the phenotypes of these diseases, underlie susceptibility to prion diseases.Citation5 Additionally, many lines of evidence from experiments with laboratory prion strains support the view that the phenotype of sCJD—its distinctive incubation time, clinical features, and brain pathology—is enciphered in the strain-specific conformation of PrPSc Citation56,Citation70–Citation73. However, in contrast to the experiments with laboratory rodent prion strains, in which the digestion of brain PrPSc with proteolytic enzyme proteinase K (PK) consistently results in a single protease-resistant domain with mass ~19 kDa, the outcome in sCJD is more complex. Distinctive glycosylation patterns and up to four PK-resistant fragments of the pathogenic prion protein (rPrPSc) found in sCJD brains are easily distinguishable on western blot (WB).Citation5,Citation71,Citation74-Citation77
The WB findings together with human PRNP gene polymorphism led Parchi, Gambetti, and colleagues to posit a clinicopathological classification of sCJD into five or six subtypes; notably, the WB characteristics of PrPSc breed true upon transmission to susceptible transgenic mice and Guinea pigs (Cavia porcellus).Citation5,Citation71,Citation74,Citation78 An alternative classification of the PrPSc types and their pairing with CJD phenotypes has been proposed by Collinge and collaborators.Citation37,Citation75,Citation76,Citation79 This classification differs from the previous one in two major aspects: first, it recognizes three (not two) PrPSc electrophoretic mobilities and second, it also identifies PrPSc isoforms with different ratios of the three PrP glycoforms.Citation37 Although the disease phenotypes of patients with sCJD are remarkably heterogeneous, 21 kDa fragments of unglycosylated PrPSc (Type 1) frequently differ from the disease duration and phenotypes associated with the 19 kDa fragments of unglycosylated PrPSc (Type 2).Citation3,Citation5,Citation71,Citation74
Cumulatively, these findings argue that the PrPSc type represents yet an additional major modifier of the phenotype in human prion diseases; accordingly, WB-based clinicopathologic classifications became an important tool in studies of prion pathogenesis in human brains and in transgenic mice models.Citation37,Citation71 Because two distinct PK cleavage sites in PrPSc Types 1 and 2 most likely stem from distinct conformations, some investigators contend that PrPSc Types 1 and 2 code distinct prion strains.Citation3,Citation71,Citation75,Citation80 However, the heterogeneity of sCJD, along with a growing number of studies including bioassays, all suggest that the range of prions causing sCJD exceeds the number of categories recognized within the current WB-based clinicopathologic schemes.Citation81-Citation83 Additionally, recent findings of the co-occurrence of PrPSc Types 1 and 2 in 40% or more sCJD cases created a conundrum and suggested that the originally observed differences were quantitative rather than qualitative.Citation84-Citation89 Finally, up to 90% of brain PrPSc in sCJD eludes WB analysis because it is destroyed by proteinase-K treatment, which is necessary to eliminate PrPC 81. Consequently, the conformation or role of this major protease-sensitive (s) fraction of PrPSc in the pathogenesis of the disease is a subject of speculation.Citation64,Citation81,Citation90 Thus, no direct structural data are available for sCJD brain PrPSc beyond the evidence that it is resistant to proteolytic digestion. Nevertheless, to determine the full spectrum of sCJD prion strains, and the conformational features in the pathogenic human prion protein governing replication of sCJD prions is fundamental for the rational development of diagnostic as well as therapeutic strategies.
Novel conformational methods derived from a conformation-dependent immunoassay (CDI)
Three obstacles have slowed progress in the research of human prions: phenotypic variability of sCJD on complex genetic background, biosafety constraints, and lack of suitable tools for studying molecular characteristics beyond WB typing. Aiming to advance our understanding of the molecular pathogenesis of human prion diseases, we developed the conformation-dependent immunoassay (CDI)Citation56,Citation81,Citation91 to determine the conformational range and strain-dependent molecular features of sCJD PrPSc, first in patients who were homozygous for codon 129 of the PRNP gene.Citation92 Even relatively minute variations in a soluble protein structure can be determined by measuring conformational stability in a denaturant such as Gdn HCl.Citation93 Utilizing this concept, we designed a procedure in which PrPSc is first exposed to denaturant Gdn HCl and then exposed to europium-labeled mAb against the epitopes hidden in the native conformation.Citation56 As the concentration of Gdn HCl increases, PrPSc dissociates and unfolds from native β-sheet-structured aggregates, and more epitopes become available to antibody binding. These experiments involve insoluble oligomeric forms of PrPSc, and denaturation of this protein is irreversible in vitro; consequently the Gibbs free energy change (ΔG) of PrPSc cannot be calculated.Citation94 Therefore we chose instead to use the Gdn HCl value found at the half-maximal denaturation ([GdnHCl]1/2) as a measure of the relative conformational stability of PrPSc. The differences in stability reveal evidence of distinct conformations of PrPSc 56, 93, 94.
To measure the concentration of different forms of PrPSc and follow the unfolding, we used europium-labeled mAb 3F4Citation95 (epitope residues 107–112) for detection and 8H4 mAb (epitope residues 175–185)Citation96 to capture human PrPSc in a sandwich CDI format.Citation81,Citation97 The analytical sensitivity and specificity of the optimized CDI for detection of PrPSc was previously reported by us and others in numerous publicationsCitation56,Citation81,Citation91,Citation98-Citation100 and has been shown to be as low as ~500 fg (~20 attomoles) of PrPSc which is similar to the sensitivity of human prion bioassay in Tg(MHu2M)5378/Prnp0/0 mice.Citation81 Because CDI is not dependent on protease treatment, it allowed us to address fundamental questions concerning the concentration and conformation of different isoforms of sCJD PrPSc, including protease-sensitive (s) and protease-resistant (r)PrPSc 92.
The dissociation and unfolding of PrPSc in the presence of increasing concentration of Gdn HCl can be described as follows:
[PrPSc]n → [sPrPSc]n → iPrP → uPrP
where [PrPSc]n are native aggregates of PrPSc, [sPrPSc]n are soluble protease-sensitive oligomers of PrPSc, iPrP is an intermediate, and uPrP is completely unfolded (denatured) PrP.Citation53,Citation57,Citation94 The CDI monitors the global transition from native aggregates to fully denatured monomers of PrPSc. In contrast, the WB-based techniques monitor either the partial solubilization of PrPSc Citation101 or conversion of rPrPSc to protease-sensitive conformersCitation72 after exposure to denaturant. As a result, the stability data on soluble protease-sensitive oligomers and intermediates of PrPSc cannot be obtained with WB techniques and may explain some markedly different values.Citation102
Structural heterogeneity of sCJD PrPSc and the role of sPrPSc in pathogenesis
Our recent finding of 6-fold difference in concentrations of PrPSc between Type 1 and Type 2 PrPSc(129M) in the frontal cortex was surprising, even though some variability was to be expected due to differences in the predominantly affected areas in distinct sCJD phenotypes.Citation81,Citation92 Moreover, the average levels of PrPSc were up to 100-fold lower than those in standard laboratory prion models such as Syrian hamsters infected with Sc237 prionsCitation56; and together with the up to 100-fold variability within each phenotypic group, these lower levels of PrPSc may explain why some sCJD cases are difficult to transmit, and why lower endpoint titers are obtained with human prions in transgenic mice expressing human or chimeric PrPC Citation37, Citation71, Citation81, Citation103.
Up to 90% of the pathogenic prion protein in sCJD is protease-sensitiveCitation81 and we found the highest concentrations in Type 2 PrPSc(129M) ().Citation92 The broad range of absolute and relative levels of rPrPSc and sPrPSc offers evidence of a broad spectrum of PrPSc molecules differing in protease sensitivity in each group with an identical polymorphism at codon 129 of the PRNP gene and an identical WB pattern. Moreover, these findings signal the existence of a variety of sCJD PrPSc conformers; and since protease sensitivity is one of the characteristics of prion strains, they also suggest that distinct sCJD prion strains exist.Citation56,Citation57,Citation81,Citation82,Citation104,Citation105
Figure 1. The relationship between duration of the disease, (left panel) concentration, and (right panel) conformational stability of sPrPSc conformers in sCJD patients with different WB patterns and PRNP gene polymorphisms.Citation92 The stability of PrPSc was determined with CDI before and after protease treatment and the change is expressed as ΔGdn HCl1/2.Citation92 The symbols are mean ± SEM in each sCJD group.
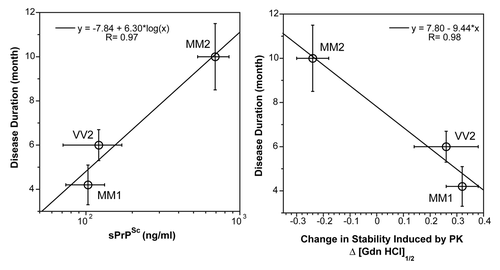
The heterogeneity of PrPSc conformations we found with CDI within sCJD patients homozygous for codon 129 plymorphism of the PRNP gene is remarkable, having a range corresponding to that of stabilities found in approximately 30 distinct strains of de-novo and natural laboratory rodent prions studied up to now.Citation56,Citation72,Citation92,Citation106 The intriguing differential effect of PK treatment with increasing stability of type 1 and decreasing stability of Type 2 PrPSc(129M) suggests that in contrast to Type 1, the protease-resistant core of Type 2 was profoundly destabilized. Together with the increased frequency of exposed epitopes in codon 129 MM samples with Type 2 rPrPSc after PK treatment, these observations may indicate one of three possibilities: that the ligand protecting the 3F4 epitope was removed by PK treatment; that epitope 108–112 was protected by the N-terminus of PrPSc; or that conformational transition resulted in more exposed 108–112 epitope. Whether the epitope hindrance in undigested PrPSc is the result of lipid, glycosaminoglycan, nucleic acid, or protein binding to the conformers unique to the MM2 sCJDF PrPSc remains to be established. Since sCJD cases with Type 2 PrPSc(129M) have remarkably extended disease durations, the molecular mechanism underlying these effects calls for detailed investigation. Cumulatively, our findings indicate that sCJD PrPSc exhibits extensive conformational heterogeneity and suggest that a wide spectrum of sCJD prions cause the disease. Whether this heterogeneity originates in a stochastic misfolding process that generates many distinct self-replicating conformationsCitation37,Citation62 or in a complex process of evolutionary selection during development of the diseaseCitation73 remains to be established.
We discovered protease-sensitive conformers of PrPSc while developing a conformation-dependent immunoassay (CDI), which does not require proteolytic degradation of ubiquitous PrPC 56. Although the original definition of sPrPSc was purely operational, considerable additional data demonstrate that (1) sPrPSc replicates in vivo and in vitro as an invariant and major fraction of PrPSc; (2) sPrPSc separates from rPrPSc in high speed centrifugation, and (3) the proteolytic sensitivity of PrPSc can reliably differentiate various prion strains.Citation56,Citation57,Citation81,Citation82,Citation104,Citation105 Accumulation of sPrPSc precedes protease-resistant product (rPrPSc) in prion infectionCitation64,Citation107; and up to 90% of PrPSc accumulating in CJD brains consists of sPrPSc Citation81. Thus, the detection by CDI of sPrPSc as a disease-specific marker is widely regarded as a more reliable basis for diagnosing prion diseases. This improved detection led to the discovery of a new human prion disorder, variably protease-sensitive prionopathy (VPSPr).Citation17,Citation56,Citation81,Citation90,Citation108 It is noteworthy that synthetic prions generated in vitro during polymerization of recombinant mouse PrP into amyloid fibers produced prions composed exclusively of sPrPSc upon inoculation into wild mice.Citation106
Our recent data indicate that the levels as well as stability of sPrPSc are a good predictor of the progression rate in sCJD ().Citation92 Despite the inevitable influence of variable genetic background and the potential difficulties in evaluating initial symptoms, the disease progression rate and incubation time jointly represent an important parameter, which is influenced by replication rate, propagation, and clearance of prions from the brain.Citation64,Citation109 The correlations among the levels of sPrPSc, the stability of sPrPSc, and the duration of the disease found in this study all indicate that sPrPSc conformers play an important role in the pathogenesis. When sPrPSc is less stable than rPrPSc, the difference in stability correlates with less accumulated sPrPSc and shorter duration of the disease. Conversely, when sPrP conformers are more stable than rPrPSc, we observed the opposite effect—more accumulated sPrPSc and extended disease duration ().
In laboratory rodent prion models, we found that levels of sPrPSc varied with the incubation time of the diseaseCitation56 and we hypothesized that the molecular mechanism of this link may be related to the replication or clearance rate of prions.Citation56,Citation64,Citation81 Our recent data on sCJD prions extend this observation and indicate that higher levels of less stable sPrPSc lead to faster progression of the disease.Citation92 These observations are in accord with the experiments on yeast prions and suggest that the stability of misfolded protein is inversely related to the replication rate.Citation110 Although the modulating effect of prion clearance in the mammalian brains is likely,Citation64 the data from both yeast and human prions lead to the hypothesis that the less stable prions replicate faster by exposing more available sites for growth of the aggregates. In mammalian prions, this effect leads to shorter incubation time and faster progression of the disease.
Future directions
Additional steps are necessary to improve our understanding of the phenotypic diversity of sCJD. One step is to determine whether the mixed WB patterns of Type 1–2 rPrPSc in the same or different anatomical areas represent a unique conformation or a mixture of conformers, and to map the distribution in the individual brain.Citation89 Additionally, novel conformational approaches using tandem protein misfolding cyclic amplification (PMCA) and CDI should allow analysis of the impact of different PrPSc polymorphisms and conformations on the replication rate of human prions. Such studies have clearly broader implications, as recent data suggest that the process of intercellular induction of protein misfolding is relevant in the pathogenesis of growing number of other neurodegenerative diseases.Citation39-Citation43
| Abbreviations: | ||
| ALS | = | amyotrophic lateral sclerosis |
| CDI | = | conformation-dependent immunoassay |
| CJD | = | Creutzfeldt-Jakob disease |
| GSS | = | Gerstmann-Sträussler-Scheinker syndrome |
| PrP | = | prion protein |
| PrPC | = | normal or cellular prion protein |
| PrPSc | = | pathogenic prion protein |
| PRNP | = | prion protein gene |
| rPrPSc | = | protease-resistant conformers of pathogenic prion protein |
| sPrPSc | = | protease-sensitive conformers of pathogenic prion protein |
| sCJD | = | sporadic Creutzfeldt-Jakob disease |
| SFI | = | sporadic fatal insomnia |
| VPSPr | = | variable protease-sensitive prionopathy |
Acknowledgments
The authors thank Pierlugi Gambetti and Witold Surewicz for encouragement and stimulating discussions. This work was supported by grants from NIA (AG-14359), NINDS (NS074317), CDC (UR8/CCU515004), and the Charles S. Britton Fund.
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Gajdusek DC. Slow-virus infections of the nervous system. N Engl J Med 1967; 276:392 - 400; http://dx.doi.org/10.1056/NEJM196702162760708; PMID: 6066787
- Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci U S A 1994; 91:2839 - 42; http://dx.doi.org/10.1073/pnas.91.7.2839; PMID: 7908444
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46:224 - 33; http://dx.doi.org/10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W; PMID: 10443888
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull 2003; 66:213 - 39; http://dx.doi.org/10.1093/bmb/66.1.213; PMID: 14522861
- Prusiner SB, ed. Prion Biology and Diseases. Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 2004.
- Holman RC, Belay ED, Christensen KY, Maddox RA, Minino AM, Folkema AM, et al. Human prion diseases in the United States. PLoS One 2010; 5:e8521; http://dx.doi.org/10.1371/journal.pone.0008521; PMID: 20049325
- Masters CL, Gajdusek DC, Gibbs CJ Jr., Bernouilli C, Asher DM. Familial Creutzfeldt-Jakob disease and other familial dementias: an inquiry into possible models of virus-induced familial diseases. In: Prusiner SB, Hadlow WJ, eds. Slow Transmissible Diseases of the Nervous System, Vol 1. New York: Academic Press, 1979:143-94.
- Jakob A. Über eine der multiplen Sklerose klinisch nahestehende Erkrankung des Zentralnervensystems (spastische Pseudosklerose) mit bemerkenswertem anatomischem Befunde. Mitteilung eines vierten Falles. Med Klin (Munich) 1921; 17:372 - 6
- Creutzfeldt HG. Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems. Z Gesamte Neurol Psychiatr 1920; 57:1 - 18; http://dx.doi.org/10.1007/BF02866081
- Masters CL, Gajdusek DC. The spectrum of Creutzfeldt-Jakob disease and virus-induced subacute spongiform encephalopathies. Recent Adv Neuropathol 1982; 2:139 - 63
- Wilson SAK. Syndrome of Jakob: Cortico-striato-spinal degeneration. London: Edward Arnold, 1940.
- Kirschbaum WR. Jakob-Creutzfeldt Disease. Amsterdam: Elsevier, 1968.
- Nevin S, McMENEMEY WH, Behrman S, Jones DP. Subacute spongiform encephalopathy--a subacute form of encephalopathy attributable to vascular dysfunction (spongiform cerebral atrophy). Brain 1960; 83:519 - 64; http://dx.doi.org/10.1093/brain/83.4.519; PMID: 13728615
- Gambetti P, Parchi P. Insomnia in prion diseases: sporadic and familial. N Engl J Med 1999; 340:1675 - 7; http://dx.doi.org/10.1056/NEJM199905273402111; PMID: 10341282
- Mastrianni J, Nixon F, Layzer R, DeArmond SJ, Prusiner SB. Fatal sporadic insomnia: fatal familial insomnia phenotype without a mutation of the prion protein gene. Neurology 1997; 48:Suppl. A296
- Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol 2008; 63:697 - 708; http://dx.doi.org/10.1002/ana.21420; PMID: 18571782
- Gajdusek DC, Zigas V. Kuru; clinical, pathological and epidemiological study of an acute progressive degenerative disease of the central nervous system among natives of the Eastern Highlands of New Guinea. Am J Med 1959; 26:442 - 69; http://dx.doi.org/10.1016/0002-9343(59)90251-7; PMID: 13626997
- Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native population. N Engl J Med 1957; 257:974 - 8; http://dx.doi.org/10.1056/NEJM195711142572005; PMID: 13483871
- Gajdusek DC. Kuru. Trans R Soc Trop Med Hyg 1963; 57:151 - 69; http://dx.doi.org/10.1016/0035-9203(63)90057-9; PMID: 13946176
- Hadlow WJ. Scrapie and kuru. Lancet 1959; 274:289 - 90; http://dx.doi.org/10.1016/S0140-6736(59)92081-1
- Gibbs CJ Jr., Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM, et al. Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 1968; 161:388 - 9; http://dx.doi.org/10.1126/science.161.3839.388; PMID: 5661299
- Gajdusek DC, Gibbs CJ Jr., Alpers M. Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature 1966; 209:794 - 6; http://dx.doi.org/10.1038/209794a0; PMID: 5922150
- Masters CL, Gajdusek DC, Gibbs CJ Jr.. Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Sträussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain 1981; 104:559 - 88; http://dx.doi.org/10.1093/brain/104.3.559; PMID: 6791762
- Tateishi J, Brown P, Kitamoto T, Hoque ZM, Roos R, Wollman R, et al. First experimental transmission of fatal familial insomnia. Nature 1995; 376:434 - 5; http://dx.doi.org/10.1038/376434a0; PMID: 7630420
- Jakob A. Über eigenartige Erkrankungen des Zentralnervensystems mit bemerkenswertem anatomischen Befunde (spastische Pseudosklerose-Encephalomyelopathie mit disseminierten Degenerationsherden). Z Gesamte Neurol Psychiatr 1921; 64:147 - 228; http://dx.doi.org/10.1007/BF02870932
- Loftus B, Rogers M. Characterization of a prion protein (PrP) gene from rabbit; a species with apparent resistance to infection by prions. Gene 1997; 184:215 - 9; http://dx.doi.org/10.1016/S0378-1119(96)00598-7; PMID: 9031631
- Klatzo I, Gajdusek DC, Zigas V. Pathology of Kuru. Lab Invest 1959; 8:799 - 847; PMID: 13665963
- Gibbs CJ Jr., Gajdusek DC. Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science 1973; 182:67 - 8; http://dx.doi.org/10.1126/science.182.4107.67; PMID: 4199733
- Lampert PW, Gajdusek DC, Gibbs CJ Jr.. Experimental spongiform encephalopathy (Creutzfeldt-Jakob disease) in chimpanzees. Electron microscopic studies. J Neuropathol Exp Neurol 1971; 30:20 - 32; http://dx.doi.org/10.1097/00005072-197101000-00004; PMID: 4925307
- Beck E, Daniel PM, Matthews WB, Stevens DL, Alpers MP, Asher DM, et al. Creutzfeldt-Jakob disease. The neuropathology of a transmission experiment. Brain 1969; 92:699 - 716; http://dx.doi.org/10.1093/brain/92.4.699; PMID: 4903507
- Sigurdsson B. Rida, a chronic encephalitis of sheep with general remarks on infections which develop slowly and some of their special characteristics. Br Vet J 1954; 110:341 - 54
- Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, et al. Search for a prion-specific nucleic acid. J Virol 2005; 79:10796 - 806; http://dx.doi.org/10.1128/JVI.79.16.10796-10806.2005; PMID: 16051871
- Riesner D. The search for a nucleic acid component to scrapie infectivity. Semin Virol 1991; 2:215 - 26
- Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 2009; 78:177 - 204; http://dx.doi.org/10.1146/annurev.biochem.78.082907.145410; PMID: 19231987
- Cobb NJ, Surewicz WK. Prion diseases and their biochemical mechanisms. Biochemistry 2009; 48:2574 - 85; http://dx.doi.org/10.1021/bi900108v; PMID: 19239250
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318:930 - 6; http://dx.doi.org/10.1126/science.1138718; PMID: 17991853
- Morales R, Abid K, Soto C. The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta 2007; 1772:681-91.
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313:1781 - 4; http://dx.doi.org/10.1126/science.1131864; PMID: 16990547
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 2009; 284:12845 - 52; http://dx.doi.org/10.1074/jbc.M808759200; PMID: 19282288
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009; 106:13010 - 5; http://dx.doi.org/10.1073/pnas.0903691106; PMID: 19651612
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 2009; 106:20051 - 6; PMID: 19892735
- Zhao W, Beers DR, Henkel JS, Zhang W, Urushitani M, Julien JP, et al. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010; 58:231 - 43; http://dx.doi.org/10.1002/glia.20919; PMID: 19672969
- Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SBH, Aebersold R, et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985; 40:735 - 46; http://dx.doi.org/10.1016/0092-8674(85)90333-2; PMID: 2859120
- Liao Y-C, Lebo RV, Clawson GA, Smuckler EA. Human prion protein cDNA: molecular cloning, chromosomal mapping, and biological implications. Science 1986; 233:364 - 7; http://dx.doi.org/10.1126/science.3014653; PMID: 3014653
- Baldwin MA, Stahl N, Burlingame AL, Prusiner SB. Structure determination of glycoinsotiol phospholipid anchors by permethylation and tandem mass spectrometry. Methods: A Companion to Methods in Enzymology 1990; 1:306-14.
- Baldwin MA, Stahl N, Reinders LG, Gibson BW, Prusiner SB, Burlingame AL. Permethylation and tandem mass spectrometry of oligosaccharides having free hexosamine: analysis of the glycoinositol phospholipid anchor glycan from the scrapie prion protein. Anal Biochem 1990; 191:174 - 82; http://dx.doi.org/10.1016/0003-2697(90)90405-X; PMID: 1981823
- Safar J, Ceroni M, Gajdusek DC, Gibbs CJ Jr.. Differences in the membrane interaction of scrapie amyloid precursor proteins in normal and scrapie- or Creutzfeldt-Jakob disease-infected brains. J Infect Dis 1991; 163:488 - 94; http://dx.doi.org/10.1093/infdis/163.3.488; PMID: 1671680
- Haraguchi T, Fisher S, Olofsson S, Endo T, Groth D, Tarentino A, et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch Biochem Biophys 1989; 274:1 - 13; http://dx.doi.org/10.1016/0003-9861(89)90409-8; PMID: 2505674
- Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990; 29:8879 - 84; http://dx.doi.org/10.1021/bi00490a001; PMID: 1980209
- Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A 2000; 97:145 - 50; http://dx.doi.org/10.1073/pnas.97.1.145; PMID: 10618385
- Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; http://dx.doi.org/10.1073/pnas.90.23.10962; PMID: 7902575
- Safar J, Roller PP, Gajdusek DC, Gibbs CJ Jr.. Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J Biol Chem 1993; 268:20276 - 84; PMID: 8104185
- Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991; 30:7672 - 80; http://dx.doi.org/10.1021/bi00245a003; PMID: 1678278
- Smirnovas V, Kim JI, Lu X, Atarashi R, Caughey B, Surewicz WK. Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J Biol Chem 2009; 284:24233 - 41; http://dx.doi.org/10.1074/jbc.M109.036558; PMID: 19596861
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 1998; 4:1157 - 65; http://dx.doi.org/10.1038/2654; PMID: 9771749
- Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G, et al. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 2002; 41:12868 - 75; http://dx.doi.org/10.1021/bi025958g; PMID: 12379130
- Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Pürro M, et al. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J 2005; 24:2472 - 80; http://dx.doi.org/10.1038/sj.emboj.7600717; PMID: 15962001
- Aguzzi A, Baumann F, Bremer J. The prion’s elusive reason for being. Annu Rev Neurosci 2008; 31:439 - 77; http://dx.doi.org/10.1146/annurev.neuro.31.060407.125620; PMID: 18558863
- Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 2007; 1772:629 - 44; PMID: 17451912
- Milhavet O, Lehmann S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Brain Res Rev 2002; 38:328 - 39; http://dx.doi.org/10.1016/S0165-0173(01)00150-3; PMID: 11890980
- Prusiner SB. Shattuck lecture--neurodegenerative diseases and prions. N Engl J Med 2001; 344:1516 - 26; http://dx.doi.org/10.1056/NEJM200105173442006; PMID: 11357156
- Tremblay P, Ball HL, Kaneko K, Groth D, Hegde RS, Cohen FE, et al. Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J Virol 2004; 78:2088 - 99; http://dx.doi.org/10.1128/JVI.78.4.2088-2099.2004; PMID: 14747574
- Safar JG, DeArmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E, et al. Prion clearance in bigenic mice. J Gen Virol 2005; 86:2913 - 23; http://dx.doi.org/10.1099/vir.0.80947-0; PMID: 16186247
- Yuan J, Xiao X, McGeehan J, Dong Z, Cali I, Fujioka H, et al. Insoluble aggregates and protease-resistant conformers of prion protein in uninfected human brains. J Biol Chem 2006; 281:34848 - 58; http://dx.doi.org/10.1074/jbc.M602238200; PMID: 16987816
- Linsell L, Cousens SN, Smith PG, Knight RS, Zeidler M, Stewart G, et al. A case-control study of sporadic Creutzfeldt-Jakob disease in the United Kingdom: analysis of clustering. Neurology 2004; 63:2077 - 83; PMID: 15596753
- Cathala F, Baron H. Clinical aspects of Creutzfeldt-Jakob disease. In: Prusiner SB, McKinley MP, eds. Prions - Novel Infectious Pathogens Causing Scrapie and Creutzfeldt-Jakob Disease. Orlando: Academic Press, 1987:467-509.
- Brown P, Cathala F, Castaigne P, Gajdusek DC. Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann Neurol 1986; 20:597 - 602; http://dx.doi.org/10.1002/ana.410200507; PMID: 3539001
- Brown P, Cathala F, Sadowsky D, Gajdusek DC. Creutzfeldt-Jakob disease in France: II. Clinical characteristics of 124 consecutive verified cases during the decade 1968--1977. Ann Neurol 1979; 6:430 - 7; http://dx.doi.org/10.1002/ana.410060510; PMID: 391141
- Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 1994; 68:7859 - 68; PMID: 7966576
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274:2079 - 82; http://dx.doi.org/10.1126/science.274.5295.2079; PMID: 8953038
- Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, et al. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci 2001; 10:854 - 63; http://dx.doi.org/10.1110/ps.39201; PMID: 11274476
- Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science 2010; 327:869 - 72; http://dx.doi.org/10.1126/science.1183218; PMID: 20044542
- Parchi P, Capellari S, Chen SG, Petersen RB, Gambetti P, Kopp N, et al. Typing prion isoforms. Nature 1997; 386:232 - 4; http://dx.doi.org/10.1038/386232a0; PMID: 9069279
- Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996; 383:685 - 90; http://dx.doi.org/10.1038/383685a0; PMID: 8878476
- Wadsworth JDF, Hill AF, Joiner S, Jackson GS, Clarke AR, Collinge J. Strain-specific prion-protein conformation determined by metal ions. Nat Cell Biol 1999; 1:55 - 9; http://dx.doi.org/10.1038/9030; PMID: 10559865
- Zou WQ, Capellari S, Parchi P, Sy MS, Gambetti P, Chen SG. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J Biol Chem 2003; 278:40429 - 36; http://dx.doi.org/10.1074/jbc.M308550200; PMID: 12917418
- Safar JG, Giles K, Lessard P, Letessier F, Patel S, Serban A, et al. Conserved properties of human and bovine prion strains on transmission to guinea pigs. Lab Invest 2011; 91:1326 - 36; http://dx.doi.org/10.1038/labinvest.2011.89; PMID: 21727894
- Hill AF, Desbruslais M, Joiner S, Sidle KCL, Gowland I, Collinge J, et al. The same prion strain causes vCJD and BSE. Nature 1997; 389:448 - 50, 526; http://dx.doi.org/10.1038/38925; PMID: 9333232
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996; 39:767 - 78; http://dx.doi.org/10.1002/ana.410390613; PMID: 8651649
- Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, et al. Diagnosis of human prion disease. Proc Natl Acad Sci U S A 2005; 102:3501 - 6; http://dx.doi.org/10.1073/pnas.0409651102; PMID: 15741275
- Uro-Coste E, Cassard H, Simon S, Lugan S, Bilheude JM, Perret-Liaudet A, et al. Beyond PrP9res) type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog 2008; 4:e1000029; http://dx.doi.org/10.1371/journal.ppat.1000029; PMID: 18389084
- Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A. Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol 2005; 4:805 - 14; http://dx.doi.org/10.1016/S1474-4422(05)70225-8; PMID: 16297838
- Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology 1999; 53:2173 - 6; PMID: 10599800
- Kovács GG, Head MW, Hegyi I, Bunn TJ, Flicker H, Hainfellner JA, et al. Immunohistochemistry for the prion protein: comparison of different monoclonal antibodies in human prion disease subtypes. Brain Pathol 2002; 12:1 - 11; http://dx.doi.org/10.1111/j.1750-3639.2002.tb00417.x; PMID: 11770893
- Head MW, Bunn TJ, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS, et al. Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991-2002. Ann Neurol 2004; 55:851 - 9; http://dx.doi.org/10.1002/ana.20127; PMID: 15174020
- Lewis V, Hill AF, Klug GM, Boyd A, Masters CL, Collins SJ. Australian sporadic CJD analysis supports endogenous determinants of molecular-clinical profiles. Neurology 2005; 65:113 - 8; http://dx.doi.org/10.1212/01.wnl.0000167188.65787.a0; PMID: 16009895
- Schoch G, Seeger H, Bogousslavsky J, Tolnay M, Janzer RC, Aguzzi A, et al. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med 2006; 3:e14; http://dx.doi.org/10.1371/journal.pmed.0030014; PMID: 16354106
- Cali I, Castellani R, Alshekhlee A, Cohen Y, Blevins J, Yuan J, et al. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain 2009; 132:2643 - 58; http://dx.doi.org/10.1093/brain/awp196; PMID: 19734292
- Cronier S, Gros N, Tattum MH, Jackson GS, Clarke AR, Collinge J, et al. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J 2008; 416:297 - 305; http://dx.doi.org/10.1042/BJ20081235; PMID: 18684106
- Safar JG, Scott M, Monaghan J, Deering C, Didorenko S, Vergara J, et al. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat Biotechnol 2002; 20:1147 - 50; http://dx.doi.org/10.1038/nbt748; PMID: 12389035
- Kim C, Haldiman T, Cohen Y, Chen W, Blevins J, Sy MS, et al. Protease-sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt-Jakob disease are indicator of progression rate. PLoS Pathog 2011; 7:e1002242; http://dx.doi.org/10.1371/journal.ppat.1002242; PMID: 21931554
- Shirley BA, ed. Protein Stability and Folding: Theory and Practice. Totowa, New Jersey: Humana Press, 1995.
- Safar J, Roller PP, Gajdusek DC, Gibbs CJ Jr.. Scrapie amyloid (prion) protein has the conformational characteristics of an aggregated molten globule folding intermediate. Biochemistry 1994; 33:8375 - 83; http://dx.doi.org/10.1021/bi00193a027; PMID: 8031772
- Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, et al. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 1987; 61:3688 - 93; PMID: 2446004
- Zanusso G, Liu D, Ferrari S, Hegyi I, Yin X, Aguzzi A, et al. Prion protein expression in different species: analysis with a panel of new mAbs. Proc Natl Acad Sci U S A 1998; 95:8812 - 6; http://dx.doi.org/10.1073/pnas.95.15.8812; PMID: 9671761
- Choi EM, Geschwind MD, Deering C, Pomeroy K, Kuo A, Miller BL, et al. Prion proteins in subpopulations of white blood cells from patients with sporadic Creutzfeldt-Jakob disease. Lab Invest 2009; 89:624 - 35; http://dx.doi.org/10.1038/labinvest.2009.30; PMID: 19434060
- Bellon A, Seyfert-Brandt W, Lang W, Baron H, Gröner A, Vey M. Improved conformation-dependent immunoassay: suitability for human prion detection with enhanced sensitivity. J Gen Virol 2003; 84:1921 - 5; http://dx.doi.org/10.1099/vir.0.18996-0; PMID: 12810888
- Thackray AM, Hopkins L, Bujdoso R. Proteinase K-sensitive disease-associated ovine prion protein revealed by conformation-dependent immunoassay. Biochem J 2007; 401:475 - 83; http://dx.doi.org/10.1042/BJ20061264; PMID: 17018021
- Jones M, Peden AH, Yull H, Wight D, Bishop MT, Prowse CV, et al. Human platelets as a substrate source for the in vitro amplification of the abnormal prion protein (PrP) associated with variant Creutzfeldt-Jakob disease. Transfusion 2009; 49:376 - 84; http://dx.doi.org/10.1111/j.1537-2995.2008.01954.x; PMID: 18980616
- Pirisinu L, Di Bari M, Marcon S, Vaccari G, D’Agostino C, Fazzi P, et al. A new method for the characterization of strain-specific conformational stability of protease-sensitive and protease-resistant PrP. PLoS One 2010; 5:e12723; http://dx.doi.org/10.1371/journal.pone.0012723; PMID: 20856860
- Choi YP, Peden AH, Gröner A, Ironside JW, Head MW. Distinct stability states of disease-associated human prion protein identified by conformation-dependent immunoassay. J Virol 2010; 84:12030 - 8; http://dx.doi.org/10.1128/JVI.01057-10; PMID: 20844046
- Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 2010; 107:12005 - 10; http://dx.doi.org/10.1073/pnas.1004688107; PMID: 20547859
- Pastrana MA, Sajnani G, Onisko B, Castilla J, Morales R, Soto C, et al. Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry 2006; 45:15710 - 7; http://dx.doi.org/10.1021/bi0615442; PMID: 17176093
- Notari S, Strammiello R, Capellari S, Giese A, Cescatti M, Grassi J, et al. Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J Biol Chem 2008; 283:30557 - 65; http://dx.doi.org/10.1074/jbc.M801877200; PMID: 18753138
- Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen HO, et al. Protease-sensitive synthetic prions. PLoS Pathog 2010; 6:e1000736; http://dx.doi.org/10.1371/journal.ppat.1000736; PMID: 20107515
- Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD, et al. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 2007; 53:325 - 35; http://dx.doi.org/10.1016/j.neuron.2007.01.005; PMID: 17270731
- Jones M, Peden AH, Prowse CV, Gröner A, Manson JC, Turner ML, et al. In vitro amplification and detection of variant Creutzfeldt-Jakob disease PrPSc. J Pathol 2007; 213:21 - 6; http://dx.doi.org/10.1002/path.2204; PMID: 17614097
- Prusiner SB, Scott MR, DeArmond SJ, Carlson G. Transmission and replication of prions. In: Prusiner SB, ed. Prion Biology and Diseases. Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 2004:187-242.
- Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature 2006; 442:585 - 9; http://dx.doi.org/10.1038/nature04922; PMID: 16810177