Abstract
Alzheimer disease (AD) is characterized by the amyloidogenic processing of the amyloid precursor protein (APP), culminating in the accumulation of amyloid-β peptides in the brain. The enzymatic action of the β-secretase, BACE1 is the rate-limiting step in this amyloidogenic processing of APP. BACE1 cleavage of wild-type APP (APPWT) is inhibited by the cellular prion protein (PrPC). Our recent study has revealed the molecular and cellular mechanisms behind this observation by showing that PrPC directly interacts with the pro-domain of BACE1 in the trans-Golgi network (TGN), decreasing the amount of BACE1 at the cell surface and in endosomes where it cleaves APPWT, while increasing BACE1 in the TGN where it preferentially cleaves APP with the Swedish mutation (APPSwe). PrPC deletion in transgenic mice expressing the Swedish and Indiana familial mutations (APPSwe,Ind) failed to affect amyloid-β accumulation, which is explained by the differential subcellular sites of action of BACE1 toward APPWT and APPSwe. This, together with our observation that PrPC is reduced in sporadic but not familial AD brain, suggests that PrPC plays a key protective role against sporadic AD. It also highlights the need for an APPWT transgenic mouse model to understand the molecular and cellular mechanisms underlying sporadic AD.
Alzheimer disease (AD) is the most prevalent form of dementia and currently affects more than 37 million people worldwide.Citation1,Citation2 Less than 1% of AD cases are known to be caused by inheritable mutations in genes encoding the amyloid precursor protein (APP), presenilin-1 (PS1) and PS2.Citation3 However, for the majority of AD cases, which are termed sporadic or late-onset, the cause and/or causes remain elusive, although aging is known to be the greatest risk factor.Citation4 AD is characterized by the accumulation in the brain of the amyloid-β (Aβ) peptide in the form of oligomers and/or fibrils. This peptide is formed by the sequential processing of APP by the β-site APP cleaving enzyme-1 (BACE1) and the PS1 or PS2 containing γ-secretase complex.Citation5 Depending on the γ-secretase cleavage site, Aβ is released as a 40 or 42 amino acid peptide, with Aβ42 exhibiting a greater propensity for aggregation.Citation6 In the normal brain, Aβ production and degradation is tightly regulated, but in the AD brain, Aβ homeostasis is altered with elevated levels of Aβ42 and/or an increased ratio of Aβ42:Aβ40.Citation7 BACE1 is the rate-limiting enzyme in Aβ production and its activity is raised in the brains of sporadic AD patients,Citation8,Citation9 therefore, understanding how this enzyme is regulated is vitally important and has become a pivotal part of AD research.
The normal cellular form of the prion protein (PrPC) converts by misfolding into the infectious form, PrPSc, which is the causative agent in the neurodegenerative transmissible spongiform encephalopathies (TSEs) including Creutzfeldt-Jacob disease (CJD).Citation10 There are a number of similarities between AD and prion diseases, including the coexistence of AD pathology in some cases of CJD.Citation11 In addition, PrPC has been shown to co-localize with Aβ in plaques,Citation12 which are present in the majority of CJD patients with associated AD-type pathology.Citation13 A systematic meta-analysis of AD genetic association studies revealed that the gene PRNP, which encodes PrPC, is a potential AD susceptibility geneCitation14 and the Met/Val polymorphism at residue 129 in PrPC has been reported to be a risk factor for early onset AD.Citation13,Citation15,Citation16 In 2007, we discovered that PrPC plays a critical role in regulating the activity of BACE1 toward the processing of wild-type APP (APPWT)Citation17 and proposed that a normal function of PrPC may be to protect against AD.Citation18 In addition to PrPC, various other proteins have been shown to alter the amyloidogenic processing of APP by the modulation of BACE1 activity, including the sorting protein-related receptor (SorLA/LR11),Citation19,Citation20 ADP ribosylation factor 6 (ARF6)Citation21 and Golgi-localized γ-ear containing ARF binding proteins (GGAs)Citation22,Citation23 among others. All of these proteins modulate BACE1 activity by altering the trafficking and localization of BACE1 in relation to APP, as a means to regulate Aβ levels.
In our recent study,Citation24 we extended our work on the role of PrPC in regulating BACE1 activity by examining the molecular and cellular mechanisms involved. We found that PrPC forms immuno-complexes with BACE1 in both mice and non-demented human brain samples. The interaction between BACE1 and PrPC was investigated using recombinant proteins by enzyme linked immunosorbent assay (ELISA) and surface plasmon resonance (SPR) to reveal a direct interaction between PrPC and the pro-domain of BACE1. As PrPC also inhibited the BACE2 mediated processing of APP, we identified a conserved sequence (LPLR) in the pro-domains of both proteins which we interrogated in BACE1 by site-directed mutagenesis. The L28Q/L30Q mutant did not traffic properly through the secretory pathway and was only present in its immature ER form, indicating that these residues are particularly important for the trafficking of BACE1. In contrast the P29G mutation resulted in comparable maturation and activity to wild-type BACE1, but the inhibitory effect of PrPC on BACE1 activity was lost, indicating that Pro29 is critical for the protein-protein interaction.Citation24
The catalytic activity of BACE1 is tightly regulated by pH, thus the opening of the active site cleft is dependent upon BACE1 localizing to acidic compartments such as endosomes, where APPWT processing primarily takes place.Citation25,Citation26 As BACE1 trafficks through the secretory pathway, the immature, prodomain containing form of the protein is processed into a mature, more catalytically active form,Citation27,Citation28 which is subsequently trafficked to the cell surface before re-internalisation into endosomes.Citation29,Citation30 Other than PrPC only two other proteins have been shown to regulate BACE1 via its prodomain, namely PS1 and SorLA.Citation20,Citation31 Whereas PS1 aids the maturation and trafficking of BACE1, SorLA regulates the BACE1 processing of APP in a manner similar to PrPC leading to a decrease in Aβ peptide generation shown in both cell culture models and SorLA knockout mice.Citation19,Citation20 SorLA mediates its inhibitory effect by retaining BACE1 in the trans Golgi network (TGN) preventing its association with its other interacting partner APP.Citation20 Interestingly, an unbiased microarray screen for genes differentially expressed in lymphoblasts of patients with sporadic AD revealed that SorLA protein levels were dramatically reduced.Citation32
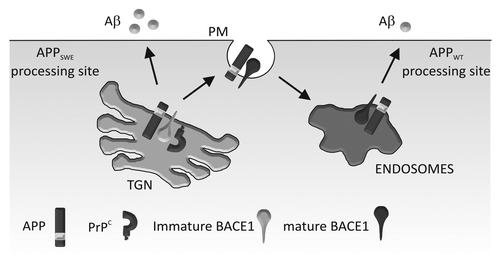
In our recent study,Citation24 we used colocalization with subcellular markers to assess the effect of PrPC on BACE1 trafficking. This revealed that PrPC caused retention of BACE1 in the TGN, reducing the trafficking of BACE1 to the cell surface and endosomes where it preferentially cleaves APPWT. We demonstrated that the reduction in BACE1 localization to endosomes only amounted to ~30%,Citation24 whereas the reduction in APP processing was > 95%.Citation17 This discrepancy can be explained by the likelihood that only a small pool of total BACE1 protein within cells may be intended for APP processing, as BACE1 has several other substrates which are cleaved in other subcellular compartments. For example, the amyloid precursor like proteins (APLPs) are cleaved within endosomal compartments,Citation33,Citation34 α-2,6-sialyltransferase (ST6Gal-1) resides in the TGN where it is cleaved by BACE1Citation35 and low density lipoprotein receptor-related protein-1 (LRP-1) and BACE1 interact at the cell surface where BACE1 releases C-terminal fragments of LRP1.Citation36 We hypothesized that PrPC sequesters BACE1 into membrane subcompartments separate to those which contain APP, and that this separation subsequently leads to changes in the downstream trafficking of BACE1 into endosomal compartments where it cleaves APPWT.Citation37 A recent study on the regulation of BACE1 internalization demonstrated that BACE1 needs to be sorted from ARF6 positive endosomes to RAB5 positive early endosomes for the proteolytic processing of APP to occur,Citation21 which suggests that a small change in the cellular localization of BACE1 early in the secretory pathway could hinder this downstream sorting process. Clearly there are several proteins, including PrPC, which are able to modulate the cellular activity of BACE1, primarily by altering its trafficking to endosomes that contain APP. It may be a combination of cellular factors/ stresses that alter one or more of these regulatory proteins that ultimately leads to de-regulation of BACE1 activity and consequently Aβ homeostasis.
In the non-amyloidogenic processing of APP, α-secretases (notably ADAM10 and other members of the A Distintegrin And Metalloproteinase (ADAM) family) cleave APP in the middle of the Aβ sequence precluding the production of intact Aβ peptides.Citation5 Overexpression of ADAM10 in transgenic mice resulted in an increase in α-secretase cleavage of APP and a reduction in Aβ production and deposition,Citation38 reinforcing the consensus view that the amyloidogenic and non-amyloidogenic APP processing pathways are reciprocally regulated. Recently, the shedding of various proteins, including PrPC, by ADAM10 was investigated in vivo using conditional neuron specific ADAM10 knockout mice.Citation39,Citation40 As well as confirming ADAM10 to be the PrPC sheddase,Citation41 they also observed an increase in PrPC protein levels and re-location of PrPC to intracellular compartments of the secretory pathway, namely the endoplasmic reticulum (ER) and Golgi upon deletion of ADAM10.Citation39 Interestingly, in the earlier study,Citation40 the same authors reported that deletion of ADAM10, as well as reducing significantly the α-secretase cleavage of APP, also somewhat surprisingly led to a reduction in sAPPβ and Aβ, suggesting that somehow altering ADAM10 levels was also impacting on the cleavage of APP by BACE1. From our recent study,Citation24 a potential explanation for why deleting ADAM10 has such an adverse effect on the cleavage of APP by BACE1 becomes apparent. The accumulation of PrPC in the secretory pathway of the ADAM10 knockout mice leads to retention of BACE1 in these compartments and subsequent reduction in the processing of APP. In addition to its role in shedding PrPC, ADAM10 has also been reported to be responsible for its endoproteolytic cleavage to generate the neuroprotective N1 fragment.Citation42-Citation44 However, the role of ADAM10 in this endoproteolytic cleavage of PrPC is controversial.Citation39,Citation41
To extend our analysis of the effect of PrPC on BACE1 we examined the effect of PrPC deletion on Aβ production and deposition in the J20 transgenic mouse model of AD.Citation24 These mice express human APP with the Swedish (K670N/M671L) and Indiana (V717F) familial mutations (APPSwe,Ind).Citation45 Surprisingly, deletion of PrPC in this transgenic mouse model had no effect on APP proteolytic processing, Aβ plaque deposition, or levels of soluble Aβ or Aβ oligomers.Citation24 Consistent with this, others have shown that PrPC deletion in the APPSwe/PS1ΔE9 and APPSwe/PS1L166P transgenic models failed to alter BACE1 processing of APP, Aβ levels and Aβ deposition,Citation46,Citation47 and overexpression of PrPC in APPSwe,Ind transgenic mice resulted in only a minor increase in Aβ plaque formation but no difference in Aβ40 or Aβ42 levels.Citation48 In addition, a recent study by Cissé et al.Citation49 showed that PrPC deletion in the J20 transgenic mice did not change hippocampal levels of Aβ. The common denominator in all of the transgenic animals used in these studies is the Swedish familial mutation in APP. BACE1 is known to cleave APPWT and APPSwe in different subcellular compartments: the cleavage of APPSwe occurs primarily in the TGN,Citation25,Citation50 whereas the cleavage of APPWT occurs in the endosomes.Citation30,Citation51,Citation52 As PrPC interacted with the prodomain containing form of BACE1 increasing its localization to the TGN, while decreasing the amount in endosomes, we considered whether the cleavage of APPWT and APPSwe by BACE1 were differentially affected by PrPC. Using HEK cells expressing either APPWT or APPSwe we found that although PrPC inhibited the activity of BACE1 toward APPWT it had no such inhibitory effect on the activity of BACE1 toward APPSwe.Citation24 Thus the different subcellular sites of action of BACE1 toward APPWT (endosomes) and APPSwe (TGN) would appear to explain the lack of effect of PrPC deletion on APP processing and Aβ deposition in the APPSwe,Ind/PrP null mice ().
Figure 1. Schematic diagram depicting PrPC regulation of BACE1 in relation to APPWT and APPSwe. PrPC interacts with the pro-domain of BACE1 in its immature form in the TGN, slowing its maturation and trafficking to endosomes (via the cell surface). In the TGN, BACE1 preferentially cleaves APPSwe ; in the endosomes, it preferentially cleaves APPWT. Our model shows that PrPC inhibits BACE1 cleavage of APPWT, but not APPSwe which leads to increased Aβ formation. PM, plasma membrane.
Of the APP mouse models currently listed on Alzforum (http://www.alzforum.org/res/com/tra/app/) 76% contain genetic familial mutations, with 57% containing the Swedish mutation and only 2% expressing human APPWT. The transgenic models incorporating familial mutations not only in APP, but also in PS1, PS2 and tau have contributed immensely to the study of AD, however, they all have their limitations and no one model completely encapsulates all aspects of the cellular and behavioral pathology of AD (reviewed in refs. Citation3, Citation53 and Citation54). Our study has highlighted a significant limitation of the transgenic models incorporating the Swedish mutation in APP. Others have recently highlighted that caution is needed when using transgenic animal models with familial AD mutations as the underlying mechanisms driving amyloid accumulation in familial AD and in sporadic AD may well be subtly different.Citation55 As less than 1% of AD cases are caused by inheritable mutationsCitation3 and with our aging population, there is a great need for a transgenic mouse model of sporadic AD that will facilitate detailed analysis of the disease process occurring in the vast majority of AD patients.
From our studies in cell and animal models we proposed that a normal function of PrPC was to regulate the production of the neurotoxic Aβ and therefore to protect against AD.Citation17,Citation18,Citation24 A previous immunohistochemical study showed that there was a decrease in PrPC in the occipital and frontal gray matter in the brains of sporadic AD patientsCitation56 and PrPC levels in cerebrospinal fluid were found to decrease with dementia severity in AD.Citation57 A brief report also showed a decrease of PrPC in the hippocampus and frontal cortex in AD,Citation58 however, the number of cases (two AD and three controls) was too small for statistical analysis and the case information presented was insufficient to draw any relevant conclusions about the link between PrPC expression and AD. Recently, we performed a larger study (with 7 AD cases and 7 age-matched controls) where we found that PrPC was decreased in the hippocampus and temporal cortex in patients with sporadic AD compared with the age-matched controls,Citation59 consistent with our proposal that PrPC could regulate the production of Aβ (). In contrast to our dataCitation59 and that of Velayos et al.,Citation58 a more recent study reported that there was no difference in PrPC levels in the hippocampus, superior frontal cortex (BA9) and superior-middle temporal cortex (BA21–22) of patients with no cognitive impairment (NCI), amnestic mild cognitive impairment (aMCI), mild AD (mAD) and AD patients.Citation60 The reasons for the differences between these studies is not immediately apparent, although Saijo et al.Citation60 did not differentiate between sporadic and familial AD cases in their cohort making direct comparison with our data difficult.
Figure 2. Schematic diagram depicting PrPC regulation of BACE1 in sporadic AD brains. In the healthy brain, PrPC interacts with BACE1 inhibiting its cleavage of APP and thereby keeping the level of Aβ in check. In sporadic AD, PrPC levels are reduced, leading to impaired BACE1 inhibition and increased Aβ formation.
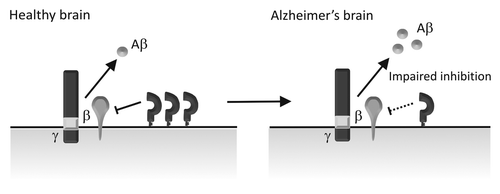
Like many of the biological changes that occur as a consequence of the pathological cascade in AD, this reduction in PrPC could simply be explained by downstream events associated with neurodegeneration. However, there was no loss of PrPC in the brains of individuals harbouring genetic mutations that cause familial AD,Citation59 suggesting that reduced expression of PrPC is not simply an effect of Aβ accumulation and the neurodegeneration in sporadic AD. Thus, this reduction in PrPC would appear to reflect a primary mechanism of disease and not merely a secondary consequence of other AD-associated changes. Although aging is the greatest risk factor for AD,Citation4 the cause of the association is unknown. The activity of BACE1 increases with age and this increase may be sufficient over time to alter the balance of Aβ generation and clearance and ultimately plaque deposition.Citation61 Using an aging series of human brains (ages 20–88), we found that PrPC decreases with age in areas of the brain affected in AD,Citation59 possibly providing a molecular mechanism for how the increase of BACE1 activity with age could affect AD progression. PrPC is known to protect neuronal cells against oxidative stress (reviewed inCitation10,Citation62), possibly in part via generation of the N1 fragment,Citation63 and aging is associated with an increase in reactive oxygen species, with oxidative stress being an early event in the pathogenesis of AD.Citation64-Citation66 Therefore, the reduction of PrPC seen in the aging and sporadic AD brain would likely also contribute to the increased susceptibility of neurons to oxidative stress, exacerbating AD pathology.Citation59
PrPC has also recently been identified as a high affinity receptor for Aβ-oligomersCitation47,Citation67-Citation69 which are considered to be the neurotoxic species in AD.Citation70,Citation71 However, there are contradictory results on whether the binding of these oligomers to PrPC is required to mediate their toxic effects and thus the functional role of the binding interactions is yet to be firmly established.Citation46,Citation72-Citation74 Although deletion of PrPC alleviated the memory deficits in the APPSwe/PS1ΔE9 mice,Citation47 ablation of PrPC did not ameliorate cognitive dysfunction in the J20 transgenic mice.Citation49 One possible reason for these discordant results is the inclusion of the PS1 transgene in the former mice which may be having a complicating effect on the interpretation of these data.
In summary, PrPC has been shown to protect neurons against oxidative stress, as well as to inhibit the β-secretase cleavage of APPWT, thereby reducing Aβ generation, and PrPC is downregulated in sporadic AD and in aging. In light of these observations, depletion of PrPC, as suggested by some as a potential treatment for AD, may have significant deleterious effects on the pathogenesis of sporadic AD. Clearly further work is required, including the development of appropriate animal models, to clarify the role of PrPC in AD.
Acknowledgments
This work is supported by the Medical Research Council of Great Britain (Grant G0802189) and Alzheimer Research UK (Grant PG2010–2).
References
- Burns A, Iliffe S. Alzheimer’s disease. BMJ 2009; 338:b158; http://dx.doi.org/10.1136/bmj.b158; PMID: 19196745
- Mount C, Downton C. Alzheimer disease: progress or profit?. Nat Med 2006; 12:780 - 4; http://dx.doi.org/10.1038/nm0706-780; PMID: 16829947
- Morrissette DA, Parachikova A, Green KN, LaFerla FM. Relevance of transgenic mouse models to human Alzheimer disease. J Biol Chem 2009; 284:6033 - 7; http://dx.doi.org/10.1074/jbc.R800030200; PMID: 18948253
- Jorm AF, Korten AE, Henderson AS. The prevalence of dementia: a quantitative integration of the literature. Acta Psychiatr Scand 1987; 76:465 - 79; http://dx.doi.org/10.1111/j.1600-0447.1987.tb02906.x; PMID: 3324647
- Vardy ER, Catto AJ, Hooper NM. Proteolytic mechanisms in amyloid-beta metabolism: therapeutic implications for Alzheimer’s disease. Trends Mol Med 2005; 11:464 - 72; http://dx.doi.org/10.1016/j.molmed.2005.08.004; PMID: 16153892
- Nunan J, Small DH. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett 2000; 483:6 - 10; http://dx.doi.org/10.1016/S0014-5793(00)02076-7; PMID: 11033346
- Pimplikar SW. Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int J Biochem Cell Biol 2009; 41:1261 - 8; http://dx.doi.org/10.1016/j.biocel.2008.12.015; PMID: 19124085
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol 2002; 59:1381 - 9; http://dx.doi.org/10.1001/archneur.59.9.1381; PMID: 12223024
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci U S A 2004; 101:3632 - 7; http://dx.doi.org/10.1073/pnas.0205689101; PMID: 14978286
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 2008; 3:11 - 40; http://dx.doi.org/10.1146/annurev.pathmechdis.3.121806.154326; PMID: 18233951
- Hainfellner JA, Wanschitz J, Jellinger K, Liberski PP, Gullotta F, Budka H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol 1998; 96:116 - 22; http://dx.doi.org/10.1007/s004010050870; PMID: 9705125
- Voigtländer T, Klöppel S, Birner P, Jarius C, Flicker H, Verghese-Nikolakaki S, et al. Marked increase of neuronal prion protein immunoreactivity in Alzheimer’s disease and human prion diseases. Acta Neuropathol 2001; 101:417 - 23; PMID: 11484812
- Del Bo R, Scarlato M, Ghezzi S, Martinelli-Boneschi F, Fenoglio C, Galimberti G, et al. Is M129V of PRNP gene associated with Alzheimer’s disease? A case-control study and a meta-analysis. Neurobiol Aging 2006; 27:770 - , e1-770, e5; http://dx.doi.org/10.1016/j.neurobiolaging.2005.05.025; PMID: 16099550
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 2007; 39:17 - 23; http://dx.doi.org/10.1038/ng1934; PMID: 17192785
- Dermaut B, Croes EA, Rademakers R, Van den Broeck M, Cruts M, Hofman A, et al. PRNP Val129 homozygosity increases risk for early-onset Alzheimer’s disease. Ann Neurol 2003; 53:409 - 12; http://dx.doi.org/10.1002/ana.10507; PMID: 12601712
- Riemenschneider M, Klopp N, Xiang W, Wagenpfeil S, Vollmert C, Müller U, et al. Prion protein codon 129 polymorphism and risk of Alzheimer disease. Neurology 2004; 63:364 - 6; PMID: 15277640
- Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, et al. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc Natl Acad Sci U S A 2007; 104:11062 - 7; http://dx.doi.org/10.1073/pnas.0609621104; PMID: 17573534
- Hooper NM, Turner AJ. A new take on prions: preventing Alzheimer’s disease. Trends Biochem Sci 2008; 33:151 - 5; http://dx.doi.org/10.1016/j.tibs.2008.01.004; PMID: 18343669
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 2005; 102:13461 - 6; http://dx.doi.org/10.1073/pnas.0503689102; PMID: 16174740
- Spoelgen R, von Arnim CA, Thomas AV, Peltan ID, Koker M, Deng A, et al. Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and beta-secretase beta-site APP-cleaving enzyme. J Neurosci 2006; 26:418 - 28; http://dx.doi.org/10.1523/JNEUROSCI.3882-05.2006; PMID: 16407538
- Sannerud R, Declerck I, Peric A, Raemaekers T, Menendez G, Zhou L, et al. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci U S A 2011; 108:E559 - 68; http://dx.doi.org/10.1073/pnas.1100745108; PMID: 21825135
- He X, Li F, Chang WP, Tang J. GGA proteins mediate the recycling pathway of memapsin 2 (BACE). J Biol Chem 2005; 280:11696 - 703; http://dx.doi.org/10.1074/jbc.M411296200; PMID: 15615712
- Wahle T, Thal DR, Sastre M, Rentmeister A, Bogdanovic N, Famulok M, et al. GGA1 is expressed in the human brain and affects the generation of amyloid beta-peptide. J Neurosci 2006; 26:12838 - 46; http://dx.doi.org/10.1523/JNEUROSCI.1982-06.2006; PMID: 17151287
- Griffiths HH, Whitehouse IJ, Baybutt H, Brown D, Kellett KA, Jackson CD, et al. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J Biol Chem 2011; 286:33489 - 500; http://dx.doi.org/10.1074/jbc.M111.278556; PMID: 21795680
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, et al. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat Med 1995; 1:1291 - 6; http://dx.doi.org/10.1038/nm1295-1291; PMID: 7489411
- Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 1994; 269:17386 - 9; PMID: 8021238
- Benjannet S, Elagoz A, Wickham L, Mamarbachi M, Munzer JS, Basak A, et al. Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J Biol Chem 2001; 276:10879 - 87; http://dx.doi.org/10.1074/jbc.M009899200; PMID: 11152688
- Creemers JW, Ines Dominguez D, Plets E, Serneels L, Taylor NA, Multhaup G, et al. Processing of beta-secretase by furin and other members of the proprotein convertase family. J Biol Chem 2001; 276:4211 - 7; http://dx.doi.org/10.1074/jbc.M006947200; PMID: 11071887
- Capell A, Steiner H, Willem M, Kaiser H, Meyer C, Walter J, et al. Maturation and pro-peptide cleavage of beta-secretase. J Biol Chem 2000; 275:30849 - 54; http://dx.doi.org/10.1074/jbc.M003202200; PMID: 10801872
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J Biol Chem 2000; 275:33729 - 37; http://dx.doi.org/10.1074/jbc.M004175200; PMID: 10924510
- Hébert SS, Bourdages V, Godin C, Ferland M, Carreau M, Lévesque G. Presenilin-1 interacts directly with the beta-site amyloid protein precursor cleaving enzyme (BACE1). Neurobiol Dis 2003; 13:238 - 45; http://dx.doi.org/10.1016/S0969-9961(03)00035-4; PMID: 12901838
- Scherzer CR, Offe K, Gearing M, Rees HD, Fang G, Heilman CJ, et al. Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch Neurol 2004; 61:1200 - 5; http://dx.doi.org/10.1001/archneur.61.8.1200; PMID: 15313836
- Eggert S, Paliga K, Soba P, Evin G, Masters CL, Weidemann A, et al. The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like cleavages: modulation of APLP-1 processing by n-glycosylation. J Biol Chem 2004; 279:18146 - 56; http://dx.doi.org/10.1074/jbc.M311601200; PMID: 14970212
- Li Q, Südhof TC. Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J Biol Chem 2004; 279:10542 - 50; http://dx.doi.org/10.1074/jbc.M310001200; PMID: 14699153
- Kitazume S, Tachida Y, Oka R, Shirotani K, Saido TC, Hashimoto Y. Alzheimer’s beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci USA 2001; 98:13554 - 9; http://dx.doi.org/10.1073/pnas.241509198; PMID: 11698669
- von Arnim CA, Kinoshita A, Peltan ID, Tangredi MM, Herl L, Lee BM, et al. The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J Biol Chem 2005; 280:17777 - 85; http://dx.doi.org/10.1074/jbc.M414248200; PMID: 15749709
- Griffiths HH, Morten IJ, Hooper NM. Emerging and potential therapies for Alzheimer’s disease. Expert Opin Ther Targets 2008; 12:693 - 704; http://dx.doi.org/10.1517/14728222.12.6.693; PMID: 18479216
- Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest 2004; 113:1456 - 64; PMID: 15146243
- Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, et al. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener 2011; 6:36; http://dx.doi.org/10.1186/1750-1326-6-36; PMID: 21619641
- Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci 2010; 30:4833 - 44; http://dx.doi.org/10.1523/JNEUROSCI.5221-09.2010; PMID: 20371803
- Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, et al. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem 2009; 284:22590 - 600; http://dx.doi.org/10.1074/jbc.M109.032599; PMID: 19564338
- Cissé MA, Sunyach C, Lefranc-Jullien S, Postina R, Vincent B, Checler F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J Biol Chem 2005; 280:40624 - 31; http://dx.doi.org/10.1074/jbc.M506069200; PMID: 16236709
- Vincent B, Paitel E, Frobert Y, Lehmann S, Grassi J, Checler F. Phorbol ester-regulated cleavage of normal prion protein in HEK293 human cells and murine neurons. J Biol Chem 2000; 275:35612 - 6; http://dx.doi.org/10.1074/jbc.M004628200; PMID: 10952979
- Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, et al. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem 2001; 276:37743 - 6; PMID: 11477090
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000; 20:4050 - 8; PMID: 10818140
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, et al. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2010; 2:306 - 14; http://dx.doi.org/10.1002/emmm.201000082; PMID: 20665634
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, et al. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 2010; 30:6367 - 74; http://dx.doi.org/10.1523/JNEUROSCI.0395-10.2010; PMID: 20445063
- Schwarze-Eicker K, Keyvani K, Görtz N, Westaway D, Sachser N, Paulus W. Prion protein (PrPc) promotes beta-amyloid plaque formation. Neurobiol Aging 2005; 26:1177 - 82; http://dx.doi.org/10.1016/j.neurobiolaging.2004.10.004; PMID: 15917101
- Cissé M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci 2011; 31:10427 - 31; http://dx.doi.org/10.1523/JNEUROSCI.1459-11.2011; PMID: 21775587
- Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “beta-secretase” site occurs in the golgi apparatus. J Biol Chem 1996; 271:9390 - 7; PMID: 8621605
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999; 286:735 - 41; http://dx.doi.org/10.1126/science.286.5440.735; PMID: 10531052
- Yan R, Han P, Miao H, Greengard P, Xu H. The transmembrane domain of the Alzheimer’s beta-secretase (BACE1) determines its late Golgi localization and access to beta -amyloid precursor protein (APP) substrate. J Biol Chem 2001; 276:36788 - 96; http://dx.doi.org/10.1074/jbc.M104350200; PMID: 11466313
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol 2008; 115:5 - 38; http://dx.doi.org/10.1007/s00401-007-0312-8; PMID: 18038275
- Kokjohn TA, Roher AE. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: understanding the paradigms, limitations, and contributions. Alzheimers Dement 2009; 5:340 - 7; http://dx.doi.org/10.1016/j.jalz.2009.03.002; PMID: 19560104
- Schieb H, Kratzin H, Jahn O, Möbius W, Rabe S, Staufenbiel M, et al. Beta-amyloid peptide variants in brains and cerebrospinal fluid from amyloid precursor protein (APP) transgenic mice: comparison with human Alzheimer amyloid. J Biol Chem 2011; 286:33747 - 58; http://dx.doi.org/10.1074/jbc.M111.246561; PMID: 21795681
- Rezaie P, Pontikis CC, Hudson L, Cairns NJ, Lantos PL. Expression of cellular prion protein in the frontal and occipital lobe in Alzheimer’s disease, diffuse Lewy body disease, and in normal brain: an immunohistochemical study. J Histochem Cytochem 2005; 53:929 - 40; http://dx.doi.org/10.1369/jhc.4A6551.2005; PMID: 16055747
- Meyne F, Gloeckner SF, Ciesielczyk B, Heinemann U, Krasnianski A, Meissner B, et al. Total prion protein levels in the cerebrospinal fluid are reduced in patients with various neurological disorders. J Alzheimers Dis 2009; 17:863 - 73; PMID: 19542614
- Velayos JL, Irujo A, Cuadrado-Tejedor M, Paternain B, Moleres FJ, Ferrer V. The cellular prion protein and its role in Alzheimer disease. Prion 2009; 3:110 - 7; http://dx.doi.org/10.4161/pri.3.2.9135; PMID: 19556894
- Whitehouse IJ, Jackson C, Turner AJ, Hooper NM. Prion protein is reduced in aging and in sporadic but not in familial Alzheimer’s disease. J Alzheimers Dis 2010; 22:1023 - 31; PMID: 20930299
- Saijo E, Scheff SW, Telling GC. Unaltered prion protein expression in Alzheimer disease patients. Prion 2011; 5:109 - 16; http://dx.doi.org/10.4161/pri.5.2.16355; PMID: 21654203
- Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC. Beta-secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol 2004; 164:719 - 25; http://dx.doi.org/10.1016/S0002-9440(10)63159-8; PMID: 14742275
- Vassallo N, Herms J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J Neurochem 2003; 86:538 - 44; http://dx.doi.org/10.1046/j.1471-4159.2003.01882.x; PMID: 12859667
- Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 2009; 284:35973 - 86; http://dx.doi.org/10.1074/jbc.M109.051086; PMID: 19850936
- Halliwell B. Oxidative stress and neurodegeneration: where are we now?. J Neurochem 2006; 97:1634 - 58; http://dx.doi.org/10.1111/j.1471-4159.2006.03907.x; PMID: 16805774
- Lee HG, Perry G, Moreira PI, Garrett MR, Liu Q, Zhu X, et al. Tau phosphorylation in Alzheimer’s disease: pathogen or protector?. Trends Mol Med 2005; 11:164 - 9; http://dx.doi.org/10.1016/j.molmed.2005.02.008; PMID: 15823754
- Zhu X, Su B, Wang X, Smith MA, Perry G. Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci 2007; 64:2202 - 10; http://dx.doi.org/10.1007/s00018-007-7218-4; PMID: 17605000
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, et al. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci 2010; 11:130; http://dx.doi.org/10.1186/1471-2202-11-130; PMID: 20946660
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, et al. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat Commun 2011; 2:336; http://dx.doi.org/10.1038/ncomms1341; PMID: 21654636
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006; 440:352 - 7; http://dx.doi.org/10.1038/nature04533; PMID: 16541076
- Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid beta-protein assembly and Alzheimer disease. J Biol Chem 2009; 284:4749 - 53; http://dx.doi.org/10.1074/jbc.R800036200; PMID: 18845536
- Forloni G, Balducci C. β-amyloid oligomers and prion protein: Fatal attraction?. Prion 2011; 5:10 - 5; http://dx.doi.org/10.4161/pri.5.1.14367; PMID: 21150333
- Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature 2010; 466:E3 - 4, discussion E4-5; http://dx.doi.org/10.1038/nature09217; PMID: 20703260
- Alier K, Ma L, Yang J, Westaway D, Jhamandas JH. Aβ inhibition of ionic conductance in mouse basal forebrain neurons is dependent upon the cellular prion protein PrPC. J Neurosci 2011; 31:16292 - 7; http://dx.doi.org/10.1523/JNEUROSCI.4367-11.2011; PMID: 22072680