Abstract
Chronic mental diseases (CMD) like the schizophrenias are progressive diseases of heterogenous but poorly understood biological origin. An imbalance in proteostasis is a hallmark of dysfunctional neurons, leading to impaired clearance and abnormal deposition of protein aggregates. Thus, it can be hypothesized that unbalanced proteostasis in such neurons may also lead to protein aggregates in schizophrenia. These protein aggregates, however, would be more subtle then in the classical neurodegenerative diseases and as such have not yet been detected. The DISC1 (Disrupted-in-schizophrenia 1) gene is considered among the most promising candidate genes for CMD having been identified as linked to CMD in a Scottish pedigree and having since been found to associate to various phenotypes of CMD. We have recently demonstrated increased insoluble DISC1 protein in the cingular cortex in approximately 20% of cases of CMD within the widely used Stanley Medical Research Institute Consortium Collection. Surprisingly, in vitro, DISC1 aggregates were cell-invasive, i.e., purified aggresomes or recombinant DISC1 fragments where internalized at an efficiency comparable to that of α-synuclein. Intracellular DISC1 aggresomes acquired gain-of-function properties in recruiting otherwise soluble proteins such as the candidate schizophrenia protein dysbindin. Disease-associated DISC1 polymorphism S704C led to a higher oligomerization tendency of DISC1. These findings justify classification of DISC1-dependent brain disorders as protein conformational disorders which we have tentatively termed DISC1opathies. The notion of disturbed proteostasis and protein aggregation as a mechanism of mental diseases is thus emerging. The yet unidentified form of neuronal impairment in CMD is more subtle than in the classical neurodegenerative diseases without leading to massive cell death and as such present a different kind of neuronal dysfunctionality, eventually confined to highly selective CNS subpopulations.
Schizophrenia
Chronic mental diseases (CMD) such as schizophrenia or the recurrent affective disorders are the most prevalent brain diseases, however an understanding of their neurobiology still remains elusive. In the absence of profound biological insight, CMD are diagnosed as mere clinical phenotypes by self-reporting of patients in a clinical interview according to internationally defined criteria DSM-IV and ICD-10 Citation1.
Among all CMD, schizophrenia leads to the most dramatic decline in cognitive abilities.Citation2 Positive symptoms (hallucinations, delusions, thought broadcasting), negative symptoms (affective flattening, social withdrawal, avolitionCitation3) and cognitive symptoms with impaired processing speed, decline in attention, verbal memory recall with largely preserved long-term memoryCitation4 together characterize the clinical picture of schizophrenia. The overall clinical conceptualization of schizophrenia has changed little since its first description where chronic aspects of disease course were emphasized in the term “dementia precox” (“premature dementia”).Citation5 This definition was later extended to include also purely acute forms without irreversible cognitive deficitsCitation6 and the most recent editions of the gold standard diagnostic manuals DSM-III and DSM IV did not require, respectively, negative or cognitive symptoms to be present.Citation7 On this background, schizophrenia with prominent negative symptoms, poor prognosis and chronicity has recently been re-introduced as a subgroup of schizophrenia with “deficit syndrome”Citation8.
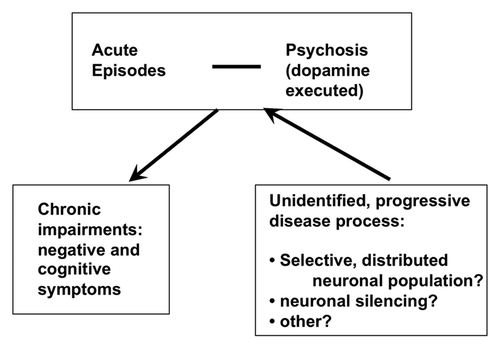
A tentative model on how positive, negative and cognitive symptoms integrate into one progressive, yet unidentified disease process intermittently leading to acute outbursts with positive symptoms (“florid” psychosis) is depicted in . There is a considerable body of evidence establishing that the term schizophrenia comprises very different clinical courses,Citation2 and therefore, likely also heterogenous biological causes.
Figure 1. Schematic drawing of the hypothesized flow of the progressive schizophrenia disease process. A yet unidentified dysfunctionality progresses through a process that does not involve massive neuronal cell death but reflect an unidentified mechanism of permanent neuronal silencing, eventually confined to a selected neuronal population. While progressing, the lesion leads intermittently to acute symptoms.
Investigations into the neurobiology of schizophrenia over the course of the last century can, with gross simplification, be summarized as follows:
(1) The search for a neuropathology in post mortem brains of clinically diagnosed schizphrenics has not yielded a specific neuropathologic signature except the slight enlargement of the third ventricles.Citation9 Even though claims have been made to signs of subtle disturbance in cortical architecture, these studies lack so far unequivocal replications.Citation9 Inconsistencies in investigations on disturbed cortical architecture in schizophrenics might be explained by the biological heterogeneity of the disease when using broad inclusion criteria, paired with low case numbers.
(2) The serendipitous discovery of the neurolepticsCitation10 and the subsequent revelation of their mechanism of actionCitation11 has established dopamine as a central player in psychosis (that is, the acute, positive symptoms of schizophrenia). Over the years, a wealth of evidence has reinforced this hypothesis and the current opinion in the field is that psychosis during schizophrenia (but not limited to it) is due to a presynaptic, hyperdopaminergic state in the striatum, while in contrast the prefrontal cortex suffers from hypodopaminergiaCitation12 (see ref. 13Citation13 for review).
(3) A neurodevelopmental component seems to be important for the development of schizophreniaCitation14 although the (as yet unidentified) abnormality during neurodevelopment must be subtle since it can remain unnoticed and become apparent only at adolescence or in conjunction with a second or third hit,Citation15 such as, for example, an exogenous or endogenous stressor.
(4) A genetic basis for schizophrenia has long been known and was initially supported by sibling and twin studies with monozygotic twins demonstrated to have a genetic risk for schizophrenia of 50% compared with unrelated individuals.Citation16,Citation17 Recent genetic linkage and their subsequent confirmation through association studies in various ethnic populations has led to the identification of candidate genes such as DISC1,Citation18 NRG1,Citation19 DTNBP1Citation20 and others.Citation21 Remarkably, these genetic studies led to the insight that diagnoses of ill individuals carrying the genetic markers crossed clinical diagnostic boundaries,Citation22,Citation23 i.e., gene carriers could show clinical phenotpyes of schizophrenia or depression, suggesting that the biological fundamentals of CMD and the clinical phenotyping may not be well aligned. The study of these candidate genes has fundamentally changed molecular psychiatry since it is now possible to model behavioral, neuropathological and biochemical phenotypes in vivo by reverse genetic engineering of mutant candidate genes in animals.Citation24,Citation25
To summarize, our knowledge has increased on acute psychotic physiology (striatal hyperdopaminergia) which can be symptomatically treated by administering dopamine antagonists, but the underlying chronic progressive process remains unknown; a lot of momentum is currently present in the field through the identification of candiate genes and the emergence of genetic animal models.
Disturbed Proteostasis as a Hallmark of Dysfunctional Neurons
Proteostasis in post-mitotic neurons is sensitive to functional disturbances with its dysequilibrium resulting in the accumulation of aggregated or insoluble proteins in the cell.Citation26 In extreme cases, this leads to massive deposition of proteins such as in the classical neurodegenerative diseases, where extracellular or intracellular proteins are deposited.Citation27,Citation28 Remarkably, the same proteins that are mutant in familial forms of these diseases have also been seen to be deposited in sporadic forms, i.e., those cases where protein aggregation cannot simply be explained by aberrant folding due to a mutation.Citation27 For example, Aβ is deposited in Alzheimer disease,Citation29 and familial APP mutations lead to early onset Alzheimer disease,Citation30 or α-synuclein is deposited in Lewy bodies in the substantia nigra in Parkinson diseaseCitation31 and some familial cases of Parkinson disease could be tracked to mutant α-synuclein.Citation32 In recent years, for most of the microscopic protein aggregates, cell-to-cell transmission has been demonstrated in vitro or in transgenic in vivo models such as for Aβ 33 and tauCitation33 in Alzheimer disease, α-synucleinCitation34 for Parkinson disease, polyglutamine protein for Huntington's diseaseCitation35 or SOD1 for amyotrophic lateral sclerosis.Citation36,Citation37
The disease relevance of the cell invasiveness of protein aggregates is still unclear. In patients with Parkinson disease who received stereotactic injections of stem cell grafts to replace degenerated substantia nigra tissue and where the graft, upon autopsy many years later, was investigated by immunohistochemistry, aggregated α-synuclein-positive inclusions were detected in the graft.Citation38 It was hypothesized that the aggregated α-synuclein-positive inclusions had been transmitted from the surrounding host tissue.Citation38
In conclusion, protein deposition and cell-to-cell transmissibility can be considered as two defining characteristics of protein conformational disorders.Citation39
DISC1opathies
We reasoned that the chronic progressive course of schizophrenia may lead to a proteostatic imbalance in affected neuronal circuitry with the disease-specific accumulation of insoluble proteins in dysfunctional neurons. We therefore tested whether candidate genes for schizophrenia, specifically those which had been shown to be mutated in familial cases of CMD were also found to be aberrantly aggregated or insoluble in sporadic cases of CMD, and in particular, those of schizophrenia.
We were particular interested in the DISC1 gene. DISC1 was identified in a Scottish pedigree where carriers of a balanced translocation mutation (1;11)(q42.1; q14.3) segregated with a range of chronic mental disease phenotypes such as schizophrenia, recurrent depression, bipolar disease, alcoholism, and adolescent conduct disorder with approximately 70% penetrance.Citation18,Citation40,Citation41 The translocation mutation leads to direct disruption of two previously unknown genes, termed DISC1 and DISC2, the latter most likely being a non-coding RNA with the potential for regulating DISC1 expression.Citation42 The translocation bisects the DISC1 gene after amino acid 597. Experimental analysis of the putative C-terminally shortened protein from residues 1–597 shows that it can act as dominant negative mutant in several biological readout systems where this mutation has been modeled,Citation43 including in in vivo mouse modelsCitation44-Citation46 (see below).
Genetic association studies in many independent populations of various ethnic backgrounds have demonstrated the DISC1 gene to be associated with a multitude of clinical disease phenotypes including schizophrenia,Citation47-Citation53 bipolar disorder,Citation54,Citation55 depression,Citation56 or autism,Citation57 (reviewed in refs. Citation42Citation and 58.
These genetic data suggest a role for DISC1 as a general vulnerability factor for adaptive behaviorCitation59 but not to a single and specific clinical phenotype. The genetic data have also received solid support from reverse genetics, i.e., studies modeling the deletion mutation by genetic engineering in mice. Three independently engineered animal models expressing the C-terminally deleted DISC1 transgene corresponding to the mutation in the original Scottish pedigree showed enlarged lateral ventricles,Citation44-Citation46 reduced neurite outgrowth,Citation46,Citation60 reduced parvalbumin-positive interneurons in inner cortical layers,Citation45,Citation46 and deficient prepulse inhibition (PPI) as prominent schizophrenia endophenotypes.Citation45,Citation46 Other strong evidence for a role of DISC1 in controling behavior comes from studies of a mouse with point mutations in mouse DISC1Citation61 and a DISC1 knockout mouse.Citation62 DISC1 knockout mice displayed a variety of subtle behavioral phenotypes including increased methamphetamine-induced hyperactivity, reduced PPI, and deficits in anxiety-specific parametersCitation62.
Thus, DISC1 is currently considered to be a top gene involved in CMD and a key player in behavioral control.
DISC1 is been called a scaffold protein interacting with more than 200 proteinsCitation63,Citation64 and localized in the postsynaptic density of the synapse,Citation65 but it has also been detected at the centrosome, in cilia, the cytoskeleton, as well as in mitochondria and the nucleus (see refs. Citation64 or Citation66 for extensive recent reviews).
When we investigated post mortem brains of patients with CMD (15 each with schizophrenia, bipolar disorder, major depressive disorder or normal controls from the Stanley Medical Research Institute's Consortium CollectionCitation67; http://www.stanleyresearch.org/dnn/Default.aspx?tabid=196), we found in approximately 20% of patients increased sarkosyl-insoluble, i.e., aggregated DISC1 in the cingular cortex (BA23) but not in normal controlsCitation68 or control patients with neurodegenerative diseases like Alzheimer disease, dementia with Lewy bodies, frontotemproal dementia and othersCitation69 where insoluble DISC1 was not detectable. The protocol used had been developed and validated using the smallest so far detected protein deposits, polyglutamine proteins.Citation68,Citation70 Interestingly, we observed the presence of insoluble DISC1 in post mortem brains of patients across clinical diagnostic boundaries corroborating the phenotypical heterogeneity of DISC1 mutation carriers in the Scottish pedigreeCitation40,Citation71 and the various clinical phenotypes genetically associating with DISC1 (see above).
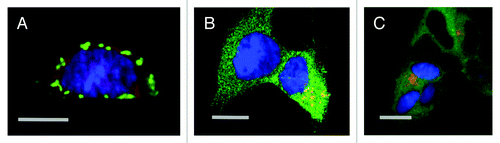
Overexpressing DISC1 in neuroblastoma cells led to the emergence of aggresomes (), that when purified and co-incubated with indicator recipient cells were taken up at low efficiency.Citation69 A highly purified recombinant DISC1 fragment expressed in E. coli demonstrated cell-invasiveness at an efficiency comparable to that of synthetic, oligomeric α-synucleinCitation69 (). Inoculation of the same recombinant DISC1 fragment into the brains of rats also leads to efficient uptake into primary neurons in vitro and in vivo (Pum, Bader, Huston, Korth, manuscript in submission).
Figure 2. Laser scanning confocal microscopy of DISC1 aggregates in neuroblastoma cells. A. Mouse neuroblastoma cells (CAD cells) permanently transfected with monomeric red fluorescent protein (mRFP) transiently transfected with untagged, full length DISC1, stained with α-DISC1 mAB 14F2Citation69 and a secondary FITC-labeled antibody. Bar 10 μm. B. Human SHSY5Y cells permanently transfected with green fluorescent protein fused to human DISC1 (598–854), incubated with recombinant human DISC1 (598–854) expressed and purified from E. coli and labeled with Dylight® (red) as described by Ottis et al.Citation69 Bar 10 μm C. Human SHSY5Y cells permanently transfected with green fluorescent protein fused to human DISC1 (598–854), and incubated with synthetic α-synuclein labeled with Dylight® (red) as described by Ottis et al.Citation69 Bar 10 μm.
These findings, the increased presence of aggregated, sarkosyl-insoluble DISC1 in brain disease and its cell-invasiveness suggested that DISC1-dependent brain disorders should be classified as protein conformational disorders, which we have tentatively termed DISC1opathiesCitation59 in analogy to other proteinopathies.
It may be argued that for a true proteinopathy like the synucleinopathies, tauopathies or other protein conformational diseases, demonstration of aggregates in tissue sections by immunohistochemistry and cell death in close proximity to those are mandatory, both of which have not been demonstrated for DISC1, so far. However, we think that coining the term DISC1opathy is justified for two reasons: 1. The definition of the term prion has been continously widened since the inclusion of non-pathogenic yeast prions, prions with proven physiological functions,Citation72,Citation73 to now comprise even so far considered non-transmissible diseases like Alzheimer disease or Parkinson disease.Citation74,Citation75 DISC1opathies are characterized by the smallest so far known protein aggregates associated to a progressive brain condition and are at the extreme of a continuum of protein conformational disorders (see ). 2. The second reason is pragmatic: the absence of an intelligible biology in the field of schizophrenia research has hindered scientific progress for a century. While heterogeneity of the biological origins of schizophrenia has always been assumed, now, the category of DISC1-dependent brain disorders (DISC1opathies) offers the opportunity of defining a brain disease subcategory as an entity that can be further characterized to molecular detail. Thus the term helps defining a long-sought, biology-based diagnostic entity and thereby enlightens a medical field where diagnostics has so far been restricted on clinical phenotyping.
Figure 3. Schematic drawing of the size continuum of protein aggregates in chronic brain diseases. The largest aggregates visible without specific immunostaining are the extracellular Aβ and prion plaques, followed by intracellular Lewy bodies. Polyglutamine proteins are visible only after specific immunostaining. At a submicroscopic level at the bottom of this inverted pyramid are DISC1 aggregates detectable only after biochemical purification.
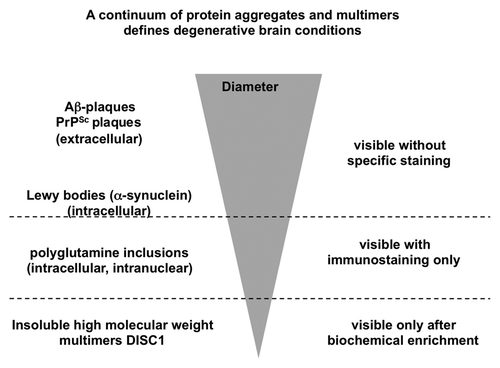
What does the cell-invasiveness of DISC1 aggregates mean? First of all, cell invasiveness of protein aggregates may be less rare than initially thought. Within the last few years, cell-invasiveness has been demonstrated for all major protein deposits of the classical neurodegenerative diseases like Aβ,Citation76,Citation77 tau,Citation33 α-synuclein,Citation34 polyglutamine proteinCitation35 or SOD1.Citation36,Citation37 But the in vivo ability of the aggregates to promote aggregation of soluble forms of itself seems to be a very inefficient process, and so far confined to transgenic animals prone to develop spontaneous aggregation of the transgenetically expressed protein at a later stage in their lives.Citation33,Citation76 It is therefore unclear whether cell-to-cell transmission can account for the observed progression of tau or α-synuclein deposition in the course of human clinical Alzheimer or Parkinson disease, respectively.Citation78,Citation79 So far, a similar neuropathological progression of protein deposits has not been reported for schizophrenia and confirmation of the presence of DISC1 aggregates or inclusions in vivo by immunohistochemistry complementing the biochemically purified insoluble DISC1 is still lacking but efforts to visualize insoluble DISC1 in post mortem brains of patients with schizophrenia are underway. We anticipate that the inclusions are likely to be subtle, and also restricted to specific subregions of the brain or neuronal subpopulations. Alternatively, it cannot be excluded that the DISC1 aggregates purified biochemically are too small to be detected by simple light microscopy using standard antibodies (see ). So far, there is neither positive nor negative evidence for increased β-sheet structures and/or amyloid in insoluble DISC1 identified in post mortem brains. It remains to be shown whether there is transmission of DISC1 aggregates in vivo in a significant manner, and whether cell-to-cell transmission is related to the pathomechanism of some CMD subtypes.
Are DISC1 aggregates infectious? The term “infectious” should not be used synonymous with “cell invasiveness.” An infectious protein cycle, like that for prions (PrPSc), requires the protein 1. to be taken up, 2. to recruit or convert fresh substrate (i.e., non aggregated protein), 3. to break the protein aggregate up into seeds, and 4. to release the material in way that it can be taken up efficiently. For leading to cellular pathology, this cycle does not have to be complete and even a protein aggregate unable to go through a full replication cycle can harm, for example, by invading a cell and recruiting otherwise soluble proteins, as we have shown for DISC1 agresomes recruiting soluble dysbindin.Citation69
The fact that only in 20% of cases with CMD (or 13% of cases with schizophrenia) increased insoluble DISC1 was detected in BA23 is consistent with the notion that CMD and the schizophrenias are heterogenous in their biological origin and thus DISC1opathies constitute only a fraction of CMD cases. The strength of the DISC1opathy concept is that it is a first step to defining a distinct subgroup of cases within CMD irrespective of their clinical phenotype, enabling a molecular analysis of major disease pathways in this subgroup. There are likely other subgroups involving different pathways, and they should definetely also be sought, but the diligent analysis of only one pathway, here the DISC1 pathway (see ), is already going to significantly advance our understanding of how subtle protein insolubility can lead to maladaptive behavior.
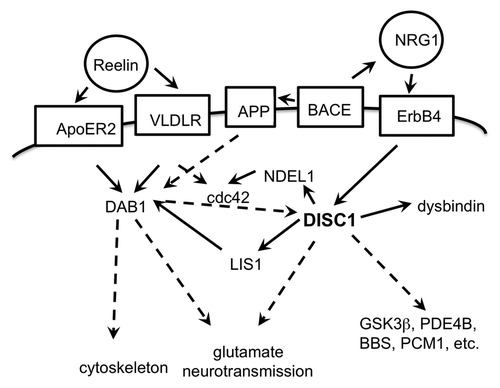
Figure 4. Schematic drawing of interactions of the DISC1 pathway. Extracellular proteins in circles, membrane proteins in boxes; direct functional connections in solid arrows, indirect functional connections (over several or unknown steps) in broken line arrows. The proteins depicted are an incomplete selection.
Similarly, the fact that increased insoluble DISC1 was present in brains of patients with different clinical diagnoses is consistent with the notion that disease-associated biology crosses clinical diagnostic boundaries.Citation22,Citation23 If diagnoses serve to define medical conditions amenable to similar therapeutic regimens, the DISC1opathy concept may group those CMD cases that might receive a future drug efficiently targeting the DISC1 pathway to correct maladaptive behavior.
The question remains how DISC1 aggregates influence cell physiology. DISC1 has been described as participating in many diverse biological functionsCitation64,Citation80 and it is currently difficult to say whether all of these functions or just a subset of them are essential for the final control over adaptive behavior. A role of DISC1 in corticogenesis has been demonstratedCitation43 consistent with the neurodevelopemental hypothesis of schizophrenia and this function was dependent on the DISC1-NDEL1 interactionCitation43,Citation81 making this interaction a prime candidate for disease-relevant experimental readouts. Other important DISC1 interactors are GSK-3β Citation82, PDE4,Citation83 BBS,Citation84,Citation85 but many more are known.Citation58,Citation63,Citation64
DISC1 aggregation leads to both loss and gain of protein interactions: DISC1 aggresomes lost interaction with NDEL1Citation68 and gained function by segregating dysbindinCitation69 in neuroblastoma cell models. It can be expected that cellular DISC1 aggregates would also co-segregate with many other proteins as is the case with other aggresomes.Citation86 These observations support the notion that abnormal protein-protein interactions from aggresomes and protein misassembly contribute to mental disease mechanisms and are a convergence point of disease pathways, as demonstrated, for example, for the DISC1 and dysbindin pathways.Citation69
Genetic studies associating DISC1 to behavioral phenotypes have included three coding polymorphisms that were identified in human DISC1: R/Q264, F/L607, and S/C704Citation42 with the C704 allele being associated with both major depressionCitation87 and schizophrenia.Citation53 DISC1 polymorphisms were also associated with reduced gray matter volume in the cingular cortex, decreased fractional anisotropy in healthy individualsCitation56 and seemed to influence cognitive decline during normal aging.Citation55 DISC1 allele C704 was also shown to lead to altered protein interactions.Citation88
Accordingly, a relation between DISC1 misassembly and behavioral phenotypes was supported by findings that the disease-associated polymorphism C704 was associated with an increased oligomerization propensity of a recombinant, C-terminal DISC1 fragment in a cell-free in vitro systemCitation89; these findings were recently corroborated by using full length recombinant DISC1.Citation90 Residue 704 lies in a C-terminal dimerization domain of DISC1 and it could be demonstrated that a well-concerted orchestration of the dimerization and oligomerization domains is required for orderly DISC1 assembly.Citation89
DISC1 interacts with other brain-disease relevant pathways, for example with the reelin pathway via LIS1Citation91 or NDEL1/cdc42Citation92 (see ). Furthermore, there is functional complementation of Alzheimer disease amyloid precursor protein (APP) deficiency by DISC1 acting downstream of DAB1.Citation81 Schizophrenia candidate gene neuregulin 1 is - like APP - the substrate of β-secretase (BACE) and regulates DISC1 expression in a BACE-dependent manner.Citation93 Thus there is evidence on the convergence of several schizophrenia candidate genes and APP as a gene relevant for a neurodegenerative disease into one pathway.
Disturbed proteostasis seems to affect some proteins more than others,Citation86 among them DISC1. The heterogeneity of CMD in general or within schizophrenia as one clinical diagnostic entity suggests the existence of multiple biological causes, and therefore it is likely that proteins other than and independent of DISC1 may also emerge as insoluble or misassembled in CMD.
To summarize, we propose that DISC1opathies are novel protein conformational disorders involved in chronic conditions of behavioral maladaptation and mental diseases. They fulfill two basic criteria of protein conformational disorders which are disease-associated protein deposition and cell-invasiveness of protein aggregates. Disease-associated polymorphisms influence DISC1 oligomerization propensity and the presence of DISC1 aggresomes changes DISC1's cellular interactions leading to loss and gain of molecular interactions. DISC1opathies are the first protein conformational disorders described in the realm of mental diseases emphasizing the importance of investigating protein aggregation in these disorders. Establishing subgroups of CMD with similar underlying biology - here DISC1-dependent disorders - is also a first step to a biological classification of CMD.
Acknowledgments
The author thanks Verian Bader, Philipp Ottis, Nick Bradshaw and Jesus Requena for technical assistance and discussions. This work was funded by the DFG (Ko 1679/3–1) and ERANET-NEURON (DISCover, BMBF 01EW1003).
References
- American Psychiatric Association APA. Diagnostic and Statistical Manual IV (DSM IV). Washington, D.C., 1994.
- an der Heiden W, Häfner H. The epidemiology of onset and course of schizophrenia. Eur Arch Psychiatry Clin Neurosci 2000; 250:292 - 303; http://dx.doi.org/10.1007/s004060070004; PMID: 11153964
- Kirkpatrick B, Fenton WS, Carpenter WT Jr., Marder SR. The NIMH-MATRICS consensus statement on negative symptoms. Schizophr Bull 2006; 32:214 - 9; http://dx.doi.org/10.1093/schbul/sbj053; PMID: 16481659
- Gur RE, Calkins ME, Gur RC, Horan WP, Nuechterlein KH, Seidman LJ, et al. The Consortium on the Genetics of Schizophrenia: neurocognitive endophenotypes. Schizophr Bull 2007; 33:49 - 68; http://dx.doi.org/10.1093/schbul/sbl055; PMID: 17101692
- Kraepelin E. Zur Diagnose und Prognose der Dementia praecox. Allg Z Psychiatr 1899; 56:246 - 63
- Bleuler E. Dementia praecox oder Gruppe der Schizophrenien. Leipzig: F. Deuticke, 1911.
- Carpenter WT Jr.. Conceptualizing schizophrenia through attenuated symptoms in the population. Am J Psychiatry 2010; 167:1013 - 6; http://dx.doi.org/10.1176/appi.ajp.2010.10060854; PMID: 20826849
- Kirkpatrick B, Buchanan RW, Ross DE, Carpenter WT Jr.. A separate disease within the syndrome of schizophrenia. Arch Gen Psychiatry 2001; 58:165 - 71; http://dx.doi.org/10.1001/archpsyc.58.2.165; PMID: 11177118
- Chua SE, McKenna PJ. A Sceptical View of the Neuropathology of Schizophrenia. In: Harrison PJ, Roberts G.W., ed. The Neuropathology of Schizophrenia. Oxford: Oxford University Press, 2000:291-337.
- Delay J, Deniker P, Harl J-M. Traitement des états d’excitation et d’agitation par une méthode médicamenteuse dérivée de l’hibernothérapie. Ann Med Psychol (Paris) 1952; 110:267 - 73; PMID: 13008201
- Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on the formation of 3-methoxytyramine and norepinephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) 1963; 20:140 - 4; http://dx.doi.org/10.1111/j.1600-0773.1963.tb01730.x; PMID: 14060771
- Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry 1991; 148:1474 - 86; PMID: 1681750
- Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr Bull 2009; 35:549 - 62; http://dx.doi.org/10.1093/schbul/sbp006; PMID: 19325164
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 1987; 44:660 - 9; http://dx.doi.org/10.1001/archpsyc.1987.01800190080012; PMID: 3606332
- Jaaro-Peled H, Hayashi-Takagi A, Seshadri S, Kamiya A, Brandon NJ, Sawa A. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci 2009; 32:485 - 95; http://dx.doi.org/10.1016/j.tins.2009.05.007; PMID: 19712980
- Kety SS. The significance of genetic factors in the etiology of schizophrenia: results from the national study of adoptees in Denmark. J Psychiatr Res 1987; 21:423 - 9; http://dx.doi.org/10.1016/0022-3956(87)90089-6; PMID: 3440955
- Gottesman II, Shields J. A critical review of recent adoption, twin, and family studies of schizophrenia: behavioral genetics perspectives. Schizophr Bull 1976; 2:360 - 401; PMID: 1034336
- Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet 2000; 9:1415 - 23; http://dx.doi.org/10.1093/hmg/9.9.1415; PMID: 10814723
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet 2002; 71:877 - 92; http://dx.doi.org/10.1086/342734; PMID: 12145742
- Schwab SG, Knapp M, Mondabon S, Hallmayer J, Borrmann-Hassenbach M, Albus M, et al. Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib-pair families with linkage and in an additional sample of triad families. Am J Hum Genet 2003; 72:185 - 90; http://dx.doi.org/10.1086/345463; PMID: 12474144
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 2005; 10:40 - 68; http://dx.doi.org/10.1038/sj.mp.4001558; PMID: 15263907
- Owen MJ, Craddock N, Jablensky A. The genetic deconstruction of psychosis. Schizophr Bull 2007; 33:905 - 11; http://dx.doi.org/10.1093/schbul/sbm053; PMID: 17551090
- Craddock N, Owen MJ. Rethinking psychosis: the disadvantages of a dichotomous classification now outweigh the advantages. World Psychiatry 2007; 6:84 - 91; PMID: 18235858
- Kellendonk C, Simpson EH, Kandel ER. Modeling cognitive endophenotypes of schizophrenia in mice. Trends Neurosci 2009; 32:347 - 58; http://dx.doi.org/10.1016/j.tins.2009.02.003; PMID: 19409625
- Arguello PA, Gogos JA. Modeling madness in mice: one piece at a time. Neuron 2006; 52:179 - 96; http://dx.doi.org/10.1016/j.neuron.2006.09.023; PMID: 17015235
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 2009; 78:959 - 91; http://dx.doi.org/10.1146/annurev.biochem.052308.114844; PMID: 19298183
- Prusiner SB. Shattuck lecture--neurodegenerative diseases and prions. N Engl J Med 2001; 344:1516 - 26; http://dx.doi.org/10.1056/NEJM200105173442006; PMID: 11357156
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science 2002; 296:1991 - 5; http://dx.doi.org/10.1126/science.1067122; PMID: 12065827
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 1985; 82:4245 - 9; http://dx.doi.org/10.1073/pnas.82.12.4245; PMID: 3159021
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991; 349:704 - 6; http://dx.doi.org/10.1038/349704a0; PMID: 1671712
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci USA 1998; 95:6469 - 73; http://dx.doi.org/10.1073/pnas.95.11.6469; PMID: 9600990
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997; 276:2045 - 7; http://dx.doi.org/10.1126/science.276.5321.2045; PMID: 9197268
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11:909 - 13; http://dx.doi.org/10.1038/ncb1901; PMID: 19503072
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 2009; 106:13010 - 5; http://dx.doi.org/10.1073/pnas.0903691106; PMID: 19651612
- Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 2009; 11:219 - 25; http://dx.doi.org/10.1038/ncb1830; PMID: 19151706
- Münch C, O’Brien J, Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci USA 2011; 108:3548 - 53; http://dx.doi.org/10.1073/pnas.1017275108; PMID: 21321227
- Grad LI, Guest WC, Yanai A, Pokrishevsky E, O’Neill MA, Gibbs E, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci USA 2011; 108:16398 - 403; http://dx.doi.org/10.1073/pnas.1102645108; PMID: 21930926
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 2008; 14:501 - 3; http://dx.doi.org/10.1038/nm1746; PMID: 18391963
- Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol 2010; 6:702 - 6; http://dx.doi.org/10.1038/nrneurol.2010.145; PMID: 21045796
- St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet 1990; 336:13 - 6; http://dx.doi.org/10.1016/0140-6736(90)91520-K; PMID: 1973210
- Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ. Schizophrenia and affective disorders--cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet 2001; 69:428 - 33; http://dx.doi.org/10.1086/321969; PMID: 11443544
- Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK. The DISC locus in psychiatric illness. Mol Psychiatry 2008; 13:36 - 64; http://dx.doi.org/10.1038/sj.mp.4002106; PMID: 17912248
- Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol 2005; 7:1167 - 78; http://dx.doi.org/10.1038/ncb1328; PMID: 16299498
- Pletnikov MV, Ayhan Y, Xu Y, Nikolskaia O, Ovanesov M, Huang H, et al. Enlargement of the lateral ventricles in mutant DISC1 transgenic mice. Mol Psychiatry 2008; 13:115; http://dx.doi.org/10.1038/sj.mp.4002144; PMID: 18202691
- Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci USA 2007; 104:14501 - 6; http://dx.doi.org/10.1073/pnas.0704774104; PMID: 17675407
- Shen S, Lang B, Nakamoto C, Zhang F, Pu J, Kuan SL, et al. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci 2008; 28:10893 - 904; http://dx.doi.org/10.1523/JNEUROSCI.3299-08.2008; PMID: 18945897
- Ekelund J, Hovatta I, Parker A, Paunio T, Varilo T, Martin R, et al. Chromosome 1 loci in Finnish schizophrenia families. Hum Mol Genet 2001; 10:1611 - 7; http://dx.doi.org/10.1093/hmg/10.15.1611; PMID: 11468279
- Hamshere ML, Bennett P, Williams N, Segurado R, Cardno A, Norton N, et al. Genomewide linkage scan in schizoaffective disorder: significant evidence for linkage at 1q42 close to DISC1, and suggestive evidence at 22q11 and 19p13. Arch Gen Psychiatry 2005; 62:1081 - 8; http://dx.doi.org/10.1001/archpsyc.62.10.1081; PMID: 16203953
- Hwu HG, Liu CM, Fann CS, Ou-Yang WC, Lee SF. Linkage of schizophrenia with chromosome 1q loci in Taiwanese families. Mol Psychiatry 2003; 8:445 - 52; http://dx.doi.org/10.1038/sj.mp.4001235; PMID: 12740602
- Thomson PA, Wray NR, Millar JK, Evans KL, Hellard SL, Condie A, et al. Association between the TRAX/DISC locus and both bipolar disorder and schizophrenia in the Scottish population. Mol Psychiatry 2005; 10:6 57 - 68; http://dx.doi.org/10.1038/sj.mp.4001669; PMID: 15838535
- Hennah W, Tuulio-Henriksson A, Paunio T, Ekelund J, Varilo T, Partonen T, et al. A haplotype within the DISC1 gene is associated with visual memory functions in families with a high density of schizophrenia. Mol Psychiatry 2005; 10:1097 - 103; http://dx.doi.org/10.1038/sj.mp.4001731; PMID: 16103888
- Cannon TD, Hennah W, van Erp TG, Thompson PM, Lonnqvist J, Huttunen M, et al. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry 2005; 62:1205 - 13; http://dx.doi.org/10.1001/archpsyc.62.11.1205; PMID: 16275808
- Qu M, Tang F, Yue W, Ruan Y, Lu T, Liu Z, et al. Positive association of the Disrupted-in-Schizophrenia-1 gene (DISC1) with schizophrenia in the Chinese Han population. Am J Med Genet B Neuropsychiatr Genet 2007; 144B:266 - 70; http://dx.doi.org/10.1002/ajmg.b.30322; PMID: 17286247
- Palo OM, Antila M, Silander K, Hennah W, Kilpinen H, Soronen P, et al. Association of distinct allelic haplotypes of DISC1 with psychotic and bipolar spectrum disorders and with underlying cognitive impairments. Hum Mol Genet 2007; 16:2517 - 28; http://dx.doi.org/10.1093/hmg/ddm207; PMID: 17673452
- Thomson PA, Harris SE, Starr JM, Whalley LJ, Porteous DJ, Deary IJ. Association between genotype at an exonic SNP in DISC1 and normal cognitive aging. Neurosci Lett 2005; 389:41 - 5; http://dx.doi.org/10.1016/j.neulet.2005.07.004; PMID: 16054297
- Hashimoto R, Numakawa T, Ohnishi T, Kumamaru E, Yagasaki Y, Ishimoto T, et al. Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum Mol Genet 2006; 15:3024 - 33; http://dx.doi.org/10.1093/hmg/ddl244; PMID: 16959794
- Kilpinen H, Ylisaukko-Oja T, Hennah W, Palo OM, Varilo T, Vanhala R, et al. Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry 2008; 13:187 - 96; http://dx.doi.org/10.1038/sj.mp.4002031; PMID: 17579608
- Bradshaw NJ, Porteous DJ. DISC1-binding proteins in neural development, signalling and schizophrenia. Neuropharmacology 2010; PMID: 21195721
- Korth C. DISCopathies: brain disorders related to DISC1 dysfunction. Rev Neurosci 2009; 20:321 - 30; http://dx.doi.org/10.1515/REVNEURO.2009.20.5-6.321; PMID: 20397618
- Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry 2008; 13:173 - 86; http://dx.doi.org/10.1038/sj.mp.4002079; PMID: 17848917
- Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 2007; 54:387 - 402; http://dx.doi.org/10.1016/j.neuron.2007.04.015; PMID: 17481393
- Kuroda K, Yamada S, Tanaka M, Iizuka M, Yano H, Mori D, et al. Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Hum Mol Genet 2011; 20:4666 - 83; http://dx.doi.org/10.1093/hmg/ddr400; PMID: 21903668
- Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, et al. Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry 2007; 12:74 - 86; http://dx.doi.org/10.1038/sj.mp.4001880; PMID: 17043677
- Soares CC, Carlyle BC, Bradshaw NJ, Porteous D. DISC1: Structure, Function, and Therapeutic Postential for Major Mental Illness. ACS Chem Neurosci 2011:dx.doi.org/10.1021/cn200062k.
- Kirkpatrick B, Xu L, Cascella N, Ozeki Y, Sawa A, Roberts RC. DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J Comp Neurol 2006; 497:436 - 50; http://dx.doi.org/10.1002/cne.21007; PMID: 16736468
- Brandon NJ, Sawa A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci 2011; 12:707 - 22; http://dx.doi.org/10.1038/nrn3120; PMID: 22095064
- Torrey EF, Webster M, Knable M, Johnston N, Yolken RH. The stanley foundation brain collection and neuropathology consortium. Schizophr Res 2000; 44:151 - 5; http://dx.doi.org/10.1016/S0920-9964(99)00192-9; PMID: 10913747
- Leliveld SR, Bader V, Hendriks P, Prikulis I, Sajnani G, Requena JR, et al. Insolubility of disrupted-in-schizophrenia 1 disrupts oligomer-dependent interactions with nuclear distribution element 1 and is associated with sporadic mental disease. J Neurosci 2008; 28:3839 - 45; http://dx.doi.org/10.1523/JNEUROSCI.5389-07.2008; PMID: 18400883
- Ottis P, Bader V, Trossbach S, Kretzschmar H, Michel M, Leliveld SR, et al. Convergence of two independent mental disease genes on the protein level: recruitment of dysbindin to cell-invasive disrupted-in-schizophrenia 1 aggresomes. Biol Psychiatry 2011; 70:604 - 10; http://dx.doi.org/10.1016/j.biopsych.2011.03.027; PMID: 21531389
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996; 87:493 - 506; http://dx.doi.org/10.1016/S0092-8674(00)81369-0; PMID: 8898202
- Blackwood DH, Muir WJ. Clinical phenotypes associated with DISC1, a candidate gene for schizophrenia. Neurotox Res 2004; 6:35 - 41; http://dx.doi.org/10.1007/BF03033294; PMID: 15184103
- Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 1995; 270:93 - 5; http://dx.doi.org/10.1126/science.270.5233.93; PMID: 7569955
- Greenwald J, Riek R. Biology of amyloid: structure, function, and regulation. Structure 2010; 18:1244 - 60; http://dx.doi.org/10.1016/j.str.2010.08.009; PMID: 20947013
- Braak H, Del Tredici K. Alzheimer’s pathogenesis: is there neuron-to-neuron propagation?. Acta Neuropathol 2011; 121:589 - 95; http://dx.doi.org/10.1007/s00401-011-0825-z; PMID: 21516512
- Soto C. Prion hypothesis: the end of the controversy?. Trends Biochem Sci 2011; 36:151 - 8; http://dx.doi.org/10.1016/j.tibs.2010.11.001; PMID: 21130657
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313:1781 - 4; http://dx.doi.org/10.1126/science.1131864; PMID: 16990547
- Morales R, Duran-Aniotz C, Castilla J, Estrada LD, Soto C. De novo induction of amyloid-β deposition in vivo. Mol Psychiatry 2011; http://dx.doi.org/10.1038/mp.2011.120; PMID: 21968933
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82:239 - 59; http://dx.doi.org/10.1007/BF00308809; PMID: 1759558
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197 - 211; http://dx.doi.org/10.1016/S0197-4580(02)00065-9; PMID: 12498954
- Brandon NJ, Millar JK, Korth C, Sive H, Singh KK, Sawa A. Understanding the role of DISC1 in psychiatric disease and during normal development. J Neurosci 2009; 29:12768 - 75; http://dx.doi.org/10.1523/JNEUROSCI.3355-09.2009; PMID: 19828788
- Young-Pearse TL, Suth S, Luth ES, Sawa A, Selkoe DJ. Biochemical and functional interaction of disrupted-in-schizophrenia 1 and amyloid precursor protein regulates neuronal migration during mammalian cortical development. J Neurosci 2010; 30:10431 - 40; http://dx.doi.org/10.1523/JNEUROSCI.1445-10.2010; PMID: 20685985
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 2009; 136:1017 - 31; http://dx.doi.org/10.1016/j.cell.2008.12.044; PMID: 19303846
- Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 2005; 310:1187 - 91; http://dx.doi.org/10.1126/science.1112915; PMID: 16293762
- Kamiya A, Tan PL, Kubo K, Engelhard C, Ishizuka K, Kubo A, et al. Recruitment of PCM1 to the centrosome by the cooperative action of DISC1 and BBS4: a candidate for psychiatric illnesses. Arch Gen Psychiatry 2008; 65:996 - 1006; http://dx.doi.org/10.1001/archpsyc.65.9.996; PMID: 18762586
- Ishizuka K, Kamiya A, Oh EC, Kanki H, Seshadri S, Robinson JF, et al. DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature 2011; 473:92 - 6; http://dx.doi.org/10.1038/nature09859; PMID: 21471969
- Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011; 144:67 - 78; http://dx.doi.org/10.1016/j.cell.2010.11.050; PMID: 21215370
- Di Giorgio A, Blasi G, Sambataro F, Rampino A, Papazacharias A, Gambi F, et al. Association of the SerCys DISC1 polymorphism with human hippocampal formation gray matter and function during memory encoding. Eur J Neurosci 2008; 28:2129 - 36; http://dx.doi.org/10.1111/j.1460-9568.2008.06482.x; PMID: 19046394
- Kamiya A, Tomoda T, Chang J, Takaki M, Zhan C, Morita M, et al. DISC1-NDEL1/NUDEL protein interaction, an essential component for neurite outgrowth, is modulated by genetic variations of DISC1. Hum Mol Genet 2006; 15:3313 - 23; http://dx.doi.org/10.1093/hmg/ddl407; PMID: 17035248
- Leliveld SR, Hendriks P, Michel M, Sajnani G, Bader V, Trossbach S, et al. Oligomer assembly of the C-terminal DISC1 domain (640-854) is controlled by self-association motifs and disease-associated polymorphism S704C. Biochemistry 2009; 48:7746 - 55; http://dx.doi.org/10.1021/bi900901e; PMID: 19583211
- Narayanan S, Arthanari H, Wolfe MS, Wagner G. Molecular characterization of disrupted in schizophrenia-1 risk variant S704C reveals the formation of altered oligomeric assembly. J Biol Chem 2011; 286:44266 - 76; http://dx.doi.org/10.1074/jbc.M111.271593; PMID: 21998303
- Brandon NJ, Handford EJ, Schurov I, Rain JC, Pelling M, Duran-Jimeniz B, et al. Disrupted in Schizophrenia 1 and Nudel form a neurodevelopmentally regulated protein complex: implications for schizophrenia and other major neurological disorders. Mol Cell Neurosci 2004; 25:42 - 55; http://dx.doi.org/10.1016/j.mcn.2003.09.009; PMID: 14962739
- Shen Y, Li N, Wu S, Zhou Y, Shan Y, Zhang Q, et al. Nudel binds Cdc42GAP to modulate Cdc42 activity at the leading edge of migrating cells. Dev Cell 2008; 14:342 - 53; http://dx.doi.org/10.1016/j.devcel.2008.01.001; PMID: 18331715
- Seshadri S, Kamiya A, Yokota Y, Prikulis I, Kano SI, Hayashi-Takagi A, et al. Disrupted-in-Schizphrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proc Natl Acad Sci USA 2010; 107:5622 - 7; http://dx.doi.org/10.1073/pnas.0909284107; PMID: 20212127