Abstract
The yeast Saccharomyces cerevisiae is a tractable model organism in which both to explore the molecular mechanisms underlying the generation of disease-associated protein misfolding and to map the cellular responses to potentially toxic misfolded proteins. Specific targets have included proteins which in certain disease states form amyloids and lead to neurodegeneration. Such studies are greatly facilitated by the extensive ‘toolbox’ available to the yeast researcher that provides a range of cell engineering options. Consequently, a number of assays at the cell and molecular level have been set up to report on specific protein misfolding events associated with endogenous or heterologous proteins. One major target is the mammalian prion protein PrP because we know little about what specific sequence and/or structural feature(s) of PrP are important for its conversion to the infectious prion form, PrPSc. Here, using a study of the expression in yeast of fusion proteins comprising the yeast prion protein Sup35 fused to various regions of mouse PrP protein, we show how PrP sequences can direct the formation of non-transmissible amyloids and focus in particular on the role of the mouse octarepeat region. Through this study we illustrate the benefits and limitations of yeast-based models for protein misfolding disorders.
Introduction
The yeast Saccharomyces cerevisiae has many attributes that make it an ideal ‘model organism’ in which to explore the molecular basis of key cellular mechanisms that operate across eukaryotic species. For example, through the application of an extensive molecular toolbox and publically available genetic resources, we have learnt much about how nascent polypeptide chains fold in the cytoplasm and in the endoplasmic reticulum (ER) and the role molecular chaperones play in ensuring that native and functional protein folds are quickly reached. The components of this toolbox allow the experimenter to rationally engineer gene expression, to monitor protein localization and to assess the impact of gene modifications on host cell phenotype through relatively straightforward manipulations.Citation1 Perhaps not surprisingly therefore, yeast has also become a favored model organism in which to explore human disease mechanisms and to screen for potential therapeutics. One particular class where recent progress has been made is with those diseases which are linked to protein misfolding and aggregation.
The last decade has seen the development of a variety of yeast-based models of human protein misfolding-linked disorders.Citation2,Citation3 These include models for Alzheimer disease (AD),Citation4,Citation5Parkinson disease (PD)Citation6 and Huntington’s disease (HD)Citation7-Citation9 all of which are associated with the misfolding and aggregation of specific proteins into amyloid structures which may or may not be linked to disease pathology. The Transmissible Spongiform Encephalopathies (TSEs) are a unique group of amyloid diseases which, unlike AD, PD and HD, are transmissible within—and in some cases between—closely related species. TSEs are often referred to as ‘prion diseases’ because of their common pathogenic mechanism that is associated with the conformational conversion of the normal soluble form of the PrPC protein into an abnormal, protease-resistant and insoluble prion form called PrPSc that is largely present in the form of high molecular weight amyloid fibrils.Citation10
The discovery that prions also exist in S. cerevisiaeCitation11 raised the possibility of dissecting the unique process by which these infectious proteins are generated and propagated, using a highly tractable and above all, safe in vivo model. Although yeast has no ortholog of the PrP protein, nevertheless there are at least eight - possibly more - proteins that can undergo a heritable change in protein conformation leading to detectable changes in cell phenotype (for recent review see ref. Citation12). In contrast to the prion form of mammalian PrP however, most yeast prions appear to be benign, although they can be associated with cellular toxicity in some contexts.Citation13-Citation15 Screening for compounds that inhibit the propagation of yeast prions has already successfully uncovered new drugs that are active against mammalian prions e.g., 6-aminophenanthridine (6AP) and Guanabenz (GA).Citation16
In this article we first briefly review the key structural features of mammalian PrP and yeast prions. This is followed by an overview of what we and others have learnt about structure-function relationships for mammalian PrP using S. cerevisiae as an experimental tool, through a combination of heterologous expression and phenotypic analysis. In so doing, we illustrate some of the approaches, benefits and limitations of such studies through an analysis of the behavior of mouse PrP fusion proteins in S. cerevisiae.
Structural Features of Mammalian PrP
Mammalian PrP is a glycoprotein, widely expressed in neuronal cells and well conserved among mammals; for example, human and mouse PrP show 88% amino acid identity. However, PrP represents a challenge as a heterologous expression target in yeast primarily because it is naturally subjected to a range of post-translational modifications (). The PrP protein is cell surface-associated and is attached to the plasma membrane by a C-terminal, glycosyl-phosphatidyl-inositol (GPI) anchor. The first 22 residues of PrP define the hydrophobic signal sequence that is cleaved during translocation of the protein into the ER. Once in the ER an intra-chain disulphide bond is formed between Cys178 and Cys213 and two N-linked oligosaccharide chains are added to residues 180 and 196 respectively. The C-terminal 23 amino acid residues are cleaved prior to the addition of the GPI anchor which is attached to Ser231 and hence the mature PrP protein that leaves the ER en route to its final destination is 209 amino acids in length and encompasses residues 23–231 (numbering based on the mouse PrP sequence).
Figure 1. Important sequence and structural features of mouse PrP and yeast Sup35 prion proteins. (A) The mouse PrP pro-protein has 253 amino-acids. Features are an N-terminal ER signal, an intra-chain disulphide bond between Cys178 and Cys213, two glycosylation sites at position 180 and 196 and a GPI anchoring signal. The mature protein is 209 amino acids long (residues 23–231). The location of three α−helices (α1–3; residues, 144–153, 172–194 and 200–224 respectively) and two β−sheets (β1–2; residues 128–131 and 161–164) are indicated. Amino acid residues 50–90 contain 5 repeats of a glycine-rich octapeptide. Amino acid residues 105–125 encompass the so-called “toxic peptide.” (B) The Saccharomyces cerevisiae Sup35 protein consists of three distinct regions: an N-terminal prion-forming domain (PrD) that contains a Q/N rich tract (QNR) and a consensus oligopeptide repeat sequence (OPR); a middle, highly charged region (M); and the functional C-terminal domain (C).
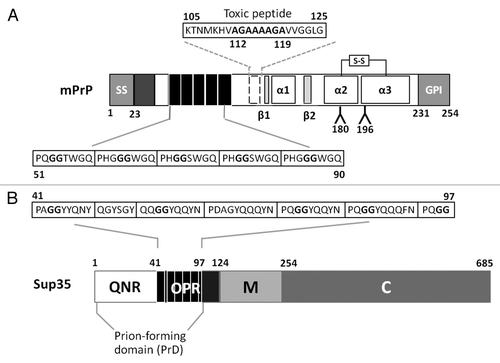
One of the most intriguing sequence features of mammalian PrP is an array of five near identical octapeptide sequences (consensus: P(Q/H)GGG(G/-)WGQ) in the N-terminal region of PrP (residues 50–90; ). These are referred to as the octarepeatsCitation17 and each octarepeat is able to bind divalent ions such as copperCitation18 with the His residues in the octarepeats acting as the primary anchor point.Citation18,Citation19 PrP also contains a highly hydrophobic transmembrane domain 1 (TM1) between residues 109–134, a region that encompasses a “toxic peptide” sequence (residues 105–125) that is highly toxic to neuronal cells.Citation20,Citation21 Within the ‘toxic peptide’ sequence lies in a hydrophobic palindrome (AGAAAAGA) extending from residues 112–119 that is associated with enhanced amyloidogenesisCitation22,Citation23and is required for the PrPC to PrPSc conversion.Citation24-Citation26
There is considerable interest in how the various structural properties of PrP contribute to de novo prion generation, propagation and subsequent transmission of PrPSc. Analysis of the primary structure of PrP has yielded important information on why certain point mutations and deletions of PrP affect the course of prion infection in mammalian models.Citation27,Citation28However, secondary and especially the tertiary structural features of PrP are of equal importance because ultimately the transition from PrPC to PrPSc occurs via one or more template-driven, conformational conversion events.Citation29
Some detailed structural information is available for mouse,Citation30,Citation31 Syrian hamster,Citation32,Citation33 and human PrPC 34, but little information is available for the corresponding PrPSc prion forms. What has emerged from these structural studies is that mammalian PrP proteins have a conserved general fold characterized by the presence of a disordered 100 residue N-terminal stretch attached to a globular C-terminal domain encompassing residues 121–231.Citation35 Within the disordered N-terminal domain, the region between residues 23 and 89 is particularly susceptible to proteinase K cleavage and remains so even in PrPSc polymers.Citation36 The C-terminal domain has two stranded anti-parallel β sheets (β1 and β2) and three α helices (α1, α2 and α3) plus the disulphide bridge which links α2 and α3Citation35(). Notably, a hydrophobic network exists between α2 and α3 and mutations in this region (e.g., Val180Ile, Phe198Ser, Val203Ile and Val210Ile in human) have been linked to familial forms of TSEs.Citation37-Citation41 As expected, these mutations have a significant effect on the dynamics and stability of PrP.Citation28
While the structures of a range of partial or full-length PrPs generally superpose,Citation34,Citation35 there is a small shift in the α2 and α3 helices when comparing the full-length protein and truncated constructs suggesting that the N-terminal domain has a small stabilizing effect on the α2 and α3 helices. In addition, comparison of the structure of the 90–231 and 29–231 fragments of Syrian hamster (Sha) PrPCitation32 revealed a slight structural divergence, implying that the octarepeat region may play a role in the general protein fold. Recent findings using trypsin-digested PrPSc also support the hypothesis that this structural divergence may be important in the context of conformational transition because the octarepeat region exposes multiple epitopes in PrPC, but none in PrPSc.Citation42
Structural Features of Yeast Prions
Fungal prion proteins show very limited amino acid sequence similarity between them or in comparison to mammalian PrP, but nevertheless are subject to the same underlying processes that drive de novo generation and propagation of the prion form. Since their discovery in S. cerevisiae,Citation11 the yeast prion proteins that give rise to the [URE3] and [PSI+] phenotypes, namely Ure2 and Sup35 respectively, have been subjected to extensive analysis in an attempt to identify the critical sequence and/or structural features that are responsible for this remarkable behavior.
Sup35 is an essential translation termination proteinCitation43,Citation44and its propensity to form transmissible prion aggregates has been linked to a specific structural feature called the prion forming domain (PrD). The Sup35-PrD encompasses the N-terminal region between residues 1 and 97Citation45-Citation47which in turn consists of two structurally and functionally distinct regions: a Gln/Asn rich region (QNR, residues 1–40) and a series of oligopeptide repeats (OPR, residues 41–97) (). The QNR region forms the core of the amyloid fibers of Sup35 while the OPR is essential for continued propagation of the [PSI+] state.Citation45,Citation48-Citation50 The oligopeptide repeat in the Sup35-OPR resembles the octarepeat of mammalian PrP including the conservation of a flexible Gly-Gly motif. However, the Sup35 oligopeptide repeats lack the His and Trp residues which mediate copper binding to the octarepeat.
The presence of oligopeptide repeats in the Sup35-PrD cannot necessarily be taken as a diagnostic feature of a yeast prion whereas the presence of a Gln/Asn-rich region (like the Sup35-QNR) generally can. Yet such Gln/Asn-rich regions are not essential for a protein to take up and propagate a prion state in yeast. For example, the HET-s prion protein from Podospora anserina (for a recent review, see ref. Citation51) has neither a predominance of Asn nor Gln residues in the C-terminal region of the protein essential for its prion properties. Furthermore, a HET-s-GFP fusion protein can be stably propagated in a prion form in S. cerevisiae.Citation52 What primary sequence features confer prion-like properties to a protein is not therefore fully established, and the critical feature could even be a secondary structural feature such as conformational flexibility and/or disorder.
Assaying Heterologous Prions in Yeast
The Sup35-PrD is absolutely required for the formation and propagation of the [PSI+] prion, but is not required for the essential Sup35 function in translation termination; this activity resides in the C region (residues 254–685).Citation47,Citation53Linking the N and C regions of Sup35 is a highly charged M region (). This distinct architecture of Sup35 has allowed the development of an effective assay that can be used to assess whether or not a particular homologous (e.g., refs. Citation54 and Citation55) or heterologous (e.g., ref. Citation56) protein sequences can confer prion-like properties to Sup35MC in yeast. The target sequence is fused in frame with the MC region of Sup35 and the ability of the resulting fusion protein to switch to a stable, heritable prion form can then be readily assessed by expressing the protein in a strain carrying the suppressible ade1–14 nonsense allele. Expression of the ade1–14 allele leads to the accumulation of a red pigment, a by-product of an associated defect in adenine biosynthesis and this allele can be used to monitor [PSI+] because the associated conformational switch generates a non-functional form of Sup35 resulting in a translation termination defect. This is turn leads to suppression of the ade1–14 allele i.e., nonsense suppression. Consequently, [PSI+] ade1–14 cells form white Ade+ colonies rather than the red Ade- colonies observed when Sup35 is fully functional, as in a [psi-] ade1–14 strain (). Intermediate levels of nonsense suppression generate a range of different colony color phenotypes ranging from pink to pink/red.
Figure 2. The expression of various Sup35/Sup35-PrP proteins in Saccharomyces cerevisiae. (A) The [PSI+] ade1–14 strain LJ14 gives rise to white colonies on rich growth medium (1/4 YEPD) indicative of nonsense suppression. Growing these cells on the same medium but supplemented with 4 mM guanidine hydrochloride (GdnHCl) results in the loss of the [PSI+] giving a non-suppressed red phenotype. Serial 1::5 dilutions of a cell suspension are shown. (B) The deletion of either the full N region (∆N, lacking amino-acid residues 5–112), the QN-rich region (∆Q, lacking amino-acid residues 5–40) or the OPR region (∆O, lacking amino-acid residues 41–97) generates a form of Sup35 that is functional, but no longer able to switch to a stable [PSI+] prion form. In each construct the N-terminal residues of Sup35 are retained (see text for details). (C) The phenotypic consequences of expressing various Sup35-mouse PrP fusion proteins in the LJ14 [PSI+] strain. See text for details of constructs shown. PrP sequences are shown in green. OCT, the mouse PrP octarepeat region; QNR, the Sup35 QN-rich region; OPR, the Sup35 oligopeptide repeat region.
![Figure 2. The expression of various Sup35/Sup35-PrP proteins in Saccharomyces cerevisiae. (A) The [PSI+] ade1–14 strain LJ14 gives rise to white colonies on rich growth medium (1/4 YEPD) indicative of nonsense suppression. Growing these cells on the same medium but supplemented with 4 mM guanidine hydrochloride (GdnHCl) results in the loss of the [PSI+] giving a non-suppressed red phenotype. Serial 1::5 dilutions of a cell suspension are shown. (B) The deletion of either the full N region (∆N, lacking amino-acid residues 5–112), the QN-rich region (∆Q, lacking amino-acid residues 5–40) or the OPR region (∆O, lacking amino-acid residues 41–97) generates a form of Sup35 that is functional, but no longer able to switch to a stable [PSI+] prion form. In each construct the N-terminal residues of Sup35 are retained (see text for details). (C) The phenotypic consequences of expressing various Sup35-mouse PrP fusion proteins in the LJ14 [PSI+] strain. See text for details of constructs shown. PrP sequences are shown in green. OCT, the mouse PrP octarepeat region; QNR, the Sup35 QN-rich region; OPR, the Sup35 oligopeptide repeat region.](/cms/asset/7b537650-52ed-4ba8-ac30-2e8765d9a10c/kprn_a_10919214_f0002.gif)
When fused to Sup35MC, only a small proportion of Gln/Asn-rich polypeptides can undergo the expected conformational change and assembly into heritable amyloids.Citation54-Citation56A recent systematic survey of the Sup35-QNR revealed that a high density of Asn residues promotes assembly of benign self-templating amyloids whereas an abundance of Gln residues promotes formation of toxic non-amyloid conformers.Citation57 Importantly, while some Gln/Asn-rich domains can facilitate the formation of protein aggregates as defined by the Sup35MC-based assay, that does not always equate to the formation of a bona fide prion state when either the fusion or the native form of the protein are examined.
Only a relatively few studies have used yeast to explore the structural features of PrP that are important for its conversion to PrPSc. Early studies showed that replacing the natural N-terminal hydrophobic signal sequence of PrP with a yeast signal sequence resulted in PrP being targeted to and post-translationally modified in the ER although it remained in its non-infectious PrPC form.Citation58 Nevertheless PrP can adopt the PrPSc-like conformation and retain its ability to propagate this form when expressed in the yeast cytosolCitation36,Citation59even though PrPSc formation does not take place in the mammalian cytoplasm. Furthermore, the environment of the yeast cytoplasm can be used to explore the nature and identity of proteins that stably interact with PrP. For example, the yeast two-hybrid system has been used to identify Sho, a putative PrP-interacting protein.Citation60
Most studies on PrP expressed in yeast have focused on exploring the role of the PrP octarepeat through studying the effects of deleting or inserting one or more octarepeats. Such studies are important because expansion of the numbers of octarepeat beyond the usual five has been found in certain familial prion diseases in humansCitation61 and the number of repeats appears to determine the type of cerebellar deposits.Citation62 Although the octarepeats are not essential for PrPSc formation per se, the expression in mice of a PrP transgene lacking the octarepeat region rendered the rodents less susceptible to scrapie infectivity.Citation63
Two independent studies using yeast as a model found that increasing the number of PrP repeats in a PrP-Sup35 chimaeric protein increased the spontaneous appearance of the [PSI+] phenotype in vivo and accelerated amyloid formation in vitro.Citation64,Citation65 This suggested that the PrP octapeptide repeats are more amyloid prone than Sup35 repeats and may also facilitate the generation of a wider range of propagatable structures through an increased conformational flexibility in the fusion proteins with expanded numbers of repeats. However, mutations associated with familial forms of the TSEs span the entire length of PrP and other primary or secondary structural features are likely to be important for protein conformation including the hydrophobic region (residues 112–119; ) that is essential for the conversion of PrPc to PrPSc 26 since deleting this region reduces the formation of PrPSc.Citation66
Yet in spite of the attractiveness of yeast as a model organism in which to explore structure-function relationships in PrP conversion, there are a number of pitfalls which the experimenter needs to be alert to. To illustrate this we describe some experiments that were designed to establish the importance of the PrP repeats in protein aggregation. The strategy we used was to replace either part or the whole of the Sup35-PrD domain of Sup35 with various regions of mouse PrP (mPrP) including a precise replacement of the Sup35 OPR region with mPrP octarepeats. These Sup35-PrP fusions still produce functional Sup35 and hence could be introduced into a haploid [PSI+] [PIN+] ade1–14 sup35::kanMX4 strain in place of a plasmid-borne wild type copy of SUP35; a process referred to as ‘plasmid shuffling’.Citation67 The resulting strains were then tested phenotypically using the suppression of ade1–14 to report aggregation-induced reduction in the levels of functional, soluble Sup35, and then biochemically to evaluate the levels and type of Sup35-PrP aggregates formed.
Assaying the Phenotypic Impact of Sup35-PrP Fusions Expressed in Yeast
To evaluate the ability of various N-terminally truncated Sup35 proteins and Sup35-PrP fusion proteins to function in translation termination we used the widely studied S. cerevisiae strain 74-D694 (ade1–14 trp1–289 his3Δ-200 ura3–52 leu2–3,112 [PSI+] [PIN+]).Citation68 For the analysis of the phenotype conferred by expressing various Sup35-PrP fusions LJ14, a derivative of 74-D694 containing a sup35::loxP-kanMX-loxP disruption, was used.Citation5 Viability of LJ14 was maintained by the presence of the SUP35 gene on a centromeric URA3-based plasmid (plasmid pYK810Citation69). The various Sup35-PrP fusions were based on plasmid pUKC1620, a single copy HIS3-based plasmid carrying the ScSUP35 gene under the control of the SUP35 promoter. The individual pUKC1620-based plasmids were transformed into LJ14 [pYK810] and, following non-selective growth, transformants that had lost the pYK810 plasmid were selected for by their ability to grow on 5-FOA-containing medium.
The phenotypic readout used was the color of the resulting colonies both before (i.e., expressing both wild type and modified Sup35) and after counter selection on 5-FOA (i.e., expressing just the modified Sup35). The starting strain LJ14 [PSI+] always gave a stable white Ade+ phenotype and this phenotype reverted to red Ade- when the cells were grown in the presence of 4mM guanidine hydrochloride (GdnHCl) (). This ‘curing’ by GdnHCl confirmed that the ade1–14 suppression phenotype was due to [PSI+] rather than a non-epigenetic cause since GdnHCl blocks the propagation of the [PSI+] prion by inhibiting the disaggregase function of the molecular chaperone Hsp104.Citation70-Citation72 A failure to eliminate the nonsense suppression phenotype with GdnHCl would indicate that there was a defect in translation termination either as a consequence of the inactivation of Sup35 through Hsp104-independent, non-heritable protein aggregation or because only low levels of soluble functional Sup35 were being produced.
Construction of Sup35-PrP Chimaeras
Previous studies have shown that the N-terminal region of Sup35 spanning residues 1–123 are essential for the propagation of [PSI+]Citation47,Citation53and subsequent deletion studies narrowed this down to residues 1–97 that encompass the QNR plus OPR regions.Citation46 All of the constructs used in our study retained the N-terminal 5 residues of Sup35 to ensure that the N-terminus was correctly N-acetylated as a preventative measure against protein degradation. In addition to creating derivatives lacking the region 5–112 (Sup35ΔN), we also created versions lacking residues 5–40 (Sup35ΔQ) and 41–97 (Sup35ΔO). Expressing these truncated versions of Sup35 resulted in the expected [psi-] phenotypes post-5-FOA selection () i.e., neither the QNR nor OPR regions alone can mediate [PSI+] prion propagation. The failure of the Sup35ΔQ protein to establish and propagate [PSI+] is most likely through a failure of Sup35 to interact with the endogenous [PSI+] prion forms of Sup35 since the QNR region mediates Sup35:Sup35 interactions.Citation48-Citation50 The failure of the Sup35ΔO derivative is because the OPR region is essential for continued propagation of [PSI+].Citation45
Three different Sup35-PrP in-frame fusions were constructed using an overlapping PCR strategyCitation73 starting with a full-length mPrP cDNA (kindly provided by Professor Adriano Agguzzi, University Hospital of Zurich) and the single copy, CEN-based plasmid pUKC1620 as templates. The full-length wild type SUP35 coding sequence in pUKC1620 also carries the 5′ (promoter) and 3′ (terminator) regions of this gene ensuring that the fusion proteins were all expressed under the control of the native SUP35 promoter. Such use of the homologous promoter avoids any phenotypic artifacts especially due to overexpression of the Sup35-PrP fusion protein.
The fusion proteins Sup35ΔNPrP152 and Sup35ΔQPrP152 were constructed to establish whether any sequences C-terminal to the mPrP octarepeat region could functionally replace either the Sup35 QNR and/or the N region in [PSI+] propagation. Sequences C-terminal to the octarepeat region are known to contribute to the conversion of human PrPC to PrPSc and include amino acid residues 93–112 and 113–120 spanning the hydrophobic region (). The region of mPrP included in our fusions spanned residues 88–240. In both cases we observed an unstable nonsense suppression phenotype that was retained when the cells expressing either plasmid were grown on 4mM GdnHCl (). A third construct (Sup35ΔNPrP191) in which we replaced the N region of Sup35 (5–112) with an extended mPrP sequence (residues 49–240) that included the octarepeat-containing region, showed a much more stable Ade+ suppression phenotype, but again this was not eliminated by growth on 4mM GdnHCl (). The failure to eliminate the suppression phenotype by growth on GdnHCl strongly suggested that the phenotype was not dependent upon continued Hsp104 function
A fourth construct analyzed (Sup35mOPR) was one which contained a precise replacement of the Sup35 OPR (residues 41–97) with the mPrP octarepeat region with all other Sup35 sequences, including the QNR, being retained. This fusion protein did not confer a nonsense suppression phenotype in the LJ14 strain, although a previous study in which an analogous construct was studied using a similar strategy, was reported to give a weak suppression phenotype.Citation65
Relating Phenotype to Levels and Aggregation State of Sup35-PrP Fusion Proteins
As a reduced level of functional Sup35 can lead to a nonsense suppression phenotype independent of the [PSI+] status of the cellCitation74 we next established whether the suppression phenotypes observed could simply reflect reduced steady-state levels of the Sup35-PrP fusion protein. Quantitative western blot analysis revealed that the steady-state levels of the Sup35ΔQPrP152 protein were similar to wild type Sup35 levels although the levels Sup35ΔNPrP152 were only approximately 40% of wild type (). In contrast, the Sup35ΔNPrP191 protein was less than 20% of the wild type protein level indicating that at least in part the nonsense suppression phenotype seen could reflect a significant reduction in the steady-state levels of the Sup35ΔNPrP191 fusion protein and this reduced levels of functional Sup35 could be further exacerbated if a fraction of the fusion protein was in an aggregated form. Deletion of the complete N region of Sup35 (5–112) or just the QNR region (5–40) resulted in an increase in the steady-state levels of the truncated protein compared with wild type Sup35 levels suggesting that the reduced protein levels were a consequence of the presence of the PrP octarepeat region in the fusion proteins. The observed differences in expression levels were not dependent on extraction methods used or due to vacuolar proteolysis (L. Jossé, unpublished data).
Figure 3. Expression and sub-cellular fractionation of various Sup35-PrP fusion proteins. (A) Western Blot analysis showing the level of expression of truncated forms of Sup35 and Sup35-PrP fusions as indicated. The equivalent amounts of protein were loaded and an anti-yeast Sup35 polyclonal antibody used to detect Sup35/Sup35-PrP fusions. The membranes were also probed with an anti-yeast phoshphoglycerate kinase (PGK) polyclonal antibody as a loading control. (B) Sub-cellular fractionation assay using high speed (100,000 xg) centrifugation to fractionate total cell lysates (T) into a soluble (S) and pelleted (high molecular weight) fraction (P). Each fraction was assayed in parallel by SDS-PAGE and western blot analysis with an anti-yeast Sup35 polyclonal antibody.
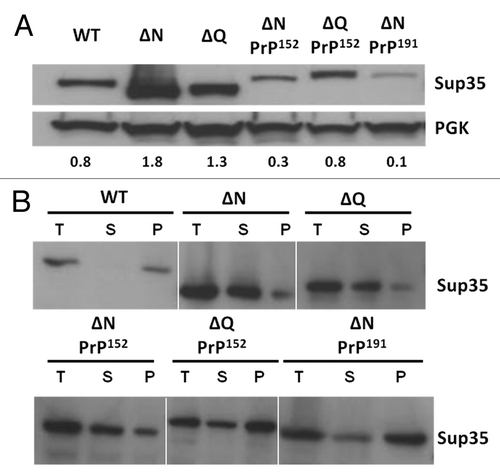
To establish whether any proportion of the Sup35-PrP fusion proteins were in an aggregated state, we used differential centrifugation to generate a high molecular weight pellet fraction and a soluble fraction. In a [PSI+] cell the majority (> 90%) of the Sup35 is typically detected in the pellet fraction (see refs. Citation75 and Citation76; see ). Deletion of either the QNR or the entire N region resulted in a shift of the bulk of the Sup35 to the soluble fraction consistent with the non-suppressed phenotype. For each of the three Sup35-PrP fusion proteins examined, a significant proportion of the protein remained in the pellet fraction indicating that each of the proteins had formed high molecular weight aggregates. The highest proportion of pelletable protein was seen with the Sup35ΔNPrP191 fusion protein consistent with the stronger suppression phenotype generated compared with the other fusions studied.
Yeast is able to produce both heritable and non-heritable amyloids that are differentiated primarily by their ability to be efficiently fragmented by the Hsp104 chaperone and its co-chaperones.Citation77,Citation78 In the case of Sup35, both types of aggregate can lead to a nonsense suppression phenotype as was observed when the N region of Sup35 was replaced either by polyglutamine-rich sequencesCitation78 or with the amyloid-prone Αβ42 region.Citation5 To establish whether a specific protein aggregate is amyloid-like, one assay that can be applied is to establish whether or not the aggregates are SDS resistant. Most if not all yeast prions form SDS-resistant polymers and their presence in vivo can be readily established using the technique of semi-denaturing detergent agarose gel electrophoresis (SDD-AGE).Citation54,Citation79
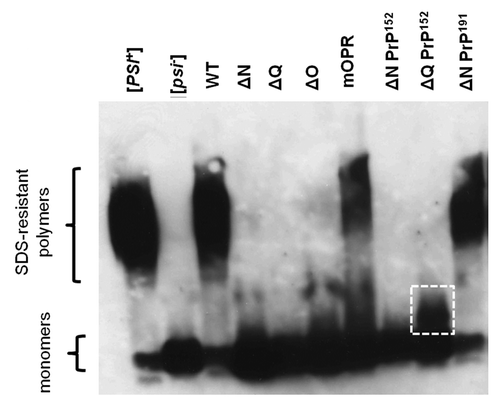
Analysis of the Sup35-based protein aggregates formed in the various strains that were phenotypically analyzed () revealed that only the Sup35ΔNPrP191 and the Sup35mOPR fusion proteins formed SDS-resistant polymers of Sup35 that could be detected by SDD-AGE, using a polyclonal antibody raised against the Sup35 MC domain (). These two fusion proteins both contained the mouse octarepeats, yet gave very different phenotypes; the Sup35ΔNPrP191 fusion gave a strong suppression phenotype while the Sup35mOPR gave no detectable suppression of the ade1–14 allele. This difference in phenotype could be explained by the relative amounts of monomer vs SDS-resistant polymer since the Sup35mOPR protein yielded a much higher level of functional monomer than the Sup35ΔNPrP191 fusion (). Interestingly, the Sup35ΔQPrP152 construct which retained the Sup35-OPR sequence did not form high molecular weight SDS-resistant aggregates although a distinct, lower molecular weight aggregate not seen in the [psi-] control, was detected ().
Figure 4. Analysis of SDS-resistant Sup35 aggregates by SDD-AGE. Western blot analysis of SDS-resistant protein aggregates containing Sup35 following fractionation of total cell extracts by electrophoresis in 1% agarose + 0.1% SDS. The locations of the high molecular weight SDS-resistant polymers and the monomeric form of the proteins are indicted. A putative, lower molecular weight SDS-resistant polymer in the ∆QPrP152 sample is indicated by the white square.
The Dependency on Hsp104 of Formation of SDS-Resistant Aggregates
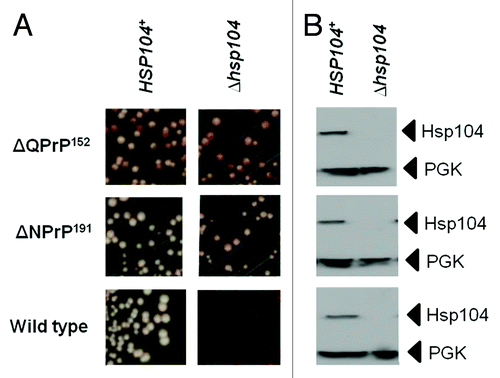
The use of GdnHCl-mediated elimination of the nonsense suppression phenotype is widely used to indicate whether the [PSI+] prion is responsible for the termination defect since continued propagation of the prion requires functional Hsp104.Citation68 However, in some strains GdnHCl can generate an antisuppression phenotype independent of the presence of [PSI+].Citation80 Consequently it is important that GdnHCl curing studies are backed up by confirmation that disrupting the HSP104 gene gives the same phenotypic effect. We generated such ∆hsp104::kanMX knockout mutants for three of the Sup35-PrP-expressing strains under test; the wild type LJ14 [PSI+] parent and the two derivatives expressing either the Sup35ΔQPrP152 or the Sup35ΔNPrP191 fusion proteins (). As expected, the wild type [PSI+] strain gave a red Ade- phenotype in the ∆hsp104::kanMX strain consistent with a failure to propagate the [PSI+] prion. However, in the case of both Sup35-PrP fusion proteins, no elimination of the nonsense suppression phenotype was observed, confirming the GdnHCl experiment () and showing that Hsp104 is not required to form the two different types of SDS-resistant aggregates produced in these two strains.
Figure 5. The nonsense suppression phenotype in strains expressing the Sup35ΔQPrP152 and Sup35ΔNPrP191 fusion proteins is not dependent on Hsp104 function. (A) The suppression of the ade1–14 nonsense allele in LJ14-derived strains expressing either the Sup35ΔQPrP152 or the Sup35ΔNPrP191 fusion proteins was assessed by plating on to 1/4YPD plates. The left panels are for the parent LJ14 strain with a wild-type HSP104 gene while the right panel are the same strains but carrying a hsp104::kanMX disruption of the HSP104 gene. (B) Confirmation of the absence of Hsp104 in the hsp104::kanMX strains by western blot analysis using an anti-yeast Hsp104 polyclonal antibody. The filter was reprobed with an anti-yeast PGK polyclonal antibody as a loading control.
Lessons Learnt from Studying the Behavior of Sup35-PrP Fusions in Yeast
The yeast S. cerevisiae is an increasingly popular and tractable model in which to explore the mechanisms which ensure correct protein folding and which negate any potentially harmful consequences of protein misfolding. In the case of PrP, this disease-associated misfolding is very poorly understood at the molecular level and studies in mammalian cells suggest that it can occur without direct input from any other cellular factors. Nevertheless, molecular facilitators of infectious PrPSc formation—both protein and nucleic acid in nature—have been reported e.g., RNA.Citation81 Some of the Sup35-PrP fusion proteins we describe here clearly have a high propensity to misfold and aggregate when expressed in yeast. This aggregation does appear to be triggered by endogenous prion form of Sup35 as the aggregation continues when cells were treated with GdnHCl which eliminates [PSI+] and most other endogenous prions. The aggregates formed however were quite distinct and depended on the fusion protein being evaluated. For example, while a portion of the Sup35ΔNPrP152 protein took up an aggregated form as judged by sedimentation analysis, these aggregates were SDS sensitive indicating they were probably not amyloid in nature. In contrast the Sup35ΔNPrP191 protein did form high molecular weight SDS-resistant aggregates while the Sup35ΔQPrP152 protein formed a distinct and much lower molecular weight SDS-resistant aggregate.
The Sup35-PrP fusion proteins used here lacked the N-terminal signal sequence that directs PrP into the ER, a requirement for its subsequent post-translational modification, especially the formation of the single disulphide bridge and the addition of sugars to residues 180 and 196. It is possible therefore that the SDS-sensitive aggregates we observed with certain Sup35-PrP fusions expressed in the cytoplasm could be due to protein misfolding as a consequence of a failure to authentically post-translationally modify the protein.
Although the range of constructs we used in our study was relatively limited, it was striking that the two Sup35-Prp fusion proteins that formed SDS-resistant polymers were those carrying the mouse PrP octarepeat region. This sequence has previously been reported to be more prone to drive amyloid formation than the Sup35-OPR peptide repeats.Citation64,Citation65 However, the weak or non-existent suppression phenotype seen in cells expressing the Sup35mOPR protein would suggest that the yeast Sup35-OPR repeats and the PrP octarepeats are not functionally equivalent—at least in the context of prion propagation in vivo in yeast. The behavior of this protein might also suggest that there may be sequence features within the Sup35-PrD that act to suppress amyloid formation perhaps functioning as so-called gatekeepers that module protein polymerisation and any associated proteotoxicity.Citation82,Citation83
Previous studies have shown that the nature and the number of oligopeptide repeat units can influence the prion state of Sup35 be they native Sup35-OPR repeatsCitation84 or introduced mouse PrP repeats.Citation64,Citation65 In our study, the addition of the PrP octarepeats to the Sup35ΔNPrP152 construct resulted in an increase in the nonsense suppression phenotype () and also generated high molecular weight SDS-resistant aggregates ( and ). However, the most likely explanation for the change in nonsense suppression phenotype is that the addition of the octarepeat region to the protein resulted in a destabilization of the protein which in turn resulted in lower steady-state levels of soluble, functional Sup35 with a resulting defect in translation termination. This raises an important caveat about the use of the nonsense suppression assay for such studies since the strength of suppression can be greatly influenced simply by the relative levels of expression of Sup35 fusions that is unrelated to the protein’s propensity to aggregate. In order to establish a link between protein aggregation and the observed nonsense suppression phenotype requires the identification of a chemical or genetic means of blocking PrP-mediated aggregation in vivo. We have tested a number of compounds including quinacrine which has been reported to efficiently inhibit PrP polymerisation and PrPSc accumulation in cultured cells,Citation85 but none eliminate the nonsense suppression phenotype linked to expression of the various Sup35-PrP fusions tested here (L. Jossé and M.F. Tuite, unpublished results).
Acknowledgments
This work was supported by the EC through the APOPIS project LSHM-CT-2003–503330 (MFT, LJ, TvdH) and the POPH/FSE program through FCT (ref: SFRH/BD/27336/2006; to RM).
References
- Feldmann H. Yeast. Molecular and Cellular Biology. Wiley-Blackwell 2010.
- Braun RJ, Büttner S, Ring J, Kroemer G, Madeo F. Nervous yeast: modeling neurotoxic cell death. Trends Biochem Sci 2010; 35:135 - 44; http://dx.doi.org/10.1016/j.tibs.2009.10.005; PMID: 19926288
- Tenreiro S, Outeiro TF. Simple is good: yeast models of neurodegeneration. FEMS Yeast Res 2010; 10:970 - 9; http://dx.doi.org/10.1111/j.1567-1364.2010.00649.x; PMID: 20579105
- Bagriantsev S, Liebman S. Modulation of Abeta42 low-n oligomerization using a novel yeast reporter system. BMC Biol 2006; 4:32; http://dx.doi.org/10.1186/1741-7007-4-32; PMID: 17002801
- von der Haar T, Jossé L, Wright P, Zenthon J, Tuite MF. Development of a novel yeast cell-based system for studying the aggregation of Alzheimer’s disease-associated Abeta peptides in vivo. Neurodegener Dis 2007; 4:136 - 47; http://dx.doi.org/10.1159/000101838; PMID: 17596708
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003; 302:1772 - 5; http://dx.doi.org/10.1126/science.1090439; PMID: 14657500
- Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol 2002; 157:997 - 1004; http://dx.doi.org/10.1083/jcb.200112104; PMID: 12058016
- Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A 2000; 97:1589 - 94; http://dx.doi.org/10.1073/pnas.97.4.1589; PMID: 10677504
- Hughes RE, Olson JM. Therapeutic opportunities in polyglutamine disease. Nat Med 2001; 7:419 - 23; http://dx.doi.org/10.1038/86486; PMID: 11283667
- Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev 2009; 89:1105 - 52; http://dx.doi.org/10.1152/physrev.00006.2009; PMID: 19789378
- Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:566 - 9; http://dx.doi.org/10.1126/science.7909170; PMID: 7909170
- Tuite MF, Serio TR. The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol 2010; 11:823 - 33; http://dx.doi.org/10.1038/nrm3007; PMID: 21081963
- Summers DW, Cyr DM. Use of yeast as a system to study amyloid toxicity. Methods 2011; 53:226 - 31; http://dx.doi.org/10.1016/j.ymeth.2010.11.007; PMID: 21115125
- McGlinchey RP, Kryndushkin D, Wickner RB. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A 2011; 108:5337 - 41; http://dx.doi.org/10.1073/pnas.1102762108; PMID: 21402947
- Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, Lindquist S, et al. Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc Natl Acad Sci U S A 2008; 105:7206 - 11; http://dx.doi.org/10.1073/pnas.0802593105; PMID: 18480252
- Bach S, Tribouillard D, Talarek N, Desban N, Gug F, Galons H, et al. A yeast-based assay to isolate drugs active against mammalian prions. Methods 2006; 39:72 - 7; http://dx.doi.org/10.1016/j.ymeth.2006.04.005; PMID: 16750390
- Goldmann W, Hunter N, Martin T, Dawson M, Hope J. Different forms of the bovine PrP gene have five or six copies of a short, G-C-rich element within the protein-coding exon. J Gen Virol 1991; 72:201 - 4; http://dx.doi.org/10.1099/0022-1317-72-1-201; PMID: 1671225
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, et al. The cellular prion protein binds copper in vivo. Nature 1997; 390:684 - 7; http://dx.doi.org/10.1038/37733; PMID: 9414160
- Millhauser GL. Copper binding in the prion protein. Acc Chem Res 2004; 37:79 - 85; http://dx.doi.org/10.1021/ar0301678; PMID: 14967054
- Agostinho P, Oliveira CR. Involvement of calcineurin in the neurotoxic effects induced by amyloid-beta and prion peptides. Eur J Neurosci 2003; 17:1189 - 96; http://dx.doi.org/10.1046/j.1460-9568.2003.02546.x; PMID: 12670307
- Belosi B, Gaggelli E, Guerrini R, Kozłowski H, Łuczkowski M, Mancini FM, et al. Copper binding to the neurotoxic peptide PrP106-126: thermodynamic and structural studies. Chembiochem 2004; 5:349 - 59; http://dx.doi.org/10.1002/cbic.200300786; PMID: 14997527
- Gasset M, Baldwin MA, Lloyd DH, Gabriel JM, Holtzman DM, Cohen F, et al. Predicted α-helical regions of the prion protein when synthesized as peptides form amyloid. Proc Natl Acad Sci U S A 1992; 89:10940 - 4; http://dx.doi.org/10.1073/pnas.89.22.10940; PMID: 1438300
- Jobling MF, Stewart LR, White AR, McLean C, Friedhuber A, Maher F, et al. The hydrophobic core sequence modulates the neurotoxic and secondary structure properties of the prion peptide 106-126. J Neurochem 1999; 73:1557 - 65; http://dx.doi.org/10.1046/j.1471-4159.1999.0731557.x; PMID: 10501201
- Hölscher C, Delius H, Bürkle A. Overexpression of nonconvertible PrPc delta114-121 in scrapie-infected mouse neuroblastoma cells leads to trans-dominant inhibition of wild-type PrP(Sc) accumulation. J Virol 1998; 72:1153 - 9; PMID: 9445012
- Muramoto T, Scott M, Cohen FE, Prusiner SB. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc Natl Acad Sci U S A 1996; 93:15457 - 62; http://dx.doi.org/10.1073/pnas.93.26.15457; PMID: 8986833
- Norstrom EM, Mastrianni JA. The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J Biol Chem 2005; 280:27236 - 43; http://dx.doi.org/10.1074/jbc.M413441200; PMID: 15917252
- Goldfarb LG, Brown P, Haltia M, Cathala F, McCombie WR, Kovanen J, et al. Creutzfeldt-Jakob disease cosegregates with the codon 178Asn PRNP mutation in families of European origin. Ann Neurol 1992; 31:274 - 81; http://dx.doi.org/10.1002/ana.410310308; PMID: 1353341
- van der Kamp MW, Daggett V. The consequences of pathogenic mutations to the human prion protein. Protein Eng Des Sel 2009; 22:461 - 8; http://dx.doi.org/10.1093/protein/gzp039; PMID: 19602567
- Surewicz WK, Apostol MI. Prion protein and its conformational conversion: a structural perspective. Top Curr Chem 2011; 305:135 - 67; http://dx.doi.org/10.1007/128_2011_165; PMID: 21630136
- Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. NMR structure of the mouse prion protein domain PrP(121-231). Nature 1996; 382:180 - 2; http://dx.doi.org/10.1038/382180a0; PMID: 8700211
- Riek R, Hornemann S, Wider G, Glockshuber R, Wüthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett 1997; 413:282 - 8; http://dx.doi.org/10.1016/S0014-5793(97)00920-4; PMID: 9280298
- Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, et al. Structure of the recombinant full-length hamster prion protein PrP(29-231): the N terminus is highly flexible. Proc Natl Acad Sci U S A 1997; 94:13452 - 7; http://dx.doi.org/10.1073/pnas.94.25.13452; PMID: 9391046
- Liu H, Farr-Jones S, Ulyanov NB, Llinas M, Marqusee S, Groth D, et al. Solution structure of Syrian hamster prion protein rPrP(90-231). Biochemistry 1999; 38:5362 - 77; http://dx.doi.org/10.1021/bi982878x; PMID: 10220323
- Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A 2000; 97:145 - 50; http://dx.doi.org/10.1073/pnas.97.1.145; PMID: 10618385
- Wüthrich K, Riek R. Three-dimensional structures of prion proteins. Adv Protein Chem 2001; 57:55 - 82; http://dx.doi.org/10.1016/S0065-3233(01)57018-7; PMID: 11447697
- Ma J, Lindquist S. De novo generation of a PrPSc-like conformation in living cells. Nat Cell Biol 1999; 1:358 - 61; http://dx.doi.org/10.1038/14053; PMID: 10559963
- Hsiao K, Dlouhy SR, Farlow MR, Cass C, Da Costa M, Conneally PM, et al. Mutant prion proteins in Gerstmann-Sträussler-Scheinker disease with neurofibrillary tangles. Nat Genet 1992; 1:68 - 71; http://dx.doi.org/10.1038/ng0492-68; PMID: 1363810
- Peoc’h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, et al. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat 2000; 15:482; http://dx.doi.org/10.1002/(SICI)1098-1004(200005)15:5<482::AID-HUMU16>3.0.CO;2-1; PMID: 10790216
- Pocchiari M, Salvatore M, Cutruzzolá F, Genuardi M, Allocatelli CT, Masullo C, et al. A new point mutation of the prion protein gene in Creutzfeldt-Jakob disease. Ann Neurol 1993; 34:802 - 7; http://dx.doi.org/10.1002/ana.410340608; PMID: 7902693
- Ripoll L, Laplanche JL, Salzmann M, Jouvet A, Planques B, Dussaucy M, et al. A new point mutation in the prion protein gene at codon 210 in Creutzfeldt-Jakob disease. Neurology 1993; 43:1934 - 8; PMID: 8105421
- Kitamoto T, Ohta M, Doh-ura K, Hitoshi S, Terao Y, Tateishi J. Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun 1993; 191:709 - 14; http://dx.doi.org/10.1006/bbrc.1993.1275; PMID: 8461023
- Yam AY, Gao CM, Wang X, Wu P, Peretz D. The octarepeat region of the prion protein is conformationally altered in PrP(Sc). PLoS One 2010; 5:e9316; http://dx.doi.org/10.1371/journal.pone.0009316; PMID: 20195363
- Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, Paushkin SV, et al. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae.. EMBO J 1995; 14:4365 - 73; PMID: 7556078
- Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov S, Kisselev L, et al. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J 1995; 14:4065 - 72; PMID: 7664746
- Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol 2004; 2:E86; http://dx.doi.org/10.1371/journal.pbio.0020086; PMID: 15045026
- Parham SN, Resende CG, Tuite MF. Oligopeptide repeats in the yeast protein Sup35p stabilize intermolecular prion interactions. EMBO J 2001; 20:2111 - 9; http://dx.doi.org/10.1093/emboj/20.9.2111; PMID: 11331577
- Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae.. Genetics 1994; 137:671 - 6; PMID: 8088512
- Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature 2007; 449:233 - 7; http://dx.doi.org/10.1038/nature06108; PMID: 17767153
- Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 2005; 435:765 - 72; http://dx.doi.org/10.1038/nature03679; PMID: 15944694
- DePace AH, Santoso A, Hillner P, Weissman JS. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 1998; 93:1241 - 52; http://dx.doi.org/10.1016/S0092-8674(00)81467-1; PMID: 9657156
- Saupe SJ. The [Het-s] prion of Podospora anserina and its role in heterokaryon incompatibility. Semin Cell Dev Biol 2011; 22:460 - 8; http://dx.doi.org/10.1016/j.semcdb.2011.02.019; PMID: 21334447
- Taneja V, Maddelein ML, Talarek N, Saupe SJ, Liebman SW. A non-Q/N-rich prion domain of a foreign prion, [Het-s], can propagate as a prion in yeast. Mol Cell 2007; 27:67 - 77; http://dx.doi.org/10.1016/j.molcel.2007.05.027; PMID: 17612491
- Ter-Avanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge-Vechtomov SG, et al. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol Microbiol 1993; 7:683 - 92; http://dx.doi.org/10.1111/j.1365-2958.1993.tb01159.x; PMID: 8469113
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146 - 58; http://dx.doi.org/10.1016/j.cell.2009.02.044; PMID: 19345193
- Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell 2000; 5:163 - 72; http://dx.doi.org/10.1016/S1097-2765(00)80412-8; PMID: 10678178
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003; 115:879 - 91; http://dx.doi.org/10.1016/S0092-8674(03)01020-1; PMID: 14697205
- Halfmann R, Alberti S, Krishnan R, Lyle N, O’Donnell CW, King OD, et al. Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol Cell 2011; 43:72 - 84; http://dx.doi.org/10.1016/j.molcel.2011.05.013; PMID: 21726811
- Li A, Dong J, Harris DA. Cell surface expression of the prion protein in yeast does not alter copper utilization phenotypes. J Biol Chem 2004; 279:29469 - 77; http://dx.doi.org/10.1074/jbc.M402517200; PMID: 15090539
- Yang W, Yang H, Tien P. In vitro self-propagation of recombinant PrPSc-like conformation generated in the yeast cytoplasm. FEBS Lett 2006; 580:4231 - 5; http://dx.doi.org/10.1016/j.febslet.2006.06.074; PMID: 16831424
- Jiayu W, Zhu H, Ming X, Xiong W, Songbo W, Bocui S, et al. Mapping the interaction site of prion protein and Sho. Mol Biol Rep 2010; 37:2295 - 300; http://dx.doi.org/10.1007/s11033-009-9722-0; PMID: 19685161
- Collinge J. Human prion diseases and bovine spongiform encephalopathy (BSE). Hum Mol Genet 1997; 6:1699 - 705; http://dx.doi.org/10.1093/hmg/6.10.1699; PMID: 9300662
- Vital C, Gray F, Vital A, Parchi P, Capellari S, Petersen RB, et al. Prion encephalopathy with insertion of octapeptide repeats: the number of repeats determines the type of cerebellar deposits. Neuropathol Appl Neurobiol 1998; 24:125 - 30; http://dx.doi.org/10.1046/j.1365-2990.1998.00098.x; PMID: 9634208
- Flechsig E, Shmerling D, Hegyi I, Raeber AJ, Fischer M, Cozzio A, et al. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 2000; 27:399 - 408; http://dx.doi.org/10.1016/S0896-6273(00)00046-5; PMID: 10985358
- Dong J, Bloom JD, Goncharov V, Chattopadhyay M, Millhauser GL, Lynn DG, et al. Probing the role of PrP repeats in conformational conversion and amyloid assembly of chimeric yeast prions. J Biol Chem 2007; 282:34204 - 12; http://dx.doi.org/10.1074/jbc.M704952200; PMID: 17893150
- Tank EM, Harris DA, Desai AA, True HL. Prion protein repeat expansion results in increased aggregation and reveals phenotypic variability. Mol Cell Biol 2007; 27:5445 - 55; http://dx.doi.org/10.1128/MCB.02127-06; PMID: 17548473
- Biasini E, Tapella L, Restelli E, Pozzoli M, Massignan T, Chiesa R. The hydrophobic core region governs mutant prion protein aggregation and intracellular retention. Biochem J 2010; 430:477 - 86; http://dx.doi.org/10.1042/BJ20100615; PMID: 20626348
- Boeke JD, Trueheart J, Natsoulis G, Fink GR. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol 1987; 154:164 - 75; http://dx.doi.org/10.1016/0076-6879(87)54076-9; PMID: 3323810
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. [psi+] Science 1995; 268:880 - 4; http://dx.doi.org/10.1126/science.7754373; PMID: 7754373
- Kikuchi Y, Shimatake H, Kikuchi A. A yeast gene required for the G1-to-S transition encodes a protein containing an A-kinase target site and GTPase domain. EMBO J 1988; 7:1175 - 82; PMID: 2841115
- Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol 2001; 40:1357 - 69; http://dx.doi.org/10.1046/j.1365-2958.2001.02478.x; PMID: 11442834
- Jung GM, Masison DC. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol 2001; 43:7 - 10; http://dx.doi.org/10.1007/s002840010251; PMID: 11375656
- Grimminger V, Richter K, Imhof A, Buchner J, Walter S. The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J Biol Chem 2004; 279:7378 - 83; http://dx.doi.org/10.1074/jbc.M312403200; PMID: 14668331
- Horton RM, Cai ZL, Ho SN, Pease LR. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 1990; 8:528 - 35; PMID: 2357375
- Salas-Marco J, Bedwell DM. GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Mol Cell Biol 2004; 24:7769 - 78; http://dx.doi.org/10.1128/MCB.24.17.7769-7778.2004; PMID: 15314182
- Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996; 273:622 - 6; http://dx.doi.org/10.1126/science.273.5275.622; PMID: 8662547
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J 1996; 15:3127 - 34; PMID: 8670813
- Kushnirov VV, Vishnevskaya AB, Alexandrov IM, Ter-Avanesyan MD. Prion and nonprion amyloids: a comparison inspired by the yeast Sup35 protein. Prion 2007; 1:179 - 84; http://dx.doi.org/10.4161/pri.1.3.4840; PMID: 19164899
- Salnikova AB, Kryndushkin DS, Smirnov VN, Kushnirov VV, Ter-Avanesyan MD. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) is caused by its non-heritable amyloids. J Biol Chem 2005; 280:8808 - 12; http://dx.doi.org/10.1074/jbc.M410150200; PMID: 15618222
- Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem 2003; 278:49636 - 43; http://dx.doi.org/10.1074/jbc.M307996200; PMID: 14507919
- Bradley ME, Bagriantsev S, Vishveshwara N, Liebman SW. Guanidine reduces stop codon read-through caused by missense mutations in SUP35 or SUP45.. Yeast 2003; 20:625 - 32; http://dx.doi.org/10.1002/yea.985; PMID: 12734800
- Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature 2003; 425:717 - 20; http://dx.doi.org/10.1038/nature01979; PMID: 14562104
- Rousseau F, Schymkowitz J, Serrano L. Protein aggregation and amyloidosis: confusion of the kinds?. Curr Opin Struct Biol 2006; 16:118 - 26; http://dx.doi.org/10.1016/j.sbi.2006.01.011; PMID: 16434184
- Wang X, Zhou Y, Ren J-J, Hammer ND, Chapman MR. Gatekeeper residues in the major curlin subunit modulate bacterial amyloid fiber biogenesis. Proc Natl Acad Sci U S A 2010; 107:163 - 8; http://dx.doi.org/10.1073/pnas.0908714107; PMID: 19966296
- Liu JJ, Lindquist S. Oligopeptide-repeat expansions modulate ‘protein-only’ inheritance in yeast. Nature 1999; 400:573 - 6; http://dx.doi.org/10.1038/22919; PMID: 10448860
- Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A 2001; 98:9836 - 41; http://dx.doi.org/10.1073/pnas.161274798; PMID: 11504948