Abstract
Insulin-degrading enzyme (IDE) is a conserved Zn2+metalloendopeptidase involved in insulin degradation and in the maintenance of brain steady-state levels of amyloid β peptide (Aβ) of Alzheimer’s disease (AD). Our recent demonstration that IDE and Aβ are capable of forming a stoichiometric and extremely stable complex raises several intriguing possibilities regarding the role of this unique protein-peptide interaction in physiological and pathological conditions. These include a protective cellular function of IDE as a “dead-end chaperone” alternative to its proteolytic activity and the potential impact of the irreversible binding of Aβ to IDE upon its role as a varicella zoster virus receptor. In a pathological context, the implications for insulin signaling and its relationship to AD pathogenesis are discussed. Moreover, our findings warrant further research regarding a possible general and novel interaction between amyloidogenic peptides and other Zn2+metallopeptidases with an IDE-like fold and a substrate conformation-dependent recognition mechanism.
Insulin-Degrading Enzyme, Amyloid Peptides and Alzheimer's Disease
Insulin-degrading enzyme (IDE) is a highly conserved and ubiquitous Zn2+ metallo peptidase involved in the degradation of insulin in vivo. It belongs to the M16 family of metallopeptidases defined by its “inverted” sequence at the catalytic site (His-X-X-Glu-His) as compared to the classical sequence (His-Glu-X-X-His) found in thermolysin, neprilysin and other members of the clan. In recent years, several unique features of IDE have attracted the attention of researchers in the amyloid field and, particularly, in the field of cerebral amyloidoses associated with neurodegeneration. IDE is capable of degrading in vitro several peptides with amyloidogenic potential in vivo such as calcitonin (thyroid amyloid), atrial natriuretic factor (ANF) (atrial amyloidosis), amylin (pancreatic islet amyloidosis associated with diabetes type 2), ABri and ADan (familial British and Danish dementias, respectively) (reviewed in refs. Citation1 and Citation2). More significantly, its genetic targeting in mice has demonstrated a pivotal role of IDE in the degradation of amyloid β (Aβ) of Alzheimer's disease (AD), the ∼5 kDa intracellular domain of the amyloid precursor protein (AICD) and insulin. In effect, IDE K.O. mice show elevated steady-state levels of Aβ and AICD in their brains as well as hyperinsulinemia and glucose intolerance.Citation3,Citation4 This physiological relevance of IDE may have its counterpart under pathological conditions such as AD, in which Aβ accumulates into oligomers and fibrils that are toxic to neurons. In support of this contention, evidence is growing that relates abnormal insulin metabolism and an increased risk for AD (reviewed in ref. Citation5). IDE activity was found to be lower in brain homogenates and cortical microvessels loaded with Aβ in AD patients as compared with age-matched controls.Citation6,Citation7 It has been proposed that oxidation may inactivate IDE by its reaction with the lipid peroxidation product 4-hydroxy-2-nonenal and the concomitant formation of protein adducts that have been detected in AD brains.Citation8,Citation9 More recently, lymphoblasts from affected members of AD families linked with the region 10q23-24 harboring the IDE gene showed lower IDE activity as compared with unaffected members,Citation10 suggesting that the genetic linkage of AD in some chromosome 10-linked AD families may be related to a systemic defect in IDE activity.
The recently solved crystal structure of an inactive mutant of human IDE (E111Q) bound to insulin B chain, Aβ1–40, amylin and glucagon has shown that backbone conformation of the substrate is a major determinant of recognition.Citation11 This involves the binding of β strands of substrate conformers which are poorly populated when the peptides are free in solution with a “template” within the catalytic chamber of IDE. In the case of Aβ1–40, it is noteworthy that the central region Lys16-Asp23 acquires an extended conformation in its IDE-bound form and displays extensive contacts between residues known to be crucial in Aβ self-assembly (Leu17-Val-Phe-Phe20) and extremely conserved amino acids in a β strand (β6) that contributes to the catalytic cleft of IDE. Additional important information derived indirectly from the crystal structure is that each IDE monomer may sway between an open and a closed conformer, based on the comparison with substrate-free E. coli pitrilysin (PDB 1Q2L). In pitrilysin, the C-terminal domain is rotated away from the N-terminal domain in such a way that the chamber is widely open to the substrate while in IDE, extensive contacts between the N and C-terminal domains keep this chamber closed. It has been proposed that, after binding, the peptide substrate remains entrapped in the IDE closed conformer during the catalytic cycle. Interestingly, a substrate-free human IDE has been recently crystallized in a closed state, suggesting that this conformer is stable and that reopening of the chamber may be a limiting step to the exit of proteolytic products.Citation12
IDE and Aβ Form an Extremely Stable Complex
Our in vitro studies on the degradation of Aβ by IDE has led to the unexpected finding that Aβ, in addition to being a substrate, can remain bound to IDE with a stability consistent with an irreversible interaction.Citation13 The resistance to dissociation included harsh treatments such as boiling in high concentration of SDS with reducing agents, or pre-incubation with formic acid or 6 M guanidine HCl at pH 3. These criteria are usually used to define a non-disulfide covalent binding between proteins, with the only exception of amyloid self-assembly. Without any evidence thus far that they are covalently bound, amyloid-assembled proteins may display such stability, especially when extracted from tissue and regardless of the primary structure of the protein involved. In these cases, although a substantial amount of amyloid subunits can be dissociated with strong denaturants (a property that has allowed protein identification), there is always a remnant that resists dissociation. Several proteins with pathogenic relevance in AD and related diseases are known to bind to Aβ in an SDS-resistant manner, including apolipoprotein E, apolipoprotein J, α2-macroglobulin, transthyretin, gelsolin and the α7-nicotinic acethylcholine receptor subunit.Citation14–Citation20 Aβ also binds in vivo with high affinity to the enzyme 17β-hydroxysteroid dehydrogenase type 10 (HSD10), also known as Aβ-binding alcohol dehydrogenase (ABAD). Such interaction takes place within the mitochondrial matrix, results in a distortion of the ABAD active site, and may mediate part of the cytotoxic effects of Aβ.Citation21,Citation22 Yet, the tight binding between IDE and Aβ seems to be unique in its extremely high stability and in that IDE is the first Aβ-degrading peptidase shown to have such property. The biochemical characterization of this IDE-Aβ stable complex (IDE-AβSCx) has revealed several important features that are pertinent to its specificity and possible biological significance. Firstly, it seems to derive from the interaction between an Aβ monomer (or a very low order oligomer in the case of shorter Aβ peptides used in our study) and a natively folded IDE dimer under physiological conditions, indicating that it is unlikely to be the product of non-specific protein-peptide aggregation. Secondly, the Aβ sequence Leu17-Asn27, comprising the key residues that drive peptide self-assembly and several interactions with the IDE binding site as mentioned above, was sufficient to form IDE-AβSCx. Thirdly, IDE-AβSCx was formed with a very slow kinetics (t1/2 ∼ 45 min) under pseudo-first order conditions (100-fold molar excess of peptide), suggestive of a conformational rearrangement. In addition, such kinetic behavior ruled out the unlikely possibility that IDE-AβSCx was a spurious product of the treatment with detergents or solvents used for its detection by SDS-PAGE or reverse-phase chromatography. Finally, the competition experiments with insulin pre-incubation, and the fragments of IDE obtained after partial digestion together with sequence and MALDI-TOF analysis supported that at least part of the catalytic domain of IDE was involved in IDE-AβSCx formation. The fact that by inhibiting IDE activity with Zn2+ chelators, the yield of IDE-AβSCx was highly increased (5–10 fold) indicates that its generation is independent of catalysis and that there are no fragments of Aβ involved. An interesting question that remains to be tackled is whether Aβ behaves as a true “suicide” inhibitor of IDE. Two technical problems have hindered the answer to this question. In the first place, the typical kinetic experiments required to determine the apparent rate constant of inhibition are hampered by the partial loss of activity of IDE when incubated alone at 37°C. Secondly, the allosteric behavior of the IDE dimer activity as a function of substrate concentration may impose an overall effect on residual activity that masks the inhibition of one Aβ-bound monomer if the second monomer in the IDE dimer remains Aβ-free and available for substrate binding. It derives from our experiments that after incubation with Aβ, only a fraction of active IDE molecules end up as IDE-AβSCx and therefore, the product is expected to be heterogeneous, containing fully occupied and Aβ-free IDE dimers together with species in which one IDE monomer is occupied and the other one remains free. Defining whether IDE-AβSCx is formed with an active versus a partially or fully inactive IDE will certainly impact on the interpretations of a possible biological role of IDE-AβSCx, particularly under pathological conditions in which Aβ may be steadily or transiently overproduced. Despite these limitations, our finding that IDE-AβSCx is prevented from forming by preincubation with an excess of substrates and that once IDE-AβSCx is completed, Aβ cannot be displaced by high affinity substrates at high concentrations suggest that at least part of the IDE binding site is irreversibly blocked in IDE-AβSCx. As a summary of our findings, a proposed model of IDE-AβSCx formation is presented in . Although the shortest Aβ that we used lacks Lys, Tyr or Cys residues that are commonly found cross-linked in oxidized proteins, a covalent binding in IDE-AβSCx cannot be ruled out. As an example of this possibility, a nucleophilic attack of the amide nitrogen of Gly67 at the carbonyl carbon of Ser65 results in the internal covalent cross-linking and cyclization of the backbone in the chromophore region of green fluorescent protein.Citation23 An alternative explanation for the high stability of IDE-AβSCx is a non-covalent binding similar to the cooperative interaction of amyloid self-assembly. Homologous amyloid formation is by far more favorable than heterologous assembly, however, cross-seeding between different amyloid proteins has been described in vitro and in vivo.Citation24,Citation25 The resistance to denaturation and slow formation of IDE-AβSCx together with the structural data on IDE-substrate binding described above prompt us to postulate that IDE-AβSCx may be the result of an amyloid-like interaction between β strands of Aβ and the catalytic domain within IDE chamber. Although in our study we did not find a significant over-representation of peptides from IDE C-terminal domain, it is possible that the anti-parallel β sheet formed by strands β26 and β27 may participate in IDE-AβSCx stabilization since residues Arg824 and Tyr831, just upstream of this sheet, are involved in substrate contacts.Citation11 Structural 3-D information at atomic resolution of a purified IDE-AβSCx is needed to support or refute this interpretation.
Possible Physiological Significance of IDE-AβSCx
The immunoprecipitation of IDE from rat and AD brains followed by Western blot with antibodies specific for Aβ carboxyl-terminal epitopes (ending at Val40 or Ala42, respectively) showed a component with the molecular mass of IDE-AβSCx, suggesting that this complex is formed in vivo.Citation13 More recently, we have extended this study to a MALDI-TOF analysis of IDE immunoprecipitated from rat brain followed by in-gel digestion with endo-Lys C. This experiment showed a minor, though clearly detectable peptide with the mass of rodent Aβ 1–28, a predicted cleavage product of Aβ at Lys28-Gly29 by endo-Lys C (). A definite biochemical proof awaits the MS/MS fragmentation analysis of this peptide after a large scale preparation yet, the available data support that IDE-AβSCx is formed in the brain under physiological conditions. This raises intriguing possibilities regarding IDE-AβSCx significance, of which we would like to discuss two examples, both from IDE and Aβ sides.
IDE-AβSCx may reflect a function of IDE alternative to enzymatic activity.
The endocytic pathway has been proposed as a main subcellular site of Aβ generation from its precursor protein by β and .-secretases while the compartment in which IDE-Aβ interaction takes place is at present unknown. IDE is a ubiquitous protein and has been detected in endosomes.Citation26 However, IDE catalytic activity is maximal at pH 7, above the pH within the increasingly acidic environment along the endosomallysosomal pathway and our experiments in vitro indicate that IDE activity falls sharply below pH 6.5 (Llovera RE and Castaño EM, unpublished observations). Therefore, it is possible that within this compartment, an interaction between Aβ and IDE may be strongly displaced toward the formation of IDE-AβSCx, as expected from the combination of a high local Aβ concentration together with a pH-dependent impairment of IDE catalytic activity. Our experiments using an inactive mutant of IDE indicate that IDE-AβSCx is effectively formed in the pH range 5.5–7, supporting this possibility (de Tullio M., unpublished observations). In yeast, the facilitation of bona fide amyloid assembly of the prion [RNQ+] by the HSP40 chaperone Sis1 prevents the formation of soluble, non-amyloid toxic aggregates of the Rnq1 protein. Interestingly, Sis1 has been shown to stably bind to Rnq1 in a 1:1 complex, suggesting that a template-based mechanism is in play. These results indicate that eukaryotic cells are capable of sequestering intracellularly “benign” amyloid-like species to prevent the accumulation of more toxic soluble aggregates as an alternative to the more conservative mechanisms of chaperone-assisted correct folding or the direct degradation of misfolded conformers.Citation27 In this scenario, and taking into account that Aβ self-aggregation would be favored in an acidic compartment by its isoelectric point (pI 5.3), IDE-AβSCx may trap monomeric Aβ irreversibly within IDE chamber in a closed state thus preventing peptide nuclei formation. Once formed, IDE-AβSCx may have several possible destinations including the degradation by lysosomal proteases, translocation to the cytosol followed by proteasomal degradation or secretion through a non-classical pathway involving multivesicular bodies and exosomes (Bulloj A, Morelli L, manuscript in preparation). We would call this a “dead-end chaperone” function by which IDE prevents the rapid intracellular formation of highly toxic soluble aggregates of Aβ by trapping peptide monomers within its cavity and allowing a more controlled disposal of amyloidogenic peptides by the cell (). These predictions may be readily tested using a cellular model in which both Aβ and IDE can be overexpressed to facilitate the detection of IDE-AβSCx and follow its fate with specific antibodies after subcellular fractionation.
IDE-AβSCx formation may provide clues to unexpected functions of Aβ in the human brain.
The recent identification of IDE as a membrane receptor that facilitates varicella zoster virus (VZV) cellular entry and cell-to-cell spread of infection,Citation28 raises a second possible physiological role for IDE-AβSCx. VZV is a ubiquitous neurotropic human herpesvirus that causes chickenpox and then remains latent for decades in cranial nerve, dorsal root and autonomic nervous system ganglia. VZV reactivation occurs most often after age 60 and usually produces shingles (zoster). Less frequently, it may also spread to blood vessels of the brain, producing a unifocal or multifocal large and small-vessel angiopathy, particularly in immunocompromised hosts (reviewed in ref. Citation29). Interestingly, cerebral amyloid angiopathy due to the deposition of Aβ in the vessel walls is not only present in most cases of AD but is also common (∼30%) among non-demented subjects where its prevalence increases with age.Citation30 IDE expression and activity in human brain cortical microvessels has been extensively characterized.Citation7 A recombinant IDE N-terminal domain (residues 42–541) binds preferentially over the C-terminal domain to glycoprotein E (gE) of VZV, known to be crucial for virus infectivity and cell-to-cell spread.Citation31 Although gE is not degraded by IDE and competition experiments between Aβ or other substrates and gE to IDE binding have not been reported yet, it is noteworthy that bacitracin, a competitive inhibitor of IDE activity prevented gE-IDE binding in a dose-dependent manner suggesting that part of the substrate binding site of IDE may be involved in the interaction with gE.Citation28 It is also meaningful that VZV may use the endocytic pathway for infection, a compartment in which, as discussed above, Aβ is generated and the pH disfavors IDE catalytic activity. In addition to a competitive mechanism between Aβ and gE for IDE binding, an irreversible interaction between Aβ and IDE may “knock-down” a potential cellular receptor for VZV, pointing at a protective role of Aβ overexpression against VZV infection of the brain vasculature in elderly humans. Although highly speculative, this contention may deserve some experimental work in light of the therapeutic trials aiming at the removal of Aβ as a disease-modifying strategy for the treatment of AD.
IDE-AβSCx Formation Under Pathological Conditions: Possible Impact on Insulin Metabolism
In the context of AD pathogenesis, the reciprocal connection between Aβ and insulin metabolisms, and both to IDE, may confer a particular relevance to IDE-AβSCx formation. Several mechanisms have been proposed to explain the growing notion that insulin resistance may influence the risk for AD. These include increased Aβ generation, decreased Aβ degradation and reduced insulin-receptor (IR) mediated signaling through PI3K and phosphorylated Akt, with downstream effects related to the impairment of neuronal survival pathways (reviewed in refs. Citation32 and Citation33). A likely consequence of altered IR signaling is a reduced expression of IDE which is downstream of PI3K-Akt, with a negative impact on Aβ and insulin turnover, as reflected by the increase in plaque deposition in mutant APP transgenic mice in which insulin resistance was induced.Citation32,Citation34 Yet, a persistent insulin stimulation may ultimately result in insulin resistance, with lower Akt phosphorylation,Citation35 and reduced IDE expression that in turn, may lead to a decreased degradation of insulin. Collectively, these evidences suggest a self-promoting cascade of events by which chronic elevated levels of insulin, as a result of a slower turn-over due to IDE deficiency, may induce insulin resistance and Aβ accumulation. In addition to abnormal insulin signaling, cellular hypoxia due to brain hypoperfusion and ischemia has been proposed to increase the risk for AD (reviewed in ref. Citation36). It has been shown that hypoxia increases Aβ generation rate due to a higher expression and activity of β-secretase and components of the γ-secretase complex.Citation37,Citation38 Under these conditions, we propose that the formation of IDE-AβSCx may be enhanced and that sequestration of active IDE may impair both insulin and Aβ turnover further amplifying the deleterious loop discussed above.
IDE-AβSCx in a Broader Biochemical Perspective
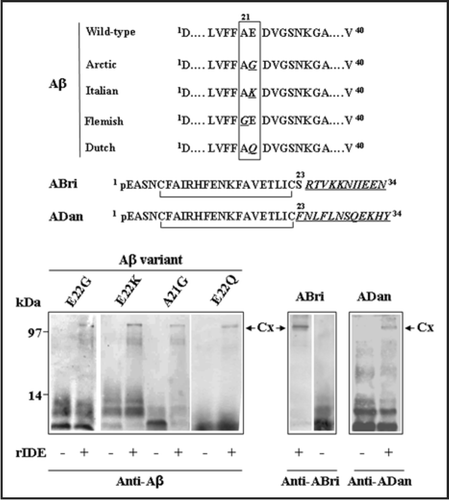
In addition to the hypothetical roles of IDE-AβSCx under physiological and pathological conditions in the brain, other issues warrant further research within a more basic context of protein-protein interactions. A prediction of our experiments on Aβ and IDE together with the conformational-dependent recognition derived from the crystal structure is that other amyloidogenic peptides—provided they are IDE substrates—have the potential to form IDE-AβSCx. We have recently extended our original observations on wild-type Aβ to genetic variants of Aβ containing pathogenic substitutions and the ABri and ADan peptides of familial British and Danish dementia in vitro, suggesting that this may be the case (). It is likely that several factors may influence the rate and amount of IDE-peptide stable complexes that can be formed in a given cell type. These include the steady-state or transient concentrations of peptides, the affinity of the IDE-peptide equilibrium and the rate of peptide aggregation, since peptides with a low critical concentration/kd ratio will tend to form lower amounts of stable complex with IDE due to a limiting availability of peptide monomers. In addition to in vitro experiments, biochemical information from amyloid-laden tissues with those amyloid peptides that have been shown to be IDE substrates in vitro (i.e., amylin in pancreatic amyloidosis of diabetes type 2 or cardiac amyloidosis due to ANF) will strongly contribute to consolidate this concept. On the other hand, it is possible that other proteases with a similar fold and a catalytic chamber such as neprilysin or presequence peptidase (recently called “cryptidases” by Malito et al.Citation39) may also tend to form highly stable complexes with amyloidogenic peptides. If this is the case, a general and novel interaction between unordered peptides with a high propensity to self-assemble into toxic β sheet oligomers and a group of neutral Zn2+ metalloendopeptidases may be proposed as a conserved (yet costly) strategy to prevent peptide aggregation under subcellular or extracellular conditions (low pH, accumulation of reactive oxygen species) in which their catalytic activity is largely disfavored.
Abbreviations
| Aβ | = | amyloid β |
| ABAD | = | amyloid β-binding alcohol dehydrogenase |
| ABri | = | amyloid Bri |
| AD | = | Alzheimer's disease |
| ADan | = | amyloid Dan |
| AICD | = | amyloid precursor protein intracellular domain |
| ANF | = | atrial natriuretic factor |
| DTT | = | dithiothreitol, endo-Lys C, endoproteinase-lysine C |
| gE | = | glycoprotein E |
| IDE | = | insulin-degrading enzyme |
| IDE-AβSCx | = | IDE-Aβ stable complex |
| IR | = | insulin receptor |
| MALDITOF | = | matrix-assisted laser desorption/ionization time-of-flight |
| MS | = | mass spectrometry |
| PI3K | = | phosphatidyl inositol 3-kinase |
| rIDE | = | recombinant IDE |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| VZV | = | varicella zoster virus |
Figures and Tables
Figure 1 A proposed model for the formation of IDE-AβSCx. Monomeric Aβ (Aβm) may follow a pathway of self-assembly to form oligomers (Aβo) or interact with the binding site (or at least a part of it) of fully active or inactive IDE conformers (IDEa and IDEi, respectively) to form a reversible IDE-Aβ encounter complex. IDEa and IDEi may be or not in equilibrium. IDE-AβSCx formation proceeds very slowly (represented by the dashed arrows) as an irreversible process that involves a conformational change in both molecules. In the case of IDEa-Aβ at neutral pH, the pathway is largely favored toward proteolysis of Aβ and free IDE (not depicted). Alternatively, it may lead to the slow formation of IDE-AβSCx.
Figure 2 Endogenous IDE-AβSCx from rat brain. Rat brain IDE was immunoprecipitated with monoclonal anti-IDE 3A2, proteins resolved on SDS-PAGE and the 120 kDa specific band digested in-gel with endo-LysC. Products were analyzed by MALDI-TOF as describedCitation13 and 51 peaks were assigned to IDE (not shown). The depicted spectrum shows a peak (peak A) with the average mass of rodent Aβ1–28 (error <1 dalton), a predicted product of endo-Lys C digestion of Aβ between Lys28-Gly29 (inset). Obs. M, observed mass; Calc. M, calculated mass. This peptide did not match to IDE sequence and was not found after digestion of a piece of gel from the 120 kDa region of a rat brain inmunoprecipitation with an unrelated antibody (not shown).
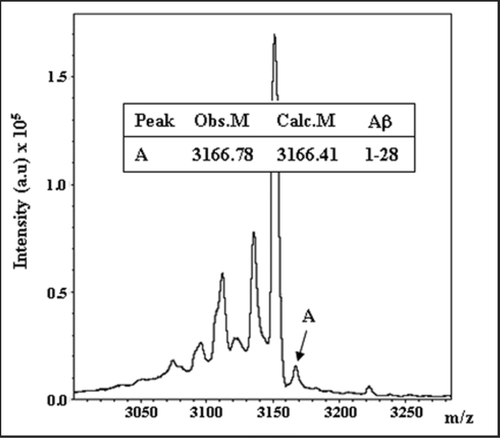
Figure 3 Proposed IDE-AβSCx formation and fate in a cellular context. IDE may interact with extracellular Aβ or with newly generated Aβ along the endocytic pathway. At neutral pH, IDE retains full catalytic activity and degrades Aβ monomers. As acidification increases along the pathway, IDE activity is reduced and more IDE-AβSCx is generated. Unbound monomeric Aβ, in turn, may aggregate readily favored by its isoelectric point. The formation of IDE-AβSCx may trap Aβ monomers irreversibly, preventing their massive oligomerization in acidic compartments. Possible destinations of IDE-AβSCx include its translocation and targeting to the proteasome, lysosomal degradation or secretion via exosomes. MVB, multivesicular bodies. The triangles on the right side depict schematically the opposite trends of IDE activity and Aβ aggregation within the intra-luminal pH range indicated by the numbers beside subcellular organelles (reviewed in ref. Citation40). For clarity, the more abundant IDE in the cytosol and other possible sites of Aβ aggregation are not depicted.
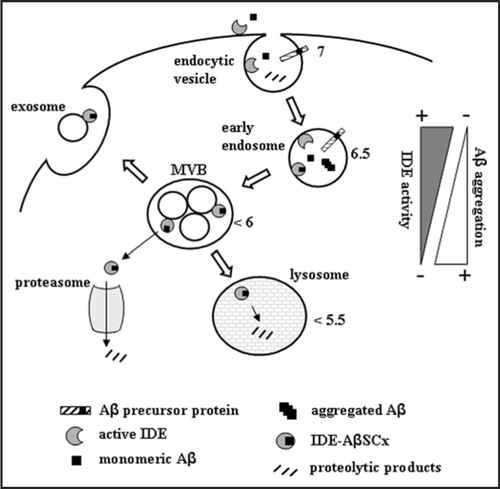
Figure 4 Stable complexes formed by IDE with different amyloid peptides. Recombinant rat IDE (rIDE) was incubated at a 25:1 molar ratio (peptide:enzyme) for 3 h at 37°C with the indicated amyloid peptides. Aβ1–40 genetic variants containing amino acid substitutions are associated with hereditary forms of AD or amyloid angiopathy and stroke. The ABri and ADan peptides are associated with familial British and Danish dementia, respectively. Top, schematic representation of the variant peptides with the amino acid substitutions in italics and underlined (the one-letter code is used). pE denotes pyroglutamic acid. The C-termini of ABri and ADan are the result of a loss of a stop codon after Ser23 or a duplication-insertion immediately before the stop codon, respectively. The N-termini are stabilized by intra-chain disulfide bonds. Bottom, Western blots with monoclonal anti-Aβ 6E10 or polyclonal anti-ABri or anti-ADan to detect Aβ, ABri or ADan peptides alone or in complex with rIDE, respectively. Before running the gel, samples were boiled in 8% SDS containing 0.1 M DTT for 5 min. The stable complexes are indicated by arrows (Cx). On the left, molecular mass standards, in kilodaltons.
Acknowledgements
This work was supported by a grant from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), PICT2006-354. We thank Dr. Jorge Ghiso (New York University School of Medicine) for his generous gift of Aβ, ABri and ADan peptides and anti-ABri and anti-ADan antibodies.
References
- Kurochkin IV. Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem Sci 2001; 26:421 - 425
- Morelli L, Llovera RE, Alonso LG, Frangione B, de Prat Gay G, Ghiso J, Castaño EM. Biochem Biophys Res Commun 2005; 332:808 - 816
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA 2003; 100:4162 - 4167
- Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL. Amyloid-β peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci USA 2003; 100:6221 - 6226
- Salkovic-Petrisic M, Hoyer S. Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: an experimental approach. J Neural Transm Suppl 2007; 72:217 - 233
- Pérez A, Morelli L, Cresto JC, Castaño EM. Degradation of soluble amyloid β-peptides 1–40, 1–42, and the Dutch variant 1–40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem Res 2000; 25:247 - 255
- Morelli L, Llovera RE, Mathov I, Lue LF, Frangione B, Ghiso J, Castaéo EM. Insulin-degrading enzyme in brain microvessels: proteolysis of amyloid β vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J Biol Chem 2004; 279:56004 - 56013
- Shinall H, Song ES, Hersh LB. Susceptibility of amyloid β peptide degrading enzymes to oxidative damage: a potential Alzheimer's disease spiral. Biochemistry 2005; 44:15345 - 15350
- Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM. Age- and region-dependent alterations in Aβ-degrading enzymes: implications for Aβ-induced disorders. Neurobiol Aging 2005; 26:645 - 654
- Kim M, Hersh LB, Leissring MA, Ingelsson M, Matsui T, Farris W, Lu A, Hyman BT, Selkoe DJ, Bertram L, Tanzi RE. Decreased catalytic activity of the insulin-degrading enzyme in chromosome 10-linked Alzheimer disease families. J Biol Chem 2007; 282:7825 - 7832
- Shen Y, Joachimiak A, Rosner MR, Tang WJ. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006; 443:870 - 874
- Im H, Manolopoulou M, Malito E, Shen Y, Zhao J, Neant-Fery M, Sun CY, Meredith SC, Sisodia SS, Leissring MA, Tang WJ. Structure of substrate-free human insulin-degrading enzyme (IDE) and biophysical analysis of ATP-induced conformational switch of IDE. J Biol Chem 2007; 282:25453 - 25463
- Llovera RE, de Tullio M, Alonso LG, Leissring MA, Kaufman SB, Roher AE, de Prat Gay G, Morelli L, Castaño EM. The catalytic domain of insulin-degrading enzyme forms a denaturant-resistant complex with amyloid β peptide: Implications for Alzheimer's disease pathogenesis. J Biol Chem 2008; 17039 - 17048
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E, high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 1993; 90:1977 - 1981
- Munson GW, Roher AE, Kuo YM, Gilligan SM, Reardon CA, Getz GS, LaDu MJ. SDS-stable complex formation between native apolipoprotein E3 and β-amyloid peptides. Biochemistry 2000; 39:16119 - 16124
- Matsubara E, Frangione B, Ghiso J. Characterization of apolipoprotein J-Alzheimer's A β interaction. J Biol Chem 1995; 270:7563 - 7567
- Hughes SR, Khorkova O, Goyal S, Knaeblein J, Heroux J, Riedel NG, Sahasrabudhe S. α2-macroglobulin associates with β-amyloid peptide and prevents fibril formation. Proc Natl Acad Sci USA 1998; 95:3275 - 3280
- Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ, Bhasin R, Silverman J, Weisgraber KH, Coyle PK, Zagorski MG, Talafous J, Eisenberg M, Saunders AM, Roses AD, Goldgaber D. Transthyretin sequesters amyloid β protein and prevents amyloid formation. Proc Natl Acad Sci USA 1994; 91:8368 - 8372
- Chauhan VP, Ray I, Chauhan A, Wisniewski HM. Binding of gelsolin, a secretory protein, to amyloid β-protein. Biochem Biophys Res Commun 1999; 258:241 - 246
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. β-amyloid (1–42) binds to δ7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem 2000; 275:5626 - 5632
- Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, Tohyama M, Ogawa S, Roher A, Stern D. An intracellular protein that binds amyloid-β peptide and mediates neurotoxicity in Alzheimer's disease. Nature 1997; 389:689 - 695
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer's disease. Science 2004; 304:448 - 452
- Barondeau DP, Kassmann CJ, Tainer JA, Getzoff ED. Understanding GFP chromophore biosynthesis: controlling backbone cyclization and modifying post-translational chemistry. Biochemistry 2005; 44:1960 - 1970
- O'Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem 2004; 279:17490 - 17499
- Yan J, Fu X, Ge F, Zhang B, Yao J, Zhang H, Qian J, Tomozawa H, Naiki H, Sawashita J, Mori M, Higuchi K. Cross-seeding and cross-competition in mouse apolipoprotein A-II amyloid fibrils and protein A amyloid fibrils. Am J Pathol 2007; 171:172 - 180
- Hamel FG, Siford GL, Jones J, Duckworth WC. Intraendosomal degradation of transforming growth factor α. Mol Cell Endocrinol 1997; 126:185 - 192
- Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, Lindquist S, Cyr DM. Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc Natl Acad Sci USA 2008; 105:7206 - 7211
- Li Q, Ali MA, Cohen JI. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 2006; 127:305 - 316
- Gilden DH, Mahalingam R, Cohrs RJ, Kleinschmidt-DeMasters BK, Forghani B. The protean manifestations of varicella-zoster virus vasculopathy. J Neurovirol 2002; 2:75 - 79
- Esiri MM, Wilcock GK. Cerebral amyloid angiopathy in dementia and old age. J Neurol Neurosurg Psychiatry 1986; 49:1221 - 1226
- Li Q, Krogmann T, Ali MA, Tang WJ, Cohen JI. The amino terminus of varicella-zoster virus (VZV) glycoprotein E is required for binding to insulin-degrading enzyme, a VZV receptor. J Virol 2007; 81:8525 - 8532
- Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J 2004; 7:902 - 904
- Lesné S, Gabriel C, Nelson DA, White E, Mackenzie ET, Vivien D, Buisson A. Akt-dependent expression of NAIP-1 protects neurons against amyloid-β toxicity. J Biol Chem 2005; 280:24941 - 24947
- Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, Frautschy SA, Cole GM. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci 2004; 24:11120 - 11126
- Egawa K, Nakashima N, Sharma PM, Maegawa H, Nagai Y, Kashiwagi A, Kikkawa R, Olefsky JM. Persistent activation of phosphatidylinositol 3-kinase causes insulin resistance due to accelerated insulin-induced insulin receptor substrate-1 degradation in 3T3-L1 adipocytes. Endocrinology 2000; 141:1930 - 1935
- Peers C, Pearson HA, Boyle JP. Hypoxia and Alzheimer's disease. Essays Biochem 2007; 43:153 - 164
- Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, Song W. Hypoxia facilitates Alzheimer's disease pathogenesis by upregulating BACE1 gene expression. Proc Natl Acad Sci USA 2006; 103:18727 - 18732
- Wang R, Zhang YW, Zhang X, Liu R, Zhang X, Hong S, Xia K, Xia J, Zhang Z, Xu H. Transcriptional regulation of APH-1A and increased γ-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J 2006; 20:1275 - 1277
- Malito E, Hulse RE, Tang WJ. Amyloid β-degrading cryptidases: insulin degrading enzyme, presequence peptidase and neprilysin. Cell Mol Life Sci 2008; [Epub ahead of print]
- Demaurex N. pH homeostasis of cellular organelles. News Physiol Sci 2002; 17:1 - 5