Abstract
The evolutionary origins of vertebrate prion genes had remained elusive until recently when multiple lines of evidence converged to the proposition that members of the prion gene family represent an ancient branch of a larger family of ZIP metal ion transporters.Citation1 A follow-up investigation which explored the mechanism of evolution in more detail led to the surprising conclusion that the emergence of the prion founder gene likely involved the reverse transcription of a spliced transcript of a LIV-1 ZIP predecessor gene.Citation2 The objective of this perspective is to discuss the possible significance of this reunion of ZIP and prion gene subfamilies for understanding the biology of the prion protein in health and disease. While a recent review article broadly introduced this area of research,Citation3 the emphasis here is to comment on some of the more pertinent concepts, experimental paradigms, ongoing developments and challenges.
The prion protein (PrP), with more than 10,000 research articles published, is one of the most intensively studied proteins in biology. It is of interest to interdisciplinary research that includes, but is not limited to, genetics, biochemistry, structural protein biology, cell biology, bioinformatics and epidemiology. The continued interest in the prion protein is borne out by its ability to cause incurable diseases in humans and animals.Citation4 In disease, the cellular prion protein (PrPC) acquires alternative and self-propagating states. Genes coding for the prion protein have only been identified in the vertebrate lineage, and are unrelated to an ever-increasing number of proteins in fungi that similarly possess the ability to acquire prion-like alternative and self-propagating states.Citation5 The origins of the prion gene family were not known until recently when, expanding on initial data which documented the binding of mouse PrP to a subset of Zrt-, Irt-like (ZIP) metal ion transporters,Citation6 the startling discovery was made that all members of the prion gene family had evolutionarily descended from the LIV-1 branch of ZIP genes.Citation1 The large family of ZIP proteins (also called Solute Carrier 39; SLC39) comprises 14 paralogs in humans which have been categorized into four subfamilies based on sequence similarities.Citation3,Citation7 The LIV-1 subfamily of ZIP zinc transporters (LZTs) contains 9 proteins, with at least some of this molecular diversity reflecting an evolutionary trend to molecular specialization, exemplified in the preferential association of individual LZTs with diverse subcellular compartments ().
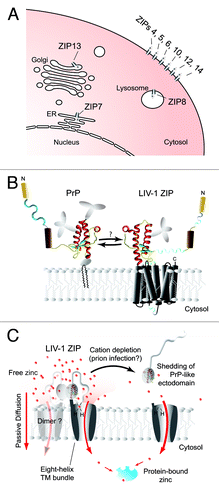
Figure 1. Localization, function and possible interaction of LZTs with the prion protein. (A) Cartoon depicting the reported predominant cellular localization of LIV-1 ZIP transporter (LZT) family members. Whereas more distantly related LZT metal ion transporters may preferentially associate with a variety of intracellular organelles, members of the phylogenetic subbranch of LZTs comprising ZIPs 5, 6 and 10 are predominantly observed at the plasma membrane. (B) Model of prion protein next to an LZT, depicted with its predicted PrP-like ectodomain. The interface that mediates the interaction between PrP and LZTs is as yet undefined. (C) Model of LZT-mediated cellular zinc import through passive diffusion and shedding of the PrP-like ectodomain in response to extracellular metal starvation. It should be noted that a similar endoproteolysis of a subset of LZTs can be observed in prion-infected mouse brains (unpublished dataCitation20). Panels B and C were modified from a Progress in Neurobiology articleCitation3 with permission from Elsevier.
In a recently published follow-up manuscript it could be documented that the emergence of the prion founder gene depended on two genomic rearrangements which occurred hundreds of millions of years apart.Citation2 The first of these generated a cysteine-flanked core domain (a central feature in all prion proteins and related LZTs) within the ectodomain of a pre-existing ZIP ancestor at a time when metazoa emerged on the planet. The true origin of this domain remains undetermined. The second event, the actual creation of the prion founder gene, can be traced back to a time before the divergence of teleosts and tetrapods, approximately a half-billion years ago and roughly coinciding with the Cambrian explosion. Surprisingly, this event appears to have been the result of a genomic insertion of a spliced and C-terminally truncated ZIP transcript. Because genomic rearrangements which involve retroinsertion of transcribed genes into a new genomic location with concomitant intron loss are most often mediated through retroviral elements, these data led to the proposition that the prion founder gene constituted a retrogene. Interestingly, evidence for a reenactment of this event, whereby the portion of an ancestral ZIP gene coding for its PrP-like ectodomain was retrotransposed, can also be found in a more recent branch of the phylogenetic tree (marsupials).Citation2 Taken together, these studies solved an enigma in prion research, the fact that despite more than 20 years of intense investigation, the evolutionary origin of the prion gene had remained elusive.
In light of persistent claims by a small number of researchers that virus-like agents play a role in the biology of prion diseases,Citation8-Citation10 it is ironic that the emergence of the prion founder gene may have involved retroviral elements. It should be self-evident, however, that the aforementioned steps proposed for the emergence of the prion founder gene have no bearing on the nature of the agent spreading contemporary prion diseases.
What significance then, if any, does the discovery of the evolutionary origins of the prion protein have? In the prion literature one can find references to “kryptonite,” “the Borg” and “martians.”Citation11,Citation12 Continuing on the science fiction theme we would like to use the analogy of an alien, newly arrived on Earth, who finds a doorknob on the ground. It is apparent that no amount of isolated investigations of this doorknob would reveal the fact that it is meant to be attached to a door, nor would it allow the alien to discover what door it used to be attached to and why. In this analogy the prion protein is, so to speak, descended from an ancient “doorknob.” The connection to ZIP proteins links the prion protein “doorknob” to a whole family of “intact doors” (the ZIP transporters) which have retained their doorknobs (the N-terminal extracellular domains of ZIP transporters). Finding the doors should be highly useful for this extraterrestrial observer intent on understanding the doorknob. It provides the ability to study the mechanisms of attached doorknobs and explore the spaces in front of and behind the doors they separate (e.g., upstream and downstream signaling pathways). As time passes, it may emerge that the detached doorknob (prion protein) no longer functions quite like the attached doorknobs (N-terminal domain of ZIP transporters). However, the fact that PrP co-purified with ZIPs 6 and 10 already suggests that the prion protein may at least have retained some of its original “doorknob” properties.Citation6
At first glance it may seem counterintuitive that proteins encoded by genes which are linked through evolution in the way proposed for LZTs and prion genes would bind to each other. However, it is not difficult to see how a physical encounter of the gene products of LZTs and the prion founder gene would have occurred. First, given that LZTs appear to be expressed in every eukaryotic cell within the metazoan lineage, so long as the retroinsertion of the prion founder gene occurred downstream from any active promoter, the premise that an LZT was co-translated within the same cell (a minimum requirement for a physical encounter) was fulfilled. It should be noted that the prion founder gene retained both an N-terminal signal sequence required for directing the nascent protein to the secretory pathway and a means by which it could be inserted into the lipid bilayer (a glycosylphosphatidylinositol (GPI) anchor), preserving its orientation and relative distance to the membrane ().Citation1 Moreover, because the prion founder retrogene would have initially coded for an expression product identical to the ectodomain of its LZT ancestor, it can be envisaged how these shared attributes would have directed both gene products to the same molecular environment, raising the possibility that they may interact with an overlapping set of binding partners. Our preliminary investigation of the localization of ZIPs has revealed that a small subpopulation within the cellular pool of LZTs may indeed associate with raft-like membrane structures which can be purified on the basis of their buoyancy during sucrose gradient centrifugations and which are known to be strongly enriched in cholesterol, sphingolipids and a subset of GPI-anchored membrane proteins, including PrP (unpublished data).
A direct interaction between members of the prion protein family and LZTs can also seem plausible if one assumes a scenario in which the LZT ancestor of the prion founder gene would have assembled into dimers or oligomers to fulfill its function as a cation importer. Thus, one can deduce that the ability of LZTs to assemble into functional dimers or oligomers could have translated into an ability of LZT predecessor and PrP founder gene products to bind to each other, if elements of the LZT ectodomain were contributing to the assembly of dimer or oligomer structures. To date, not much is known about the molecular organization of a majority of ZIP transporters, including the subbranch of LZTs comprising ZIPs 5, 6 and 10 from which the prion founder gene was descended. However, recent observations made with knockdown and metal chelation experiments, which queried the expression, stability and maturation of a subset of LZTs, were suggestive of a functional heterodimerization involving ZIP6 and ZIP10 (unpublished data). The aforementioned scenario further gains strength from the observation that ZIP13, a more distant LZT member of the ZIP family, appears to assemble into functional dimers.Citation13 In light of the cumulative sequence divergence which can be observed among contemporary members of the prion gene subfamily and LZTs, the physiological significance of their shared origins has likely been fading with evolutionary time. However, if confirmed, the aforementioned model may not only serve as an explanation for weak interactions observed between contemporary prion proteins and LZTs (), but may also further our understanding of why members of the mammalian prion protein family—PrP, Sho and Doppel—can be shown to interact.Citation6,Citation14
It is to be expected that more conclusive insights into this aspect of ZIP protein biology will emerge from detailed structural investigations of LZTs. To date, no high-resolution structural model exists for any ZIP protein. This contrasts an abundance of NMR and X-ray data available for a large range of prion protein orthologs and their derivatives, including recombinant constructs featuring point mutations or larger deletions.Citation15,Citation16 Incidentally, however, no PrP ortholog of fish has so far yielded a structure, despite considerable similarity and an undisputed phylogenetic linkage of fish and mammalian prion protein sequences.Citation17,Citation18 The failure to obtain a high-resolution structure may be a reflection of fish prion proteins differing in some way from their mammalian cousins that renders them refractory to tried-and-true in vitro folding strategies.Citation19 This piece of information is relevant and might serve as a harbinger indicating that attempts to solve the structure of the PrP-like ectodomains of LZTs may not be a trivial undertaking, because the branch of LZTs which aligns best to contemporary prion sequences exhibits the closest sequence similarity to fish rather than mammalian prion proteins.Citation1
If LZT ectodomains do indeed acquire a PrP-like fold, then it will be valuable to investigate the purpose of this fold in relation to their cation channel activity. Our recent work suggests that a subset of LZT ectodomains can be shed into the medium in response to divalent cation starvation () (unpublished data, see ref. Citation20). These observations are consistent with earlier data indicating that a rare zinc deficiency disease, acrodermatitis enteropathica, characterized by an inability of affected individuals to absorb zinc when dietary levels of this metal are limited, is linked to mutations in the gene coding for ZIP4 which appear to interfere with the zinc starvation-induced ectodomain shedding of the ZIP4 cellular cation import channel.Citation21 Taken together, these data suggest that the PrP-like ectodomain present in a subset of LZTs serves a function as a negative regulator of divalent cation uptake.Citation3 There is robust evidence for the ability of the prion protein to coordinate divalent cations, with copper exhibiting the strongest binding and levels of available cations influencing the coordination geometry observed.Citation22 Attempts to uncover the functional significance of the metal binding characteristics of the prion protein have been less successful. Our preliminary observations suggest that the prion protein may, similar to LZTs, undergo endoproteolysis in response to divalent metal starvation. More specifically, when N2a mouse neuroblastoma cells are grown in medium starved of transition metals for a prolonged time period, a diminution of full-length PrPC levels can be observed. Strikingly, this effect can be rescued by the addition of zinc but not copper (unpublished data, see ref. Citation20). In light of the evolutionary relationship of ZIPs and PrP, a research direction to pursue could be the determination of whether this zinc starvation-induced cleavage of PrP (1) occurs in the context of its ability to interact with LZTs, and (2) similar to LZT ectodomain shedding, serves a function in cellular cation homeostasis.
Due to the molecular complexity and heterogeneity of the LIV-1 subfamily of ZIP transporters, investigations into the functional relationship of LZTs and prion proteins may prove to be far from straightforward. To date, knockout studies of the most closely related LZTs are not available, yet if work on other ZIPs is indicative of the results to be expected, it is likely that crosstalk, functional redundancies, spatial overlap and complex posttranslational modifications will complicate their investigation.Citation23 That this is not merely a theoretical scenario is evident from our observation that N2a cells actively transcribe at least five out of six LZTs which were probed (unpublished data, see ref. Citation20). To understand the functional relationship of LZTs and the prion protein, we have begun to investigate the phylogenetic subbranch most closely related to PrP comprising ZIPs 5, 6 and 10. Interestingly, when expressed at similar levels and with identical C-terminal tags in neuroblastoma cells, the degree to which members within this LZT subbranch co-localize with the endogenous prion protein differs, with heterologously expressed ZIP5 demonstrating the most pronounced overlap in its cellular distribution (unpublished data). Assuming that this co-localization is reflective of direct binding of the prion protein to LZTs, it may not only raise the possibility to map the molecular interface between these proteins but may also help to investigate whether the interaction alters the divalent cation uptake biology of the respective LZTs. If so, one may find that despite PrP’s own preference for binding copper over zinc, its physiological cation biology may bear the cumulative signature of the ZIP transporters it interacts with and as such may primarily affect zinc rather than copper influx to the cell.
From a prion research perspective, the most pressing question remains whether LZTs play a role in the molecular etiology underlying prion diseases. On the basis of data which did not detect an influence of the brain-expressed PrP paralog Sho on PrP conversionCitation24,Citation25 (despite a selective and progressive reduction of Sho protein levels in prion diseaseCitation26), one may anticipate that other members of the prion/ZIP gene family similarly play no critical role in prion disease. However, given that Sho lacks both the cysteine-flanked core and C-terminal channel domains present in LZTs, the Sho data available to date may not be a suitable proxy for predicting whether LZTs play a role in the disease-associated conversion and/or the cell-to-cell spread of prion disease. It will be interesting to investigate whether there is an aspect to the physiological function of the PrP-like ectodomain of LZTs that may rely on an intrinsic ability to populate alternative folding states. In light of the likely function of the ectodomain in the gating and regulation of LZTs, it is, for example, conceivable that this domain can acquire alternative folds as part of an adaptive response to altering cation levels, similar to an observation made for the disordered domain within the prion protein.Citation27 If so, the study of ZIPs may help to define the cellular triggers and molecular constraints which promote conformational changes, which may in turn further our understanding of the profound changes in the physicochemical properties of PrP in prion diseases. Studies which employ hybrid PrP-LZT constructs may represent an experimental approach that would be informative in this context.
Recent biochemical data suggest that both the occurrence of prion strains and the species barrier phenomenon observed in prion diseases may relate to a conformational selection process which can be simulated with defined components in vitro.Citation28-Citation30 It is likely that this selection process would be influenced in vivo by PrP’s physiological milieu and its protein-protein interactions. In fact, it has repeatedly been speculated that other cellular factors which bind to the prion protein or reside in its proximity during its passage through the secretory pathway may contribute to these phenomena in vivo. In particular, the endoproteolytically-shed PrP-like LZT ectodomains may be plausible candidates in this context and could also act as possible modulators of prion disease spread; not only because they contain exclusively structural elements present in PrPC, but also because their lack of transmembrane domains would be expected to confer added mobility.
Finally, if PrPC modulates or contributes to a cation biology dictated by LZTs, changes to its expression level and/or fold, as seen in prion disease, would be expected to affect such a physiological role. This effect could manifest either as (1) an ablation of such a function, for example if PrPSc would no longer be able to interact with LZTs, or as (2) a poisoning, if PrPSc’s interaction with LZTs was retained but acquired a non-physiological quality. Needless to say that both scenarios could contribute to the symptoms of prion diseases and their impact would be expected to correlate with the severity of the underlying physiological perturbation.
Acknowledgments
Work on the projects described herein was funded through support from PrioNet Canada and the W. Garfield Weston Foundation.
References
- Schmitt-Ulms G, Ehsani S, Watts JC, Westaway D, Wille H. Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS One 2009; 4:e7208; http://dx.doi.org/10.1371/journal.pone.0007208; PMID: 19784368
- Ehsani S, Tao R, Pocanschi CL, Ren H, Harrison PM, Schmitt-Ulms G. Evidence for retrogene origins of the prion gene family. PLoS One 2011; 6:e26800; http://dx.doi.org/10.1371/journal.pone.0026800; PMID: 22046361
- Ehsani S, Huo H, Salehzadeh A, Pocanschi CL, Watts JC, Wille H, et al. Family reunion--the ZIP/prion gene family. Prog Neurobiol 2011; 93:405 - 20; http://dx.doi.org/10.1016/j.pneurobio.2010.12.001; PMID: 21163327
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Wickner RB, Shewmaker F, Edskes H, Kryndushkin D, Nemecek J, McGlinchey R, et al. Prion amyloid structure explains templating: how proteins can be genes. FEMS Yeast Res 2010; 10:980 - 91; http://dx.doi.org/10.1111/j.1567-1364.2010.00666.x; PMID: 20726897
- Watts JC, Huo H, Bai Y, Ehsani S, Jeon AH, Shi T, et al. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog 2009; 5:e1000608; http://dx.doi.org/10.1371/journal.ppat.1000608; PMID: 19798432
- Taylor KM, Morgan HE, Smart K, Zahari NM, Pumford S, Ellis IO, et al. The emerging role of the LIV-1 subfamily of zinc transporters in breast cancer. Mol Med 2007; 13:396 - 406; http://dx.doi.org/10.2119/2007-00040.Taylor; PMID: 17673939
- Diringer H. Proposed link between transmissible spongiform encephalopathies of man and animals. Lancet 1995; 346:1208 - 10; http://dx.doi.org/10.1016/S0140-6736(95)92905-3; PMID: 7475665
- Lantos PL. From slow virus to prion: a review of transmissible spongiform encephalopathies. Histopathology 1992; 20:1 - 11; http://dx.doi.org/10.1111/j.1365-2559.1992.tb00909.x; PMID: 1531331
- Manuelidis L. Transmissible encephalopathy agents: virulence, geography and clockwork. Virulence 2010; 1:101 - 4; http://dx.doi.org/10.4161/viru.1.2.10822; PMID: 21178425
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions and prionoids. Neuron 2009; 64:783 - 90; http://dx.doi.org/10.1016/j.neuron.2009.12.016; PMID: 20064386
- Prusiner SB. The prion diseases. Sci Am 1995; 272:48-51, 4-7.
- Bin BH, Fukada T, Hosaka T, Yamasaki S, Ohashi W, Hojyo S, et al. Biochemical characterization of human ZIP13 protein: a homo-dimerized zinc transporter involved in the spondylocheiro dysplastic Ehlers-Danlos syndrome. J Biol Chem 2011; 286:40255 - 65; http://dx.doi.org/10.1074/jbc.M111.256784; PMID: 21917916
- Jiayu W, Zhu H, Ming X, Xiong W, Songbo W, Bocui S, et al. Mapping the interaction site of prion protein and Sho. Mol Biol Rep 2010; 37:2295 - 300; http://dx.doi.org/10.1007/s11033-009-9722-0; PMID: 19685161
- Hornemann S, Christen B, von Schroetter C, Pérez DR, Wüthrich K. Prion protein library of recombinant constructs for structural biology. FEBS J 2009; 276:2359 - 67; http://dx.doi.org/10.1111/j.1742-4658.2009.06968.x; PMID: 19348007
- Wüthrich K, Riek R. Three-dimensional structures of prion proteins. Adv Protein Chem 2001; 57:55 - 82; http://dx.doi.org/10.1016/S0065-3233(01)57018-7; PMID: 11447697
- Premzl M, Gamulin V. Comparative genomic analysis of prion genes. BMC Genomics 2007; 8:1; http://dx.doi.org/10.1186/1471-2164-8-1; PMID: 17199895
- Premzl M, Gready JE, Jermiin LS, Simonic T, Marshall Graves JA. Evolution of vertebrate genes related to prion and Shadoo proteins--clues from comparative genomic analysis. Mol Biol Evol 2004; 21:2210 - 31; http://dx.doi.org/10.1093/molbev/msh245; PMID: 15342797
- Christen B, Wüthrich K, Hornemann S. Putative prion protein from Fugu (Takifugu rubripes). FEBS J 2008; 275:263 - 70; http://dx.doi.org/10.1111/j.1742-4658.2007.06196.x; PMID: 18070106
- Ehsani S, Salehzadeh A, Huo H, Reginold W, Pocanschi CL, Ren H, et al. LIV-1 ZIP ectodomain shedding in prion-infected mice resembles cellular response to transition metal starvation. Under review
- Kambe T, Andrews GK. Novel proteolytic processing of the ectodomain of the zinc transporter ZIP4 (SLC39A4) during zinc deficiency is inhibited by acrodermatitis enteropathica mutations. Mol Cell Biol 2009; 29:129 - 39; http://dx.doi.org/10.1128/MCB.00963-08; PMID: 18936158
- Millhauser GL. Copper and the prion protein: methods, structures, function and disease. Annu Rev Phys Chem 2007; 58:299 - 320; http://dx.doi.org/10.1146/annurev.physchem.58.032806.104657; PMID: 17076634
- Dufner-Beattie J, Huang ZL, Geiser J, Xu W, Andrews GK. Mouse ZIP1 and ZIP3 genes together are essential for adaptation to dietary zinc deficiency during pregnancy. Genesis 2006; 44:239 - 51; http://dx.doi.org/10.1002/dvg.20211; PMID: 16652366
- Westaway D, Genovesi S, Daude N, Brown R, Lau A, Lee I, et al. Down-regulation of Shadoo in prion infections traces a pre-clinical event inversely related to PrP(Sc) accumulation. PLoS Pathog 2011; 7:e1002391; http://dx.doi.org/10.1371/journal.ppat.1002391; PMID: 22114562
- Watts JC, Stöhr J, Bhardwaj S, Wille H, Oehler A, Dearmond SJ, et al. Protease-resistant prions selectively decrease Shadoo protein. PLoS Pathog 2011; 7:e1002382; http://dx.doi.org/10.1371/journal.ppat.1002382; PMID: 22163178
- Watts JC, Drisaldi B, Ng V, Yang J, Strome B, Horne P, et al. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J 2007; 26:4038 - 50; http://dx.doi.org/10.1038/sj.emboj.7601830; PMID: 17703189
- Chattopadhyay M, Walter ED, Newell DJ, Jackson PJ, Aronoff-Spencer E, Peisach J, et al. The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J Am Chem Soc 2005; 127:12647 - 56; http://dx.doi.org/10.1021/ja053254z; PMID: 16144413
- Colby DW, Prusiner SB. De novo generation of prion strains. Nat Rev Microbiol 2011; 9:771 - 7; http://dx.doi.org/10.1038/nrmicro2650; PMID: 21947062
- Sweeting B, Khan MQ, Chakrabartty A, Pai EF. Structural factors underlying the species barrier and susceptibility to infection in prion disease. Biochem Cell Biol 2010; 88:195 - 202; http://dx.doi.org/10.1139/O09-172; PMID: 20453922
- Weissmann C, Li J, Mahal SP, Browning S. Prions on the move. EMBO Rep 2011; 12:1109 - 17; http://dx.doi.org/10.1038/embor.2011.192; PMID: 21997298