Abstract
Prion diseases are caused by a conformational modification of the cellular prion protein (PrPC) into disease-specific forms, termed PrPSc, that have the ability to interact with PrPC promoting its conversion to PrPSc. In vitro studies demonstrated that anti-PrP antibodies inhibit this process. In particular, the single chain variable fragment D18 antibody (scFvD18) showed high efficiency in curing chronically prion-infected cells. This molecule binds the PrPC region involved in the interaction with PrPSc thus halting further prion formation. These findings prompted us to test the efficiency of scFvD18 in vivo. A recombinant Adeno-Associated Viral vector serotype 9 was used to deliver scFvD18 to the brain of mice that were subsequently infected by intraperitoneal route with the mouse-adapted scrapie strain RML. We found that the treatment was safe, prolonged the incubation time of scrapie-infected animals and decreased the burden of total proteinase-resistant PrPSc in the brain, suggesting that scFvD18 interferes with prion replication in vivo. This approach is relevant for designing new therapeutic strategies for prion diseases and other disorders characterized by protein misfolding.
Introduction
Prion diseases, or transmissible spongiform encephalopathies (TSE), are neurodegenerative disorders of humans and animals that are sporadic or inherited in origin and can be transmitted.Citation1 TSE are characterized by spongiform degeneration of the neuropil, neuronal loss and gliosis.Citation2 They are caused by conformational modifications of the prion protein (PrP) from a normal cellular isoform (PrPC) to insoluble and protease-resistant, disease-specific species termed PrPSc. The interaction of PrPSc with PrPC drives the conversion of PrPC into abnormal species leading to generation of infectious prions.Citation1 Accordingly, reagents binding either PrP conformer may halt PrPSc formation by inhibiting this interaction.
To date, no therapies for prion diseases exist, and the development of new therapeutic strategies is of utmost importance. In Alzheimer disease (AD), both passive and active immunization for Aβ protein was found to be effective in preventing disease and cognitive deficits in mouse models.Citation3,Citation4 Neutralization of prion infectivity after incubation with anti-PrP antibodies indicated a potential usefulness of antibody therapy for prion diseases.Citation5 Active immunization with recombinant PrP delayed the onset of experimental scrapie in mice but the therapeutic effect was poor.Citation6,Citation7 Passive immunization with anti-PrP monoclonal antibodies (mAbs) have a much more effective anti-prion activity in vivo, but only after intraperitoneal infection, reflecting the fact that these antibodies have short half-life and poor diffusion from vessels to the central nervous system (CNS) because of the blood-brain barrier (BBB).Citation8 To translate this therapeutic strategy from experimental to human conditions, the anti-PrP immunoreagents have to permeate the BBB, which is preferably achieved by monovalent antibody fragments since divalent ones were found to be neurotoxic.Citation9
Intracerebral delivery of anti-PrP antibodies could be an alternative or additional approach. Solforosi and coworkers tested in vivo several antibodies recognizing specific epitopes within the sequences 95–105 and 133–157 of PrPC.Citation10 However, when inoculated in the hippocampus of C57Bl/10 mice, mAb anti-PrP 95–105 caused extensive neuronal loss, while anti-PrP 133–157 did not. These findings were challenged by a recent study by Klohn and colleagues reporting that anti-PrP antibodies to an epitope within the 90–110 sequence (ICSM 35) as well as those used by Solforosi et al. failed to trigger neuronal apoptosis.Citation11
To minimize the neurotoxic effect, we treated mice with the single chain variable fragment antibody D18 (scFvD18) that specifically recognizes residues 132–156 of PrPC. Since this is the putative region of PrPC-PrPSc interaction, it can be argued that D18 operates mechanistically by directly blocking or modifying this interaction. This monovalent antibody has been previously tested in vitro and inhibited prion replication in cultured cells.Citation12 In 2007 Wuertzer and colleagues demonstrated that scFvD18, administered intracerebrally by using the Adeno-Associated Virus 2, delayed the onset of scrapie in mice intraperitoneally (i.p.) infected with the RML strain.Citation13
In the last few years, different AAV serotypes have been identified and AAV9 showed greater intracerebral diffusion and transduction efficiency than AAV2.Citation14,Citation15 Furthermore, AAV9 vector crosses the BBB and has the potential advantage to overcome pre-existing humoral immunity against the prevalent human serotypes 2. Thus we engineered the scFvD18 into the AAV9 vector (AAV9-scFvD18) which was intracerebrally inoculated in mice followed by i.p. infection with RML prion strain. The treatment efficiently reduced the accumulation of protease-resistant PrP and significantly delayed the onset of disease.
Results
Distribution of AAV9 in the CNS
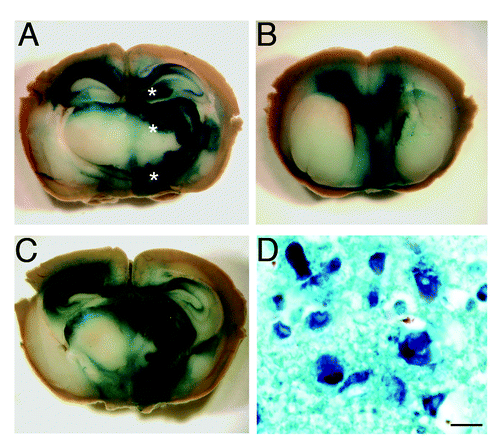
We first investigated the distribution of AAV9 vector in the CNS of 6 week-old CD1 mice using β galactosidase as reporter gene. Groups of three animals each were examined 1 mo, 2 mo and 3 mo after stereotaxical injection of AAV9-LacZ (β-gal) into the right hypothalamus, thalamus and hippocampus. The distribution of β-gal in the brain was assessed by X-gal histochemical staining. The highest level of expression was detected one month after the inoculation within the regions of injection, and also in directly surrounding areas such as the deep layers of the cerebral cortex, the corpus callosum and the septal nuclei (). Then, β-gal signal began to decrease gradually showing weak signal 3 mo after the administration. The results indicated that AAV9 vector was able to spread to the controlateral hemisphere, especially at the level of the hippocampus, hypothalamus and corpus callosum. The examination of serial sections of the whole brain indicated that the β-gal positivity also spread rostrocaudally (> 4 mm) from the inoculation sites. Microscopically, it was mainly localized in the cytoplasm of neurons ().
Figure 1. Distribution of β gal activity in the CNS of AAV9-LacZ injected mice. The brain of a mouse one month after the inoculation of AAV9-LacZ shows the presence of intense blue staining in the caudal diencephalon (A, C) and septum (B) indicating that the vector spread into these brain areas. (*) Sites of AAV9-LacZ inoculation. The blue reaction product corresponds to sites of β gal activity visualized by X-gal stain. Microscopy of the thalamus (D) showing both enzymatic activity (X-gal, blue) and the immunohistochemical stain for β gal reporter gene (dark blue). The enzyme is localized inside neurons and is mainly perinuclear. (a-c) Images taken by Zeiss Stereomicroscope. Scale bar in d = 1μm.
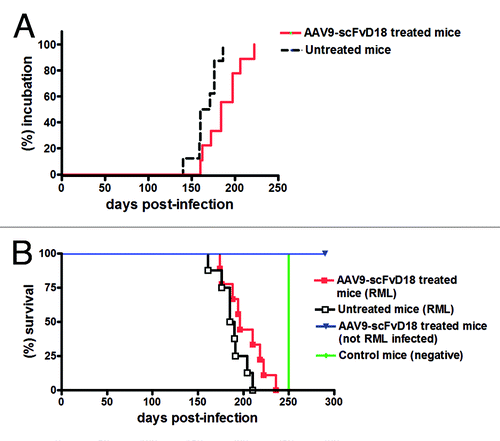
Effect of AAV9-scFvD18 treatment on experimental scrapie: incubation and survival times
To assess the onset of neurological symptoms, the monitoring of mice started 80 d after RML inoculation. The clinical picture of the disease was similar in all groups of infected animals. In the first stage the animals were hypoactive and apathetic with loss of interest in exploration of the cage. Then, they become hyperactive in the open field, as a result of an altered response to novel environment. In the late stage of the disease, the behavior was characterized by anxiety and fear, and the animals walked in close proximity to the wall of the cage. Behavioral scoreCitation16 showed that scFvD18 treated animals exhibited the clinical signs of disease with a delay of ~20 d compared with the untreated group. In particular, the mean incubation time of treated animals was 187 ± 7 d (means ± standard error of the mean [S.E.M.]) while that of control mice was 166 ± 5 d (). Thus, treated animals had significantly longer incubation time compared with untreated mice (p = 0.0186, Logrank test) and showed less hindlimb clasping behavior in the late stage of disease. Finally, the survival time of treated mice was slightly longer than that of untreated animals (202 ± 7 d vs. 188 ± 5 d) without reaching statistical significance (p = 0.085, Logrank test) ().
Figure 2. Effect of AAV9-scFvD18 treatment on incubation and survival times of experimental scrapie and control mice. Incubation time of AAV9-scFvD18 treated and untreated mice intraperitoneally infected with 10% RML (A). The group of animals subjected to the treatment showed clinical signs of disease later than the untreated one (187 ± 7 vs. 166 ± 5 d; p = 0.0186, Logrank test). Survival time of AAV9-scFvD18 treated and untreated mice intraperitoneally infected with 10% RML (B). The survival time of treated animals was slightly longer than that of untreated mice (202 ± 7 vs. 188 ± 5; p = 0.085, Logrank test). AAV9-scFvD18 treated mice without scrapie were sacrificed at the end of their predicted lifespan (~700 d), while control mice have been sacrificed when all RML-infected animals succumbed to disease (~250 d).
Effect of AAV9-scFvD18 treatment on experimental scrapie: neuropathological changes and PrP depositon
Histological examination of mice culled in the early stage of disease (166 d after prion infection, d.p.i.) revealed less extensive neuropathological changes in AAV9-scFvD18 treated mice compared with their untreated counterpart (). In particular, we found less spongiosis and gliosis in the septum, thalamus, hippocampus and adjacent cerebral cortex (). The burden of total proteinase K (PK)-resistant PrPSc, assessed by immunohistochemistry and western blot analysis, revealed lower levels of PrP in the brain of scFvD18 treated mice. The difference was most evident in the thalamus, hippocampus and caudate/putamen nuclei (). Accordingly, densitometric analysis of western blots () confirmed a statistically significant reduction of PrP in scFvD18 treated mice (p = 0.0005, t-test) () in the presence of similar patterns of PK-resistant PrPSc migration. The distribution of reactive astrocytes was similar to the targeting of spongiosis and PK- resistant PrPSc deposition ().
Figure 3. Neuropathological changes in scrapie infected mice at an early stage of disease (166 d.p.i). The lesion profiles (A) were determined on H&E stained sections, by scoring the vacuolar changes in nine standard brain regions. The septum, thalamus, hippocampus and adjacent cerebral cortex of AAV9-scFvD18 treated mice showed lower spongiform changes than that of untreated animals. The difference is most evident in the septum, hippocampus, and thalamus (H&E) (B). Scale bar = 2μm (all microphotographs are at the same magnification).
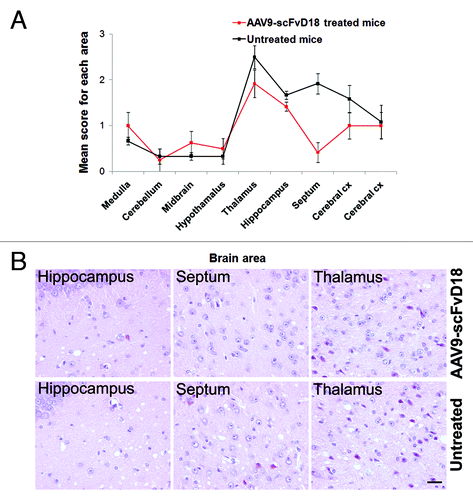
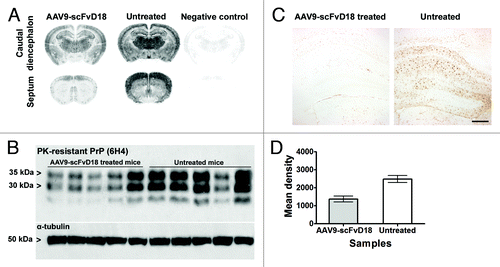
Figure 4. PK-resistant PrPSc deposition in scrapie infected mice at an early stage of disease (166 d.p.i). Histoblots with anti-PrP antibody (6H4) showed lower PK-resistant PrPSc deposition in the brain of AAV9-scFvD18 treated mice than that of untreated animals (A). Western blot analysis (B) with anti-PrP antibody (6H4) of PK-digested brain homogenates from AAV9-scFvD18 treated and untreated mice and the relevant densitometric analysis (D) demonstrate statistically significant lower levels of PK-resistant PrPSc in the brain of AAV9-scFvD18 treated mice (p = 0.005; double tailed, unpaired, t-test). Astrocytic activation (C) parallel the PK-resistant PrPSc deposition, and is much more severe in untrated animals. Scale bar = 20 μm (all microphotographs are at the same magnification).
On the contrary, neuropathological examination of mice culled at terminal stage revealed similar pathological changes in AAV9-scFvD18 treated and untreated animals, in particular spongiform changes () and astrogliosis.
Figure 5. Neuropathological and biochemical changes in scrapie infected mice at terminal stage. The most severe spongiform alterations were found into the hippocampus, thalamus and septum (H&E), without significant difference between AAV9-scFvD18 treated and untreated animals (A). Scale bar = 2μm (all microphotographs are at the same magnification). The hippocampus, enthorinal cortex, caudate/putamen nuclei and thalamus of scFvD18 treated mice (B) disclosed less PK-resistant PrPSc immunoreactivity (6H4) than untreated mice (arrows indicate areas with most evident differences). Western blot analysis (C) with anti-PrP antibody (6H4) of PK-digested brain homogenates from AAV9-scFvD18 treated and untreated mice and the relevant densitometric analysis (D) demonstrate statistically significant lower levels of PK-resistant PrPSc amount in the brain of AAV9-scFvD18 treated mice (p = 0.0154; double tailed, unpaired, t-test).
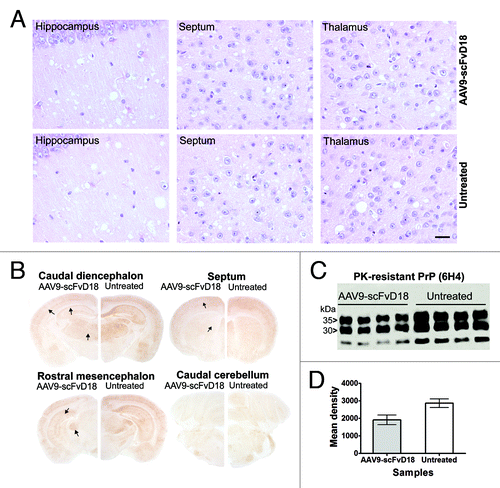
Intriguingly, also at the terminal stage of the disease, immunohistochemical () and western blot analysis () disclosed less PK-resistant PrPSc in the brain of scFvD18 treated mice compared with untreated mice. The difference was evident in the hippocampus, thalamus and caudate/putamen nuclei (). Densitometric analysis of western blot documented statistically significant lower levels of PK-resistant PrPSc in the brain of treated mice (p = 0.0154, t-test) ().
Assessing neurotoxicity following AAV9 injection
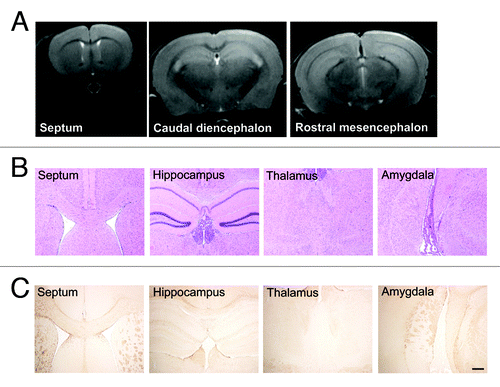
To study the tolerability of the vector, groups of animals intracerebrally inoculated with AAV9 empty vector, AAV9-LacZ or AAV9-scFvD18 were monthly subjected to magnetic resonance imaging (MRI) acquisitions and histological analysis was performed at different times after inoculation. T2-weighted MRI images confirmed that neither the inoculation of the vector nor the viral particles themselves elicited inflammatory or neurotoxic effects (). H&E stain () of septum, hippocampus, thalamus and amygdala shows normal appearance of these structures, and GFAP immunostain () of the same areas rules out astrocytic activation related to the injection. These results confirmed the lack of structural and neuropathological changes and adverse effects related to both AAV9 inoculation and β galactosidase or scFvD18 expression.
Figure 6. MRI and histological analysis of AAV9-scFvD18 treated mice. T2-weighted MRI images (A) of animals of the groups (i) and (ii) were monthly acquired and confirmed that neither the inoculation of the vector nor the viral particles themselves had neurotoxic effects. H&E stain (B) of septum, hippocampus, thalamus and amygdala shows normal appearance of these structures, and GFAP immunostain (C) of the same areas rules out astrocytic activation related to the injection. Scale bar = 25 μm (all microphotographs are at the same magnification).
Discussion
Aim of the study was to evaluate the therapeutic efficacy of the monovalent anti-PrP antibody scFvD18 intracerebrally delivered by AAV9 vector in mice experimentally infected with RML scrapie.
At first, we showed that AAV9 intracerebral inoculation is safe and the vector widely diffuses in the CNS in compliance with previous reports showing retrograde axonal transport.Citation17 The highest intracerebral transgene expression was reached one month after AAV9 administration. These characteristics make the method suitable for the delivery of anti-prion immunoreagents. MRI () and neuropathological analyses () confirmed that the expression of the monovalent antibody scFvD18 directed against residues 132–156 of PrP was well tolerated by mice.
In RML infected mice, the bilateral injection of AAV9-scFvD18 lead to a delay in the appearance of clinical signs, and a reduction of PK-resistant PrPSc content, indicating an effect on PrP accumulation.
Spongiform degeneration and astrogliosis were less severe in the group of AAV9-scFvD18 treated vs. untreated animals at an intermediate stage of the disease, but this difference faded at the terminal stage. Control experiments excluded the appearance of brain changes related to the surgical stress or the AAV9 injection.
It is conceivable that scFvD18 exerted an effective activity against PK-resistant PrPSc deposition and neurodegeneration at an early stage, when the brain harbored a low amount of PrPSc. Progressive neuronal loss and increasing PrP accumulation in the brain could have gradually reduced the protective effects of scFvD18, leading to a weakening of therapeutic efficacy. This is a possible explanation for the fact that the treatment with scFvD18 significantly delayed the onset of clinical symptoms in mice, while the survival time was also longer compared with the untreated animals but the difference did not reach statistical significance. It is also noteworthy, in this regard, that the presence of the scFvD18 encoding DNA sequence in the brain was confirmed by real-time PCR even 8 mo after the inoculation (data not shown).
The reduction of cerebral PK-resistant PrPSc in treated mice without significant modification of the spongiosis and gliosis as well as of the survival time is in compliance with the tenet that PK-resistant PrPSc levels in brain do not directly correlate with the severity of the neuropathological alterations and therefore the course of the disease. Previous studies argued about a lack of correspondence between infectivity titers or disease severity and amount of PK-resistant PrPSc.Citation18,Citation19 In particular, Barron and coworkers demonstrated that tissues with very low levels of PK-resistant PrPSc could be highly infective.Citation20 In this regard, the expression of scFvD18 could have hampered the formation of PK-resistant PrPSc without interfering with an alternative PK-sensitive PrPSc isoform.
Although the molecular diagnosis of prion diseases relies upon detection of PK-resistant fragments of PrPSc, it has become clear that PK-sensitive pathological isoforms may have a significant role in the pathogenesis of prion disease.Citation21 Moreover, several biochemical studies have suggested that cofactors, such as non-proteinaceous chaperones or RNA, are required to produce infectious prions, possibly by forming physical complexes with PrP.Citation22,Citation23 So, PrP itself may not be the only responsible for the propagation of the disease.
The efficacy of scFvD18 to interfere with prion propagation in vivo is susceptible to further improvement, considering that systemic administration of AAV9 leads to transduction in the spleen and in astrocytes throughout the entire CNS, with limited neuronal involvement.Citation24,Citation25 Therefore it is conceivable to combine AAV9-scFvD18 intracerebral treatment with systemic delivery to inhibit PrPSc invasion of peripheral organs and CNS simultaneously, to further enhance the effect of the treatment. Indeed, in vitro studies showed that both neurons and astrocytes could sustain prion propagation, even if the role of infected astrocytes remains controversial.Citation26
In summary, our study suggests that intracerebral delivery of an scFv gene with the AAV9 vector is effective in delaying the appearance of symptoms in a mouse model of prion disease reducing the amount of PK-resistant PrPSc in the CNS without eliciting toxic effects or inflammatory responses. Despite the ability of AAV9 to cross the BBB, we choose the intracerebral route of administration to obtain the highest neuronal transduction efficiency and to avoid any variable associated with their peripheral infusion and their passage across the BBB.Citation27
Moreover, studies of AAV9 vectors may be also useful for the therapeutic approach to other neurodegenerative disorders. Currently, experimental AAV2-based therapies for AD,Citation28 amyotrophic lateral sclerosis,Citation29 Huntington’s disease,Citation30 and Parkinson disease have been proposed, peaking a phase I clinical trial for Parkinson disease.Citation31,Citation32 However, the diffusion and transduction efficiency of the AAV2 vectors is limited and could be improved by using the AAV9 serotype as delivery system.
In this scenario, any novel therapeutic intervention, that could significantly delay the onset of these incurable and fatal disorders, including those based on AAV9-scFvD18, should be explored in search for a cure.
Materials and Methods
Ethics statement
Mice were housed in groups of 2–5 animals in individually ventilated cages, daily fed and water provided ad libitum. Lighting was on an automatic 12 h basis. Regular veterinary care was daily performed for assessment of animal health.
Animal facility is licensed and inspected by the Italian Ministry of Health. Current animal husbandry and housing practices comply with the Council of Europe Convention ETS123 (European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes; Strasbourg, 18.03.1986); Italian Legislative Decree 116/92, Gazzetta Ufficiale della Repubblica Italiana 10, 18 February 1992; and with the 86/609/EEC (Council Directive of 24 November 1986 on the approximation of laws, regulations and administrative provisions of the Member States regarding the protection of animals used for experimental and other scientific purposes).
The study, including its Ethics aspects, was approved by the Italian Ministry of Health (Permit Number: NP-02–08). All surgery was performed under tribromoethanol anesthesia, and all efforts were made to minimize suffering.
AAV vector preparation
AAV9-scFvD18 and AAV9-LacZ vectors were prepared by the AAV Vector-Unit (AVU) core facility at the International Centre for Genetic Engineering and Biotechnology in Trieste (http://www.icgeb.org/avu-core-facility.html). In brief, infectious AAV9 vector particles were generated in HEK293 cells by cotransfecting each vector plasmid (pAAV-scFvD18 and pAAV-LacZ) together with the packaging plasmid (pAAV9–2) and helper plasmid (pHELPER; Stratagene), expressing AAV and adenovirus helper functions, respectively.Citation33 Viral stocks were obtained by CsCl2 gradient centrifugation; rAAV titers, determined by measuring the copy number of viral genomes in pooled, dialyzed gradient fractions, as described previously (see ref. Citation34), were in the range of ~10 11/1012 genome copies/ml.
Surgical procedures
Six week-old CD1 mice (35–40 g) were anesthetized with tribromoethanol (100 µl/10 g) i.p.-administered. Surgical procedures were performed under sterile conditions. All instruments (i.e., blades, scissors, forceps, needles) were autoclaved. The animals remained insensate throughout the approximately 30 min surgery.
AAV9-β galactosidase (AAV9-LacZ) delivery to the brain for AAV9 diffusion studies
Anesthetized mice were placed in a stereotaxic instrument on a mouse and neonatal rat adaptor. A small incision was made in the skin in order to locate the Bregma point and the needle of the syringe (Hamilton Syringes, G26S) inserted into the brain. Once the needle was inserted, the engineered AAV9-LacZ vectors (5.37 × 1011 genome copies/ml) were injected at a rate of 1 µl/2 min by manual pressing of the syringe plunger. The inoculation was performed in three areas of the right hemisphere of the brain following specific stereotaxic coordinates (Mouse Brain, in Stereotaxic Coordinates, by Keith B. J. Franklin and George Paxinos, Academic Press, ISBN 0–12–266070–6): hypothalamus (1.8 caudal; + 0.5 lateral; 5.3 depth), thalamus (1.8 caudal; + 0.5 lateral; 3.5 depth) and hippocampus (1.8 caudal; + 0.5 lateral; 1.8 depth) with 2 µl of AAV9-LacZ per site (with a total of 6 µl per brain).
β-galactosidase assay
Groups of three animals each were examined 1 mo, 2 mo and 3 mo after inoculation. Mice were deeply anesthetized with intraperitoneal injection of Tiletamine-Zolazepam, (0.5 mg/10 g) and intracardially perfused with 0.5% glutaraldehyde fixative and brain harvested for histological analysis. Collected samples were immersed in 0.5% glutaraldehyde for 6 h at 4°C and cut in coronal slides (~1 mm thick). Enzymatic activity was revealed by X-gal histochemical staining. X-gal dilution buffer (5mM K4Fe(CN)6, 5mM K4Fe(CN)6, 2mM MgCl2, diluted in PBS) was freshly prepared and warmed to 37°C prior to use. X-gal stock solution (4% in dimethylformamide) was prepared diluting 0.2 g of X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside; Boehringer Mannheim) in 5 ml of N, N dimethylformamide. X-gal stock solution was then diluted 1:40 in warmed dilution buffer. Coronal slices were then rinsed 3 times in PBS, immersed in X-gal solution (37°C, overnight in the dark) and observed under stereomicroscope (Zeiss). Finally they were dehydrated and embedded in paraffin. Ten μm thick sections were cut and counterstained with eosin. Some sections were also immunostained for β-gal.
AAV9-scFvD18 delivery to the brain for therapeutic studies
Following the same procedures adopted for AAV9-LacZ inoculation, the AAV9 empty vector (1.2 × 1012 genome copies/ml) or the engineered AAV9-scFvD18 vector (1.4 × 1012 genome copies/ml) were injected at a rate of 1 µl/2 min by manual pressing of the syringe plunger. To improve both diffusion and therapeutic efficiencies, the inoculation was performed in both left and right hemisphere of the brain following the same stereotaxic coordinates used for the diffusion studies: hypothalamus (1.8 caudal; ± 0.5 lateral; 5.3 depth) thalamus (1.8 caudal; ± 0.5 lateral; 3.5 depth) and hippocampus (1.8 caudal; ± 0.5 lateral; 1.8 depth) with 2 µl of AAV9-scFvD18 per site (with a total of 12 µl per brain).
AAV9 tolerability
Groups of animals were intracerebrally inoculated with AAV9 empty vector, AAV9-LacZ or AAV9-scFvD18 (following their relative stereotaxical coordinates) and were monthly monitored by MRI acquisition (see below) and histological analysis to detect eventual toxic or inflammatory effects related to the injection.
Homogenate preparation and administration
Under sterile condition, 10% RML-infected brain homogenate (wt/vol) was prepared from pools of terminally RML-sick CD1 mouse brains using sterile saline solution (NaCl, B. Braun 0.9%), while 10% mock homogenate (wt/vol) was obtained from pools of healthy CD1 mouse brains.
Mice were infected one month after AAV9-scFvD18 delivery administering 100 μl of RML intraperitoneally.
Animal groups
Five groups of 15 animals were obtained: (i) treated with AAV9-empty vector, (ii) treated with AAV9-scFvD18, (iii) treated with AAV9-scFvD18 and subsequently i.p. infected with RML, (iv) infected with RML, (v) negative controls. Groups (i) and (ii) were monthly monitored by MRI acquisition (see below) and histological analysis to detect eventual toxic or inflammatory effects related to the injection
Behavioral testing
Behavioral monitoring was performed weekly and included spontaneous locomotor activity in the open field, nest construction test, reactivity to external stimuli and inverted screen test.Citation17,Citation35 The incubation time was calculated as the period between the day of prion infection and the appearance of clinical signs of the disease (decrease of locomotor and social activities, of grooming and of food consumption, tail rigidity, kyphosis, uncoordinated movements, awkwardness of gait, attenuation of righting reflex, hind limb clasping). In the late stages of disease, tremor, hind limb paralysis and ataxia appeared. At the agonic state, characterized by superficial breathing and lateral decubitus, the mice were sacrificed and survival time was calculated as the period between the inoculation and the day of death.
Magnetic Resonance Imaging (MRI)
Magnetic resonance imaging (MRI) was performed on a 7 Tesla horizontal-bore scanner (BioSpec 70/30 USR, Bruker, Ettlingen, Germany) equipped with 200 mT/m gradients, by a mouse brain dedicated surface coil. Mice were anesthetized with isofluorane at a dose approximately of 2.5 l/min, modulated according to the breathing frequency. The animals were positioned on an apposite bed inside the magnet and were monitored throughout the procedure for breathing frequency and body temperature with specific probes. For each mouse, high resolution T2-weighted images were acquired by a T2-weighted High Resolution Turbo Spin Echo sequence. The following parameters were employed: 26 axial slices, slice thickness 0.60 mm without gap; TR 3500 ms; TE 54 ms; field-of-view = 22 × 22 mm2, data matrix = 256 × 256; this yielded a 86 × 86 μm2 in plane resolution.
Sacrifice
To study the presence of differences in PK-resistant PrPSc deposition, five mice each of groups (iii) and (iv), randomly selected at the beginning of the experiment, were euthanized when the first symptoms of disease appeared within the group (iv) (166 d post infection). Conversely, other clinically affected mice (n = 10 per group) were sacrificed at the agonic stage of disease. Animals of the groups (i) and (ii) were monitored for the entire predicted lifespan and then culled and subjected to the autopsy, while negative controls animals of the group (v) were sacrificed when all RML-infected mice of group (iv) succumbed to disease. Mice were euthanized for harvest of tissues by intraperitoneal injection of Tiletamine-Zolazepam, (0.5 mg/10 g). Under deep anesthesia, intracardiac injection of Embutramide (0.2 ml) was performed. Brains and other organs were removed, half prepared for biochemical studies and half processed for histological analysis.
Neuropathology
Brains and other organs were fixed in Carnoy,Citation36 dehydrated and embedded in paraplast.
7-μm thick serial sections were stained with hematoxylin-eosin (H&E) and thioflavine S, or immunostained with monoclonal antibodies to PrP (6H4, 1:1000; Prionics), myelin (CNPase, 1:800; Sigma Aldrich) and polyclonal antibodies to astrocyte activation (GFAP, 1:2000; Dako), β-galactosidase (β-gal, 1:1000; USBiological) and apoptosis (Caspase-3, 1:500; Millipore). Before PrP immunostaining, the sections were sequentially treated with proteinase K (10 μg/ml, 5 min) and guanidine isothiocyanate (3M, 20 min), and non-specific binding was prevented using ARK kit (Dako). Immunoreactions were visualized using 3–3′-diaminobenzidine (DAB, Dako) as chromogen. Spongiform profiles were determined on H&E-stained sections, by scoring the vacuolar changes in nine standard gray matter area as described.Citation37
Histoblot were performed as previously reported using 6H4 (1:10000, overnight).Citation38
Biochemistry
Ten percent (wt/vol) brain homogenates from frozen tissues were prepared in lysis buffer. The cleared lysate was digested with 50 μg/ml of proteinase K (1 h, 37°C). Reactions were terminated by the addition of phenylmethanesulfonyl fluoride (PMSF, 5mM). 100 µg of total proteins were loaded on 12.5% polyacrylamide gels, transferred to polyvinylidene fluoride membranes and probed with antibody to PrP (6H4, 1:5000; Prionics) and α-tubulin (α-tub, 1:4000; Sigma Aldrich). The immunoreactions were visualized by enhanced chemiluminescence system (Amersham).
Statistical analysis
Statistical analysis was performed using the GraphPad-Prism 4.0 software. Kaplan-Meier survival curves were plotted, and differences in incubation or survival times between groups of mice were compared using the Logrank test. Densitometric analysis of PK-resistant PrPSc immunoblot data were performed using a double-tailed unpaired t-test (Lucia measurement software).
| Abbreviations: | ||
| TSE | = | Transmissible Spongiform Encephalopathy |
| AD | = | Alzheimer Disease |
| mAb | = | monoclonal antibody |
| CNS | = | Central Nervous System |
| BBB | = | Blood Brain Barrier |
| scFv | = | single chain variable Fragment |
| i.p. | = | intraperitoneally |
| β-gal | = | β galactosidase |
| PK | = | proteinase-K |
| MRI | = | Magnetic Resonance Imaging |
| PMSF | = | phenylmethanesulfonyl fluoride |
Disclosure of Potential Conflicts of Interest
The authors have declared that no competing interests exist.
Acknowledgments
This work was supported by the Italian Ministry of Health and Prion Encephalopathies Italian Association AIEnP.
References
- Prusiner SB. Molecular biology of prion diseases. Science 1991; 252:1515 - 22; http://dx.doi.org/10.1126/science.1675487; PMID: 1675487
- Jendroska K, Heinzel FP, Torchia M, Stowring L, Kretzschmar HA, Kon A, et al. Proteinase-resistant prion protein accumulation in Syrian hamster brain correlates with regional pathology and scrapie infectivity. Neurology 1991; 41:1482 - 90; PMID: 1679911
- Schenk D, Seubert P, Ciccarelli RB. Immunotherapy w 421 ith beta-amyloid for Alzheimer's disease: a new frontier. DNA Cell Biol 2004; 20:679 - 81; http://dx.doi.org/10.1089/10445490152717532
- Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T. Immunization with a nontoxic/nonfibrillar amyloid-β homologous peptide reduces Alzheimer’s disease-associated pathology in transgenic mice. Am J Pathol 2001; 159:439 - 47; http://dx.doi.org/10.1016/S0002-9440(10)61715-4; PMID: 11485902
- Gabizon R, McKinley MP, Groth D, Prusiner SB. Immunoaffinity purification and neutralization of scrapie prion infectivity. Proc Natl Acad Sci U S A 1988; 85:6617 - 21; http://dx.doi.org/10.1073/pnas.85.18.6617; PMID: 3137571
- Sigurdsson EM, Brown DR, Daniels M, Kascsak RJ, Kascsak R, Carp R, et al. Immunization delays the onset of prion disease in mice. Am J Pathol 2002; 161:13 - 7; http://dx.doi.org/10.1016/S0002-9440(10)64151-X; PMID: 12107084
- Polymenidou M, Heppner FL, Pellicioli EC, Urich E, Miele G, Braun N, et al. Humoral immune response to native eukaryotic prion protein correlates with anti-prion protection. Proc Natl Acad Sci U S A 2004; 101:Suppl 2 14670 - 6; http://dx.doi.org/10.1073/pnas.0404772101; PMID: 15292505
- White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003; 422:80 - 3; http://dx.doi.org/10.1038/nature01457; PMID: 12621436
- Müller-Schiffmann A, Korth C. Vaccine approaches to prevent and treat prion infection : progress and challenges. BioDrugs 2008; 22:45 - 52; http://dx.doi.org/10.2165/00063030-200822010-00005; PMID: 18215090
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sánchez-Alavez M, Sugama S, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004; 303:1514 - 6; http://dx.doi.org/10.1126/science.1094273; PMID: 14752167
- Klöhn PC, Farmer M, Linehan JM, O’Malley C, Fernandez de Marco M, Taylor W, et al. PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science 2012; 335:52; http://dx.doi.org/10.1126/science.1215579; PMID: 22223800
- Campana V, Zentilin L, Mirabile I, Kranjc A, Casanova P, Giacca M, et al. Development of antibody fragments for immunotherapy of prion diseases. Biochem J 2009; 418:507 - 15; http://dx.doi.org/10.1042/BJ20081541; PMID: 19000036
- Wuertzer CA, Sullivan MA, Qiu X, Federoff HJ. CNS delivery of vectored prion-specific single-chain antibodies delays disease onset. Mol Ther 2008; 16:481 - 6; http://dx.doi.org/10.1038/sj.mt.6300387; PMID: 18180775
- Burger C, Nash K, Mandel RJ. Recombinant adeno-associated viral vectors in the nervous system. Hum Gene Ther 2005; 16:781 - 91; http://dx.doi.org/10.1089/hum.2005.16.781; PMID: 16000060
- Mandel RJ, Manfredsson FP, Foust KD, Rising A, Reimsnider S, Nash K, et al. Recombinant adeno-associated viral vectors as therapeutic agents to treat neurological disorders. Mol Ther 2006; 13:463 - 83; http://dx.doi.org/10.1016/j.ymthe.2005.11.009; PMID: 16412695
- Guenther K, Deacon RM, Perry VH, Rawlins JN. Early behavioural changes in scrapie-affected mice and the influence of dapsone. Eur J Neurosci 2001; 14:401 - 9; http://dx.doi.org/10.1046/j.0953-816x.2001.01645.x; PMID: 11553290
- Cearley CN, Wolfe JH. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol Ther 2006; 13:528 - 37; http://dx.doi.org/10.1016/j.ymthe.2005.11.015; PMID: 16413228
- Lasmézas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, et al. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 1997; 275:402 - 5; http://dx.doi.org/10.1126/science.275.5298.402; PMID: 8994041
- Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, et al. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J 1999; 18:6855 - 64; http://dx.doi.org/10.1093/emboj/18.23.6855; PMID: 10581259
- Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, et al. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J Biol Chem 2007; 282:35878 - 86; http://dx.doi.org/10.1074/jbc.M704329200; PMID: 17923484
- Cronier S, Gros N, Tattum MH, Jackson GS, Clarke AR, Collinge J, et al. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J 2008; 416:297 - 305; http://dx.doi.org/10.1042/BJ20081235; PMID: 18684106
- Gauczynski S, Hundt C, Leucht C, Weiss S. Interaction of prion proteins with cell surface receptors, molecular chaperones, and other molecules. Adv Protein Chem 2001; 57:229 - 72; http://dx.doi.org/10.1016/S0065-3233(01)57024-2; PMID: 11447692
- Supattapone S. Biochemistry. What makes a prion infectious?. Science 2010; 327:1091 - 2; http://dx.doi.org/10.1126/science.1187790; PMID: 20185716
- Akache B, Grimm D, Pandey K, Yant SR, Xu H, Kay MA. The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8, 2, 3, and 9. J Virol 2006; 80:9831 - 6; http://dx.doi.org/10.1128/JVI.00878-06; PMID: 16973587
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol 2009; 27:59 - 65; http://dx.doi.org/10.1038/nbt.1515; PMID: 19098898
- Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science 2002; 298:556 - 62; http://dx.doi.org/10.1126/science.298.5593.556; PMID: 12386325
- Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther 2011; 19:1058 - 69; http://dx.doi.org/10.1038/mt.2011.72; PMID: 21487395
- Wang YJ, Gao CY, Yang M, Liu XH, Sun Y, Pollard A, et al. Intramuscular delivery of a single chain antibody gene prevents brain Aβ deposition and cognitive impairment in a mouse model of Alzheimer’s disease. Brain Behav Immun 2010; 24:1281 - 93; http://dx.doi.org/10.1016/j.bbi.2010.05.010; PMID: 20595065
- Henriques A, Pitzer C, Dittgen T, Klugmann M, Dupuis L, Schneider A. CNS-targeted viral delivery of G-CSF in an animal model for ALS: improved efficacy and preservation of the neuromuscular unit. Mol Ther 2011; 19:284 - 92; http://dx.doi.org/10.1038/mt.2010.271; PMID: 21139572
- Southwell AL, Ko J, Patterson PH. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease. J Neurosci 2009; 29:13589 - 602; http://dx.doi.org/10.1523/JNEUROSCI.4286-09.2009; PMID: 19864571
- Mochizuki H, Mizuno Y. Gene therapy for Parkinson’s disease. J Neural Transm Suppl 2003; 65:205 - 13; PMID: 12946058
- Muramatsu S, Fujimoto K, Kato S, Mizukami H, Asari S, Ikeguchi K, et al. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol Ther 2010; 18:1731 - 5; http://dx.doi.org/10.1038/mt.2010.135; PMID: 20606642
- Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol 2004; 78:6381 - 8; http://dx.doi.org/10.1128/JVI.78.12.6381-6388.2004; PMID: 15163731
- Zentilin L, Marcello A, Giacca M. Involvement of cellular double-stranded DNA break binding proteins in processing of the recombinant adeno-associated virus genome. J Virol 2001; 75:12279 - 87; http://dx.doi.org/10.1128/JVI.75.24.12279-12287.2001; PMID: 11711618
- Outram GW. Changes in drinking and feeding habits of mice with experimental scrapie. J Comp Pathol 1972; 82:415 - 27; http://dx.doi.org/10.1016/0021-9975(72)90041-2; PMID: 4630696
- Giaccone G, Canciani B, Puoti G, Rossi G, Goffredo D, Iussich S, et al. Creutzfeldt-Jakob disease: Carnoy’s fixative improves the immunohistochemistry of the proteinase K-resistant prion protein. Brain Pathol 2000; 10:31 - 7; http://dx.doi.org/10.1111/j.1750-3639.2000.tb00240.x; PMID: 10668893
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 1968; 78:301 - 11; http://dx.doi.org/10.1016/0021-9975(68)90006-6; PMID: 4970192
- Taraboulos A, Jendroska K, Serban D, Yang SL, DeArmond SJ, Prusiner SB. Regional mapping of prion proteins in brain. Proc Natl Acad Sci U S A 1992; 89:7620 - 4; http://dx.doi.org/10.1073/pnas.89.16.7620; PMID: 1354357