Abstract
Perturbations of calcium homeostasis have been associated with several neurodegenerative disorders. A common polymorphism (rs2986017) in the CALHM1 gene, coding for a regulator of calcium homeostasis, is a genetic risk factor for the development of Alzheimer disease (AD). Although some authors failed to confirm these results, a meta-analysis has shown that this polymorphism modulates the age at disease onset. Furthermore, a recent association study has explored the genetic variability of CALHM1 gene and two adjacent paralog genes (CALHM3 and CALHM2) in an Asian population. Since several lines of evidence suggest that AD and prion diseases share pathophysiologic mechanisms, we investigated for the first time the genetic variability of the gene cluster formed by CALHM1 and its paralogs in a series of 235 sporadic Creutzfeldt-Jakob disease (sCJD) patients, and compared the genotypic and allelic frequencies with those presented in 329 controls from the same ancestry. As such, this work also represents the first association analysis of CALHM genes in sCJD. Sequencing analysis of the complete coding regions of the genes demonstrated the presence of 10 single nucleotide polymorphisms (SNP) within the CALHM genes. We observed that rs4918016-rs2986017-rs2986018 and rs41287502-rs41287500 polymorphic sites at CALHM1 were in linkage disequilibrium. We found marginal associations for sCJD risk at CALHM1 polymorphic sites rs41287502 and rs41287500 [coding for two linked missense mutations (p.(Met323Ile); (Gly282Cys)], and rs2986017 [p.(Leu86Pro)]. Interestingly, a TGG haplotype defined by the rs4918016-rs2986017-rs2986018 block was associated with sCJD. These findings underscore the need of future multinational collaborative initiatives in order to corroborate these seminal data.
Introduction
Calcium homeostasis has key roles in different intracellular and extracellular processes, and therefore is essential for cell physiology. Perturbations of calcium regulation has been described in several neurodegenerative disorders, such as Alzheimer (AD), amyotrophic lateral sclerosis, Parkinson, Huntington’s and prion diseases.Citation1-Citation11
In 2008, Dreses-Werringloer and collaborators identified a polymorphism (rs2986017) associated with AD in the gene CALHM1 coding for a novel regulator of calcium homeostasis called CALHM1, which appears to be involved in the metabolism of amyloid β precursor protein (APP).Citation12 Although some authors have failed to confirm these results, a meta-analysis has shown that this polymorphism modulates the age at disease onset.Citation13 Additionally, a recent association study explored the genetic variability of CALHM1 gene and two adjacent paralog genes CALHM3 and CALHM2 (coding for CALHM3 and CALHM2, respectively) in an Asian population; although this study failed to find an association of these genes with AD.Citation14
Several lines of evidence suggest that AD and prion diseases share pathophysiologic mechanisms.Citation15-Citation23 In this paper, we investigated the genetic variability of the gene cluster formed by CALHM1 and its paralogs in a Caucasian population of Spanish origin, and explored the potential association of these genes with sporadic Creutzfeldt-Jakob disease (sCJD).
Results
This study included 235 sCJD patients (50.6% women) and 329 control subjects (60.2% women). Age at study inclusion for control individuals followed a non-normal distribution with a mean of 73.2 y and standard deviation of 11.0 y. Age at onset for the sCJD group followed a non-normal distribution with a mean of 67.2 ± 10.8 y.
Significant differences for gender distribution were found between sCJD patients and the control group (p = 0.05). Analysis of age distribution yielded statistically significant differences for sCJD patients (p < 0.001) compared with the control group (67.2 ± 10.8 vs. 73.2 ± 11.0). Potential effects of the differential gender and age distributions on the calculation of odds ratios were minimize by the use of logistic regression algorithms controlled age and gender. Homozygosis at polymorphic codon 129 in the PRNP gene was shown to be strongly associated with sCJD (OR = 4.87, 95% CIs = 3.21–7.39, p = 8.49 x 10−14) in agreement with a previous report for this population.Citation22
By sequencing analysis of the complete coding regions of the CALHM genes, we found 8 single nucleotide polymorphisms (SNP) in CALHM1, 2 SNP in CALHM3, and none in CALHM2 (). All of these SNPs were already identified in The Single Nucleotide Polymorphism Database (dbSNP) of Nucleotide Sequence Variation (www.ncbi.nlm.nih.gov/projects/SNP/index.html).
Table 1. Polymorphisms found in CALHM1 and CALHM3 genes coding regions
After tagging analysis of sequence data of cases and controls in Haploview, we opted for rs2986017 as tag SNP for rs2986018 (D´ = 1.0, r2 = 0.80, LOD = 307.7) and rs41287502 as tag SNP for rs41287500 (D´ = 1.0, r2 = 1.0, LOD = 134.61) (see ). For further analysis, only those SNPs with a minor allele frequency (MAF) above of 1% were selected. Linkage disequilibrium plot indicated that rs4918016, rs2986017 and rs2986018 within CALHM1 formed a haplotype block. The distribution of CALHM genotypes are shown in . All polymorphisms were in Hardy-Weinberg equilibrium, with the exception of rs41287502 in sCJD (p = 0.03). The association analysis with sCJD population of polymorphisms at CALHM genes cluster is shown in .
Figure 1. Standard (D´/LOD) linkage disequilibrium plot of the coding regions of CALHM1, CALHM2 and CALHM3. Haplotype blocks were generated using the confidence interval method by Haploview v4.2 software.Citation24 Exon (E) structure of these genes is depicted above the linkage disequilibrium plot. Coding regions are indicated by black boxes, while untranslated 5′and 3′exonic regions are represented by gray boxes.
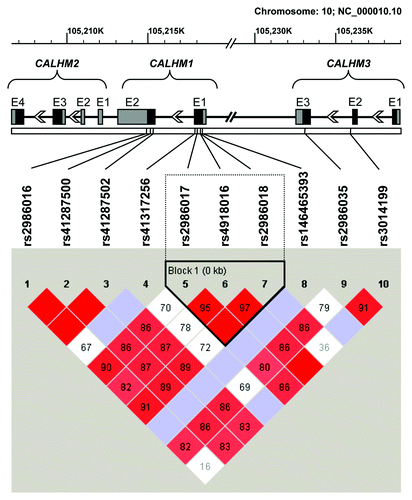
Table 2. Genotypic frequencies and odds ratios for the association of CALHM1 and CALHM3 SNPs with sCJD (minor allele carriers vs. non-carriers)
A Bonferroni correction for six comparisons yielded an adjusted significance level of p = 0.008. Based on this adjusted p value, we observed several non statistically significant trends for CALHM genes variants (). Thus, we found a dose-dependent risk tendency associated with CALHM1 rs41287502 T-allele carriers (OR = 2.21, 95% CIs = 1.12–4.34, p = 0.02) that was mainly driven by PRNP M129M individuals (OR = 3.56, 95% CIs = 1.22–10.36, p = 0.02). Rs41287502 T-allele carriers presented also a longer disease duration (p = 0.04). While we could not observe a relevant tendency for rs2986017 minor allele T-carriers, we found a risk tendency in a recessive model associated with CALHM1 rs2986017 TT-genotype (OR = 2.09, 95% CIs = 1.09–4.01, p = 0.03), which was mainly driven by individuals with earlier onset (OR = 6.66, 95% CIs = 1.38–32.15, p = 0.02).
At the CALHM3 gene, we also observed a risk tendency for rs3014199 G-allele carriers (OR = 1.72, 95% CIs = 0.98–3.01, p = 0.06) that was driven by earlier onset subjects (OR = 3.05, 95% CIs = 1.18–7.84, p = 0.02).
Interestingly, we also found that the TGG haplotype at polymorphic sites rs2986017, rs4918016 and rs2986018 defining a haplotype block () was associated with sCJD (OR = 1.88, χ2 = 5.61, p = 0.018). We performed a permutation test for haplotypes to obtain a measure of significance corrected for multiple testing bias. After 105 permutations, we observed that only 4.5% permutations exceeded the χ2 value yielding a corrected p value of 0.045. Moreover, a TG haplotype at the polymorphic sites of rs2986017 and rs2986018 within the defined haplotype block was clearly associated with the disease (OR = 1.94, χ2 = 6.45, p = 0.011, corrected p = 0.025).
Discussion
CALHM1 gene encodes for a cerebral calcium channel component controlling cytosolic calcium homeostasis, CALHM1. The CALHM1 rs2986017 polymorphism [p.(Leu86Pro)] has been associated with increased risk for late-onset AD in some, but not all reports.Citation12,Citation25-Citation34 Other studies had described its association with an earlier age at onset of AD.Citation12,Citation13,Citation26,Citation30 Shibata and collaborators have recently extended the AD risk association analysis to other adjacent CALHM1 paralogs, CALHM2 and CALHM3 with negative results for an Asian population.Citation14 We have investigated the genetic variability of the coding regions of the gene cluster formed by CALHM1, CALHM2 and CALHM3 in a Caucasian population of Spanish origin of sCJD cases and healthy controls, representing the first association analysis of CALHM genes in sCJD.
Of the 10 SNP in the CALHM genes here described (), two of them (rs2986017 and rs4918016) have been studied on several populations with minor allele frequencies (MAF) similar to those found in our population in Caucasians (for a review see refs. Thirteen and 25), and relatively different MAF in Asian populations.Citation14,Citation32 The rs2986018 and rs2986035 SNPs have only been described in Asian populations, showing lower MAF for rs2986018 on these populations compared with ours.Citation14,Citation32
Based on an adjusted significance level for multiple comparisons, we observed several marginal association findings that may be of relevance. We found that the polymorphic sites rs41287502 and rs41287500 at CALHM1 [both resulting in an amino acid change (p.[Met323Ile]; [Gly282Cys])] were in linkage disequilibrium (D´ = 1.0, r2 = 1.0, LOD = 134.61) and associated with a sCJD risk trend (OR = 2.21, p = 0.02 for all individuals; OR = 3.56, 95%, p = 0.02 for M129M individuals), and longer disease duration (p = 0.04). Whether these two linked missense mutations at CALHM1 gene alters the functionality of the protein remains to be explored.
Additionally, we observed a risk trend in a recessive model associated with CALHM1 rs2986017 TT-homozygous genotype, which was mainly driven by individuals with earlier sCJD onset, similarly to previous studies in AD.Citation12,Citation13 Interestingly, this SNP together with rs4918016 and rs2986018 define a haplotype block; and the haplotype TGG (formed by the lower frequency alleles) was found clearly associated with sCJD even after correction for multiple comparison bias.
Although our analysis comprises a big proportion of cases diagnosed in Spain in the last years, the number of cases studied is necessarily limited due to the rarity of the disease. Thus, international joint initiatives will be required to ensure universal validity to the primary data here presented.
Materials and Methods
Subjects
Patient populations included 235 sCJD cases [including 185 (78.7%) neuropathologically verified definite patients, and 50 (21.3%) probable cases] and 329 subjects with normal cognitive status measured by Mini Mental test. All subjects were Caucasians of Spanish origin. Samples for sCJD cases were obtained from patients with suspected prion diseases, submitted for diagnostic purposes under the guidelines of the Spanish National Referral and Surveillance system. Genetic cases of human prion diseases were excluded after complete DNA sequencing of PRNP coding region. Control samples were obtained with the adequate understanding and written consent of subjects, family members or legal guardians, as appropriate. The study was approved by the Bioethics and Animal Welfare Committee from the Instituto de Salud Carlos III, Madrid, Spain; and by the Ethical Research Committees from the CIEN Foundation, and Universidad Autónoma de Madrid, Madrid, Spain. All the data were analyzed anonymously, and clinical investigations have been conducted according to the principles expressed in the Declaration of Helsinki.
The sCJD group (n = 235) included 185 patients, and 50 probable sCJD cases.
DNA isolation and analysis
Total DNA was isolated from peripheral blood or cerebral tissue following standard procedures. The analysis of the polymorphism at codon 129 of the PRNP gene (rs1799990) was performed by DNA sequencing using specific primers.Citation35
The complete coding sequences of CALHM1, CALHM2 and CALHM3 genes were analyzed by DNA sequencing using specific primers designed according to the corresponding human reference sequences (see Table S1). The amplification reactions were performed with 50 ng of genomic DNA and 1 unit of Taq DNA Polymerase (Applied Biosystems) in a volume of 25 μl. The final concentrations of other reactants were: 1x Taq DNA Polymerase Buffer, 0.1 mM dNTPs, 1.5 mM MgCl2 and 0.1 μM of each primer. The PCR cycling conditions were as follows: initial denaturation at 96°C for 3 min followed by 35 cycles of 94°C for 30 sec, 63°C for 30 sec and 72°C for 1 min and a final extension at 72°C for 10 min. A 2 μl aliquot of the amplification reaction was sequenced using 0.1µM of the above primers and the BigDye® Terminator v1.1 Cycle Sequencing Kit in an ABI PRISM® 377 Analyzer (Applied Biosystems).
Haplotype analysis
Haplotypes assignation, linkage disequilibrium plot, and haplotypes association analysis were performed by using Haploview v4.2 software. Tag SNPs were selected by Haploview Tagger tool by using the “aggressive tagging: 2-marker haplotypes” routine with an r-square threshold of 0.8. Haplotype blocks were generated using the confidence interval method.Citation24
Statistical methods
Statistical analyses of nominal or categorical variables were performed by Fisher’s exact text or Pearson’s chi-square test. Quantitative variables (age at onset, disease duration) were analyzed by non-parametric statistical hypothesis contrast with Mann-Whitney U test. Additionally, logistic regression models controlled by age (as a linear variable) and gender were used to compare genotypic and allelic frequencies and to calculate association adjusted odds ratio (OR) and 95% confidence intervals (CIs). The Hardy-Weinberg test for genotype frequency distributions was performed on the observed genotype frequencies for population, with significance based on a standard observed-expected chi-square distribution with one degree of freedom. Deviations from normality of quantitative variables were checked by the Kolmogorov-Smirnov statistic with Lilliefors’ significance. All statistical analyses were performed with the GraphPad 4 or PASWStatistics 18 softwares.
Additional material
Download Zip (210.5 KB)Acknowledgments
We thank to all physicians and the epidemiological and clinical coordinators for assistance and notifying cases to the CJD Spanish Registry, as well as to F. Avellanal, J. Almazán, M. Ruiz, and E. Alcalde for their collaboration with the Spanish CJD surveillance system. We are thankful to the Biobank from the Hospital Universitario Fundación Alcorcón for providing neuropathological data to the CJD Spanish Registry of an important number of sCJD cases. This work was made possible by the generous participation of patients, control subjects and their families.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
Conceived and designed the experiments: OC MJB MC. Performed the experiments: OC MJB RH IS. Analyzed the data: OC MC. Contributed materials: MJB JC AFG PMM AL MJR AR JdP IF MC. Wrote the manuscript: OC JdP MC. Critical revision of the manuscript: MJB JC AL IF.
Funding
This work was supported by grants FIS 05/0912 from the Ministerio de Ciencia e Innovación, DGSP from Ministerio de Sanidad, Política Social e Igualdad (MC), the DGSP of the Spanish National Health Ministry (MC), and the Spanish CIBERNED (Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas) network (MJB, AL, JdP, IF). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Note
Supplemental material can be found at: http://www.landesbioscience.com/journals/prion/article/20785/
References
- Kawahara M. Disruption of calcium homeostasis in the pathogenesis of Alzheimer’s disease and other conformational diseases. Curr Alzheimer Res 2004; 1:87 - 95; http://dx.doi.org/10.2174/1567205043332234; PMID: 15975072
- Mattson MP. Calcium and neurodegeneration. Aging Cell 2007; 6:337 - 50; http://dx.doi.org/10.1111/j.1474-9726.2007.00275.x; PMID: 17328689
- Green KN, LaFerla FM. Linking calcium to Abeta and Alzheimer’s disease. Neuron 2008; 59:190 - 4; http://dx.doi.org/10.1016/j.neuron.2008.07.013; PMID: 18667147
- Marambaud P, Dreses-Werringloer U, Vingtdeux V. Calcium signaling in neurodegeneration. Mol Neurodegener 2009; 4:20; http://dx.doi.org/10.1186/1750-1326-4-20; PMID: 19419557
- Sorgato MC, Bertoli A. From cell protection to death: may Ca2+ signals explain the chameleonic attributes of the mammalian prion protein?. Biochem Biophys Res Commun 2009; 379:171 - 4; http://dx.doi.org/10.1016/j.bbrc.2008.12.026; PMID: 19101513
- Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev 2010; 131:517 - 26; http://dx.doi.org/10.1016/j.mad.2010.05.003; PMID: 20493207
- Surmeier DJ, Guzman JN, Sanchez-Padilla J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell Calcium 2010; 47:175 - 82; http://dx.doi.org/10.1016/j.ceca.2009.12.003; PMID: 20053445
- Camandola S, Mattson MP. Aberrant subcellular neuronal calcium regulation in aging and Alzheimer’s disease. Biochim Biophys Acta 2011; 1813:965 - 73; http://dx.doi.org/10.1016/j.bbamcr.2010.10.005; PMID: 20950656
- Fedrizzi L, Carafoli E. Ca2+ dysfunction in neurodegenerative disorders: Alzheimer’s disease. Biofactors 2011; 37:189 - 96; http://dx.doi.org/10.1002/biof.157; PMID: 21698698
- Giacomello M, Hudec R, Lopreiato R. Huntington’s disease, calcium, and mitochondria. Biofactors 2011; 37:206 - 18; http://dx.doi.org/10.1002/biof.162; PMID: 21674644
- Peggion C, Bertoli A, Sorgato MC. Possible role for Ca2+ in the pathophysiology of the prion protein?. Biofactors 2011; 37:241 - 9; http://dx.doi.org/10.1002/biof.161; PMID: 21698700
- Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 2008; 133:1149 - 61; http://dx.doi.org/10.1016/j.cell.2008.05.048; PMID: 18585350
- Lambert JC, Sleegers K, González-Pérez A, Ingelsson M, Beecham GW, Hiltunen M, et al. The CALHM1 P86L polymorphism is a genetic modifier of age at onset in Alzheimer’s disease: a meta-analysis study. J Alzheimers Dis 2010; 22:247 - 55; PMID: 20847397
- Shibata N, Kuerban B, Komatsu M, Ohnuma T, Baba H, Arai H. Genetic association between CALHM1, 2, and 3 polymorphisms and Alzheimer’s disease in a Japanese population. J Alzheimers Dis 2010; 20:417 - 21; PMID: 20164573
- Gambetti P, Russo C. Human brain amyloidoses. Nephrol Dial Transplant 1998; 13:Suppl 7 33 - 40; http://dx.doi.org/10.1093/ndt/13.suppl_7.33; PMID: 9870435
- Price DL, Borchelt DR, Sisodia SS. Alzheimer disease and the prion disorders amyloid beta-protein and prion protein amyloidoses. Proc Natl Acad Sci U S A 1993; 90:6381 - 4; http://dx.doi.org/10.1073/pnas.90.14.6381; PMID: 8101988
- Checler F, Vincent B. Alzheimer’s and prion diseases: distinct pathologies, common proteolytic denominators. Trends Neurosci 2002; 25:616 - 20; http://dx.doi.org/10.1016/S0166-2236(02)02263-4; PMID: 12446128
- Aguzzi A, Haass C. Games played by rogue proteins in prion disorders and Alzheimer’s disease. Science 2003; 302:814 - 8; http://dx.doi.org/10.1126/science.1087348; PMID: 14593165
- Armstrong RA, Lantos PL, Cairns NJ. Overlap between neurodegenerative disorders. Neuropathology 2005; 25:111 - 24; http://dx.doi.org/10.1111/j.1440-1789.2005.00605.x; PMID: 15875904
- Barnham KJ, Cappai R, Beyreuther K, Masters CL, Hill AF. Delineating common molecular mechanisms in Alzheimer’s and prion diseases. Trends Biochem Sci 2006; 31:465 - 72; http://dx.doi.org/10.1016/j.tibs.2006.06.006; PMID: 16820299
- Debatin L, Streffer J, Geissen M, Matschke J, Aguzzi A, Glatzel M. Association between deposition of beta-amyloid and pathological prion protein in sporadic Creutzfeldt-Jakob disease. Neurodegener Dis 2008; 5:347 - 54; http://dx.doi.org/10.1159/000121389; PMID: 18349519
- Calero O, Bullido MJ, Clarimón J, Frank-García A, Martínez-Martín P, Lleó A, et al. Genetic cross-interaction between APOE and PRNP in sporadic Alzheimer’s and Creutzfeldt-Jakob diseases. PLoS One 2011; 6:e22090; http://dx.doi.org/10.1371/journal.pone.0022090; PMID: 21799773
- de Pedro-Cuesta J, Mahillo-Fernández I, Rábano A, Calero M, Cruz M, Siden A, et al, EUROSURGYCJD Research Group. Nosocomial transmission of sporadic Creutzfeldt-Jakob disease: results from a risk-based assessment of surgical interventions. J Neurol Neurosurg Psychiatry 2011; 82:204 - 12; http://dx.doi.org/10.1136/jnnp.2009.188425; PMID: 20547628
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of haplotype blocks in the human genome. Science 2002; 296:2225 - 9; http://dx.doi.org/10.1126/science.1069424; PMID: 12029063
- Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, et al. A polymorphism in CALHM1 influences Ca+2 homeostasis, Abeta levels, and Alzheimer’s Disease Risk. Cell 2008; 135:994 - 6
- Boada M, Antúnez C, López-Arrieta J, Galán JJ, Morón FJ, Hernández I, et al. CALHM1 P86L polymorphism is associated with late-onset Alzheimer’s disease in a recessive model. J Alzheimers Dis 2010; 20:247 - 51; PMID: 20164592
- Cui PJ, Zheng L, Cao L, Wang Y, Deng YL, Wang G, et al. CALHM1 P86L polymorphism is a risk factor for Alzheimer’s disease in the Chinese population. J Alzheimers Dis 2010; 19:31 - 5; PMID: 20061624
- Bertram L, Schjeide BM, Hooli B, Mullin K, Hiltunen M, Soininen H, et al. No association between CALHM1 and Alzheimer’s disease risk. Cell 2008; 135:993 - 4, author reply 994-6; http://dx.doi.org/10.1016/j.cell.2008.11.030; PMID: 19070563
- Beecham GW, Schnetz-Boutaud N, Haines JL, Pericak-Vance MA. CALHM1 polymorphism is not associated with late-onset Alzheimer disease. Ann Hum Genet 2009; 73:379 - 81; http://dx.doi.org/10.1111/j.1469-1809.2009.00509.x; PMID: 19472444
- Minster RL, Demirci FY, DeKosky ST, Kamboh MI. No association between CALHM1 variation and risk of Alzheimer disease. Hum Mutat 2009; 30:E566 - 9; http://dx.doi.org/10.1002/humu.20989; PMID: 19191331
- Sleegers K, Brouwers N, Bettens K, Engelborghs S, van Miegroet H, De Deyn PP, et al. No association between CALHM1 and risk for Alzheimer dementia in a Belgian population. Hum Mutat 2009; 30:E570 - 4; http://dx.doi.org/10.1002/humu.20990; PMID: 19191332
- Inoue K, Tanaka N, Yamashita F, Sawano Y, Asada T, Goto Y. The P86L common allele of CALHM1 does not influence risk for Alzheimer disease in Japanese cohorts. Am J Med Genet B Neuropsychiatr Genet 2010; 153B:532 - 5; PMID: 19655363
- Nacmias B, Tedde A, Bagnoli S, Lucenteforte E, Cellini E, Piaceri I, et al. Lack of implication for CALHM1 P86L common variation in Italian patients with early and late onset Alzheimer’s disease. J Alzheimers Dis 2010; 20:37 - 41; PMID: 20164602
- Tan EK, Ho P, Cheng SY, Yih Y, Li HH, Fook-Chong S, et al. CALHM1 variant is not associated with Alzheimer’s disease among Asians. Neurobiol Aging 2011; 32:546. e11 - 2; http://dx.doi.org/10.1016/j.neurobiolaging.2009.05.008; PMID: 19545933
- Calero O, Hortigüela R, Albo C, de Pedro-Cuesta J, Calero M. Allelic discrimination of genetic human prion diseases by real-time PCR genotyping. Prion 2009; 3:146 - 50; http://dx.doi.org/10.4161/pri.3.3.9339; PMID: 19684471