Abstract
Mouse models of Alzheimer disease (AD) have been generated based on Amyloid-β Precursor Protein (AβPP) and the Presenilin (PSEN) gene mutations associated with familial AD (FAD). Such models have provided valuable insights into AD pathogenesis and represent an important research tool for the discovery of potential treatments. To model amyloid deposition in AD, we generated a new mouse line based on the presence of two copies of the genomic region encoding human wild-type AβPP as well as a mutation (L166P) in the murine Psen1. By ~6 months of age, these mice have begun to develop cerebral Aβ pathology with a significant increase in the levels of AβPP C-terminal fragments and Aβ42, as well as increase Aβ42/Aβ40 ratio. Since in the brain and other tissues of these mice, wild-type human AβPP mRNA and protein levels are comparable to those of endogenous AβPP, this model may allow studies about the role of AβPP isoforms in the pathogenesis of AD. This animal model may be suitable to test drugs aimed at inhibiting expression or altering splicing and processing of AβPP, without artifacts associated with the presence of mutations in AβPP or overexpression due to the use of exogenous promoters. These features of the new model are of critical importance in assessing the success of therapeutic interventions.
The neuropatologic hallmarks of Alzheimer disease (AD) are the accumulation of the insoluble 4 kDa amyloid-β (Aβ) peptide in brain parenchyma and vessel walls, the intraneuronal accumulation of neurofibrillary tangles (NFTs) composed of tau paired helical filaments (PHFs), and the extensive neuronal loss.Citation1 Aβ peptides are generated in the “amyloidogenic pathway” by the proteolysis of the amyloid-β precursor protein (AβPP) by the β-site AβPP-cleaving enzyme 1 (BACE1)Citation2 and the γ-secretase complex (3, 4). Cleavage by α-secretase (ADAM, a disintegrin and metalloproteinase) and γ-secretase in the “non-amyloidogenic pathway” prevents Aβ generation.Citation1-Citation4 Missense mutations in AβPP located at or near the sites of proteolysis by β- and γ-secretase have been used to develop mouse models of AD (5). The mouse lines most frequently used express a mutation at the β-secretase cleavage site (Swedish double mutation, AβPP K670N/M671L) or a combination of the Swedish double mutation with a mutation in the γ-secretase site or within the Aβ sequence.Citation5-Citation9 There are only a few animal models based on the expression of a human AβPP sequence without mutations.Citation10,Citation11 In these models, high levels of expression were obtained by using strong exogenous promoters without significant amyloid deposition. Our laboratory recently reportedCitation12 amyloid deposition in mice expressing the entire wild-type (WT) AβPP gene using its endogenous regulatory elements.
Amyloid Deposition in Mice Expressing Human Wild-Type AβPP Gene
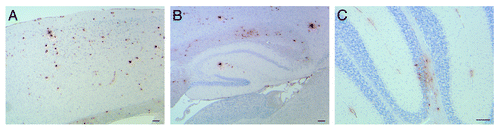
To further understand the mechanism(s) involved in Aβ generation and deposition associated with Presenilin 1 (PSEN1) mutations, we generated a novel knock-in model based on a Leu to Pro mutation at codon 166 in the murine Psen1. This mutation was chosen since is one of the most aggressive familial AD (FAD) mutations identified so far, originally found in a patient with onset of AD at 24 years of age.Citation13 Clinically, the disease in the patient was characterized by progressive ataxia and spasticity soon followed by cognitive deficits. Neuropathologic findings included neuritic and cotton wool plaques, and cerebral amyloid angiopathy (CAA), as well as the presence of NFTs and severe neuronal loss. In addition, pyramidal tract degeneration and severe cerebellar Aβ deposits were present (Boyer et al., manuscript in preparation). The PSEN1 L166P mutation leads to a significant increase in Aβ production in cell culture,Citation14,Citation15 and very severe amyloid deposition in a double transgenic mouse model.Citation16 However, Psen1-L166P knock-in mice do not develop plaque deposits, with mice analyzed up to 24 mo of age. The lack of amyloid deposition in the Psen1-L166P knock-in model may be in part due to differences in AβPP processing between mice and humans, and/or the three amino acids difference between the murine and the human Aβ sequence.Citation17 Plaque deposits can be observed when Psen1 knock-in models are crossed with FAD mutant AβPP mice, with the Psen1 mutations causing earlier and more extensive plaque formation.Citation5,Citation6 As a result, in an attempt to be able to observe Aβ deposition driven by the Psen1-L166P mutation without using exogenous promoters or mutant AβPP sequences, we crossed the Psen1-L166P knock-in mice with mice carrying the whole human WT AβPP gene (without FAD-associated mutations), previously generated using a yeast artificial chromosome (YAC)Citation17 to produce APP YAC x Psen1-L166P mice.Citation12 Expression of the WT AβPP gene in APP YAC transgenic mice do not lead to amyloid deposition.Citation18 APP YAC transgenic mice were chosen because they express human AβPP mRNA and protein at levels comparable to endogenous AβPP in the brain and other tissues, with similar relative levels of alternative splice human AβPP and murine Aβpp products and have all regulatory elements in its chromosomal environment.Citation18 Double homozygous APP YAC x Psen1-L166P mice begin to show Th-S positive amyloid deposition in the cerebral cortex between 5 and 6 mo of age. Parenchymal amyloid deposition involved the neocortex, the hippocampus, and the cerebellum (). Th-S positive amyloid plaques were surrounded by numerous dystrophic neurites and glial inflammation represented by reactive astrocytes and activated microglia. Diffuse Aβ deposits were detected using antibodies against the Aβ peptide. Immunohistochemical studies also showed the presence of intracellular Aβ deposits. CAA was observed in the cerebrum and cerebellum (). Although no obvious neuronal loss was observed, current work in the lab is aimed at assessing neuronal loss in the model as well as cognitive and motor deficits.
Figure 1. Amyloid deposition in APP YAC x Psen1-L166P mice. Sections of a 20-mo-old APP YAC x Psen1-L166P (+/+) mouse showing parenchymal amyloid deposition in the cerebral cortex (A), the hippocampus (B) and the cerebellum (C). Amyloid deposition was also observed in cerebral vessels and in pial (leptomeningeal) vessels. Immunohistochemistry using antibody 4G8 was performed as described (12). Scale bars = μ100.
Enhanced Production of Aβ42 Peptides Drives Amyloid Deposition
Amyloid deposition was not observed in heterozygous APP YAC x Psen1-L166P+/− or in APP YAC x Psen1-L166P−/− mice at the oldest age analyzed (24 months). However, replacement of the WT Psen1 allele led to amyloid deposition in APP YAC x Psen1-L166P+/+ mice, in agreement with previous work done with Psen1-M146V knock-in mice where a reduction of γ-secretase activity rather than an increase in Aβ42 levels was proposed to drive amyloid deposition in the model.Citation19 As in other animal models, the presence of a Psen1 knock-in mutation seems to enhance amyloidogenic processing of AβPP.Citation6-Citation9,Citation20 Analysis of Aβ40 and Aβ42 levels in mouse brain by ELISA showed that in APP YAC x Psen1-L166P+/− mice, the replacement of one WT allele for a mutant allele led to a significant increase in Aβ42 levels, a significant reduction in Aβ40 levels and a significant increase in Aβ42/Aβ40 ratios in the neocortex and the hippocampus. The increase in levels of Aβ42 and the decrease in those of Aβ40 were high in APP YAC x Psen1-L166P +/+ mice, and seemed to correlate with the number of WT Psen1 alleles replaced by the mutant L166P allele. In comparing with APP YAC x Psen1-L166P−/− mice, we did not observe a significant increase in total Aβ (Aβ40 + Aβ42) in APP YAC x Psen1-L166P+/− mice but it was significant (between 2 and 2.5 times) in APP YAC x Psen1-L166P+/+ mice. A detailed biochemical analysis aimed at identifying and characterizing the Aβ peptide species present in APP YAC x Psen1-L166P (+/+) mice, including post-translational modifications such as pyroglutamyl cyclization and N-terminal degradation which produces more hydrophobic Aβ species in humansCitation8,Citation21 is currently in progress.
γ-Secretase Processing and the Psen1-L166P Mutation
In the amyloidogenic pathway, AβPP undergoes successive proteolysis by BACE1 and the γ-secretase complex, while in the non-amyloidogenic pathway by α-secretase and γ-secretase.Citation1-Citation3 After α- or β-cleavage, the carboxyl terminal fragments (CTFs) of AβPP known as CTFα and CTFβ, respectively, remain membrane-associated and are further cleaved by γ-secretase, a protease complex comprised of presenilin, nicastrin, anterior pharynx defective 1, and PS enhancer 2.Citation1,Citation4 Levels of CTFs seem to inversely correlate with total γ-secretase activity.Citation22 Western blot analysis of brain samples showed that while full-length levels of AβPP remained constant, the levels of CTFβ and CTFα were significantly increased in both, cerebral cortex and hippocampus of APP YAC x Psen1-L166P (+/+) mice.Citation12 These data support the view that enhanced amyloid deposition may result from the removal of the WT Psen1 allele and impaired γ-secretase activity of the Psen1 L166P allele.Citation19 Decreased in γ-secretase activity may affect not only AβPP but it may also alter the processing of other essential substrates.Citation23 Although we did not detect any significant changes in Psen1 and Notch1 processing or in the expression of the Notch1 target genes Hes3 and Hes5 between Psen1-L166P−/− and Psen1-L166P+/+ mice, we observed lack of follicular development in female Psen1-L166P+/+ mice. We propose that these pathologic changes may be associated with abnormal Notch1 signaling as a consequence of defective γ-secretase activity.Citation12
Conclusions
Transgenic mouse models have proven useful for modeling various aspects of Aβ pathology in AD. However, none of these models recapitulates all aspects of AD. Mostly used are transgenic mice expressing mutant AβPP under the control of a strong heterologous promoter, however this feature adds to the complexity of evaluating the pathogenic significance of transgene-induced abnormalities. To circumvent this complexity, we created a mouse model in which the mutated gene is under the control of its own promoter. In order to accomplish this, we used gene targeting to modify the murine Psen1 gene on a transgenic mouse carrying two copies of the entire human WT AβPP gene. The latter possessed all the transcriptional regulatory elements required for proper spatial and temporal expression of AβPP. In the new mice (APP YAC x Psen1-L166P), expression of the Psen1-L166P mutation at normal levels under its endogenous control mechanism has a significant effect on AβPP processing and Aβ deposition. The APP YAC x Psen1-L166P model demonstrates that neither a strong promoter nor mutations in AβPP are needed to drive amyloid pathology. This model may be a unique tool, which might be ideal to explore a possible relationship between abnormal expression or splicing of AβPP and Aβ deposition. Moreover, it may be particularly suitable for testing therapies aimed at modifying secretase cleavage of AβPP in a cellular environment which is not affected by artifacts due to the presence of amino acid variations in the AβPP sequence. Such a difference may be of critical importance to assess therapeutic strategies for AD.
Acknowledgments
We thank Debra Lucas and Rose Richardson for technical assistance and Dr. Jose Bonnin for helpful comments. This study was supported by grants from the National Institute on Neurological Disorders and Stroke NS050227 (R.V.) and NS14426 (B.G.) and the National Institute on Aging AG037338 (R.V.) and AG10133 (B.G.).
References
- Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Sci Transl Med 2011; 3:77sr1
- Cole SL, Vassar R. The role of amyloid precursor protein processing by BACE1, the beta-secretase, in Alzheimer disease pathophysiology. J Biol Chem 2008; 283:29621 - 5; http://dx.doi.org/10.1074/jbc.R800015200; PMID: 18650431
- Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain 2011; 4:3; http://dx.doi.org/10.1186/1756-6606-4-3; PMID: 21214928
- Wolfe MS. The gamma-secretase complex: membrane-embedded proteolytic ensemble. Biochemistry 2006; 45:7931 - 9; http://dx.doi.org/10.1021/bi060799c; PMID: 16800619
- Hall AM, Roberson ED. Mouse models of Alzheimer's disease. [Epub ahead of print] Brain Res Bull 2011; 28; PMID: 22142973
- Crews L, Rockenstein E, Masliah E. APP transgenic modeling of Alzheimer’s disease: mechanisms of neurodegeneration and aberrant neurogenesis. Brain Struct Funct 2010; 214:111 - 26; http://dx.doi.org/10.1007/s00429-009-0232-6; PMID: 20091183
- Götz J, Ittner LM. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci 2008; 9:532 - 44; http://dx.doi.org/10.1038/nrn2420; PMID: 18568014
- Kokjohn TA, Roher AE. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: understanding the paradigms, limitations, and contributions. Alzheimers Dement 2009; 5:340 - 7; http://dx.doi.org/10.1016/j.jalz.2009.03.002; PMID: 19560104
- Ashe KH, Zahs KR. Probing the biology of Alzheimer’s disease in mice. Neuron 2010; 66:631 - 45; http://dx.doi.org/10.1016/j.neuron.2010.04.031; PMID: 20547123
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 2004; 7:954 - 60; http://dx.doi.org/10.1038/nn1302; PMID: 15311281
- Higgins LS, Catalano R, Quon D, Cordell B. Transgenic mice expressing human β-APP751, but not mice expressing β-APP695, display early Alzheimer’s disease-like histopathology. Ann N Y Acad Sci 1993; 695:224 - 7; http://dx.doi.org/10.1111/j.1749-6632.1993.tb23056.x; PMID: 8239286
- Vidal R, Sammeta N, Garringer HJ, Sambamurti K, Miravalle L, Lamb BT, et al. The Psen1-L166P-knock-in mutation leads to amyloid deposition in human wild-type amyloid precursor protein YAC transgenic mice. [Epub ahead of print] FASEB J 2012; 29; PMID: 21908716
- Murrell J, Evans RM, Boyer PJ, Piccardo P, Towfighi J, Ghetti B. Alzheimer disease with onset in adolescence due to a novel mutation in Presenilin 1 (L166P).. [Abstract 187] J Neuropathol Exp Neurol 2000; 59
- Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, et al. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci U S A 2002; 99:8025 - 30; http://dx.doi.org/10.1073/pnas.112686799; PMID: 12048239
- Bentahir M, Nyabi O, Verhamme J, Tolia A, Horré K, Wiltfang J, et al. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem 2006; 96:732 - 42; http://dx.doi.org/10.1111/j.1471-4159.2005.03578.x; PMID: 16405513
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 2006; 7:940 - 6; http://dx.doi.org/10.1038/sj.embor.7400784; PMID: 16906128
- Jankowsky JL, Younkin LH, Gonzales V, Fadale DJ, Slunt HH, Lester HA, et al. Rodent A beta modulates the solubility and distribution of amyloid deposits in transgenic mice. J Biol Chem 2007; 282:22707 - 20; http://dx.doi.org/10.1074/jbc.M611050200; PMID: 17556372
- Lamb BT, Sisodia SS, Lawler AM, Slunt HH, Kitt CA, Kearns WG, et al. Introduction and expression of the 400 kilobase amyloid precursor protein gene in transgenic mice [corrected]. Nat Genet 1993; 5:22 - 30; http://dx.doi.org/10.1038/ng0993-22; PMID: 8220418
- Wang R, Wang B, He W, Zheng H. Wild-type presenilin 1 protects against Alzheimer disease mutation-induced amyloid pathology. J Biol Chem 2006; 281:15330 - 6; http://dx.doi.org/10.1074/jbc.M512574200; PMID: 16574645
- Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, et al. FAD mutant PS-1 gene-targeted mice: increased A β 42 and A β deposition without APP overproduction. Neurobiol Aging 2002; 23:335 - 48; http://dx.doi.org/10.1016/S0197-4580(01)00330-X; PMID: 11959395
- Miravalle L, Calero M, Takao M, Roher AE, Ghetti B, Vidal R. Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry 2005; 44:10810 - 21; http://dx.doi.org/10.1021/bi0508237; PMID: 16086583
- Wang R, Wang B, He W, Zheng H. Wild-type presenilin 1 protects against Alzheimer disease mutation-induced amyloid pathology. J Biol Chem 2006; 281:15330 - 6; http://dx.doi.org/10.1074/jbc.M512574200; PMID: 16574645
- Sambamurti K, Greig NH, Utsuki T, Barnwell EL, Sharma E, Mazell C, et al. Targets for AD treatment: conflicting messages from γ-secretase inhibitors. J Neurochem 2011; 117:359 - 74; http://dx.doi.org/10.1111/j.1471-4159.2011.07213.x; PMID: 21320126