Abstract
Mammalian prions with significant levels of specific infectivity can be formed in vitro from mixtures of prion protein (PrP) and cofactor molecules, but not from PrP alone. We recently isolated and identified the essential membrane phospholipid phosphatidylethanolamine (PE) as an endogenous cofactor for prion propagation in vitro.Citation1 In this article, we discuss the potential role of PE and other essential cofactor molecules as a molecular link between the processes of prion formation and prion-induced neurodegeneration.
The mechanisms by which mammalian prions propagate and cause disease in the brains of infected animals are currently unknown, and the relationship between the two processes is also unclear. Our recent work identifying the membrane phospholipid phosphatidylethanolamine (PE) as an endogenous cofactor for prion propagation in vitroCitation1 suggests the possibility of a unifying mechanism in which prion formation and disease pathogenesis may be interconnected through the involvement of essential membrane lipids such as PE.
There has been significant interest in studying the mechanism by which mammalian prions replicate infectivity because prions do not adhere the central dogma of molecular biology (i.e., genetic information being encoded exclusively by replicating nucleic acids) despite their remarkable abilities to infect normal hosts and exhibit phenotypic strain variation in a manner reminiscent of viruses. Critical early studies showed that prion propagation requires the cellular conformer of the prion protein (PrPC),Citation2,Citation3 and that during propagation PrPC undergoes conformational change into a misfolded conformer termed PrPSc 4, 5. Later, a number of in vitro PrPSc propagation studies indirectly suggested that additional molecules might also be required for mammalian prion propagation, since PrPSc levels and/or prion infectivity could be amplified from crude brain homogenate but not a preparation of pure PrPC or recombinant (rec)PrP molecules.Citation6-Citation15 Employing a reductionist biochemical approach, we subsequently produced chemically defined hamster prions by using a substrate cocktail containing PrPC, copurified lipid molecules and synthetic RNA molecules.Citation16 Remarkably, we found that hamster prions with moderate levels of specific infectivity could be produced from these components de novo, showing for the first time that infectious prions could be produced spontaneously in a rigorously prion-free environment, an event that must occur in sporadic Creutzfeldt Jakob disease.Citation16 Subsequent studies showed that while RNA and other polyanionic molecules facilitate the propagation of hamster prions, they appear to play no role in the propagation of various strains of mouse or vole prions.Citation17 This surprising species specificity led us to seek the endogenous cofactor responsible for prion propagation in other animal species by using a variety of biochemical characterization and enrichment methods, resulting in the isolation and identification of PE as the essential molecule.Citation1 PE has a polar head group, is found in all living cells and is particularly enriched in nervous tissue. Within membranes, PE creates a more viscous bilayer than phosphatidylcholine. The prion promoting ability of PE was definitively confirmed by using synthetic PE as a solitary cofactor to generate infectious recombinant prions.Citation1
As an in vitro prion propagation cofactor, PE displays several unique properties. When reconstituted with either PrPC or recPrP substrate alone (i.e., without RNA or any other cofactors), PE facilitates the propagation of prions from every animal species tested, including deer chronic wasting disease (CWD) and sheep scrapie.Citation1 The conversion of recPrP to recPrPSc in vitro is ~10-fold higher in terms of percent conversion when facilitated by PE alone compared with the previously described combination of phosphatidylglycerol and RNA. Treatment of brain homogenate with phospholipase abolishes the in vitro propagation of both hamster and mouse prions, whereas RNase only inhibits hamster prion propagation.Citation1 Thus, PE appears to be an endogenous cofactor that can by itself efficiently facilitate the in vitro propagation of prions from multiple animal species. It is currently unknown whether any prion cofactor identified biochemically, including PE, plays a physiological role during the process prion replication in vivo. However, because PE facilitates (and phospholipase inhibits) the conversion of prions from multiple species in vitro, it seems reasonable to hypothesize that PE and other phospholipids may play a widespread role in prion formation in the brains of infected animals. A practical obstacle to testing the hypothesis that phospholipids participate in prion conversion in vivo is that membrane lipids are highly regulated, essential and difficult to manipulate. New approaches will need to be developed in order to manipulate phospholipid levels in living cells and animals.
PrPC is located on the extracellular leaflet of the plasma membrane, and PrPSc formation is believed to occur either on the cell surface or within an endocytic compartmentCitation18 (). PE is distributed asymmetrically across the plasma membrane; ~80% is located within the intracellular leaflet and ~20% is located in the extracellular leaflet of erythrocytes.Citation19 Phospholipid asymmetry is maintained by ATP-dependent flippase enzymes, and is disrupted by processes such as apoptosis and cellular aging.Citation20 It is interesting to speculate that age-dependent disruption of phospholipid distribution might play a role in the pathogenesis of prion disease and other neurodegenerative disorders by allowing cofactor molecules to become more accessible to surface-expressed pathogenic proteins such as PrP (). This hypothetical scenario offers an intriguing molecular explanation for the increased incidence of neurodegenerative diseases among the elderly.
Figure 1. Hypothetical model of PE in prion formation and pathogenesis. Prion Formation: the relative paucity of PE on the extracellular leaflet of the plasma membrane of cells is actively maintained by a flippase enzyme, whose activity declines with age, allowing more PE to be localized on the extracellular surface where it is accessible to PrPC. Pathogenesis: If extracellular PE becomes consumed during the process of prion formation by binding irreversibly to PrPSc, its depletion from the membrane surface will cause biophysical changes in the membrane itself and dysregulation of surface-expressed signaling proteins (SP) that normally bind PE, resulting in cell death.
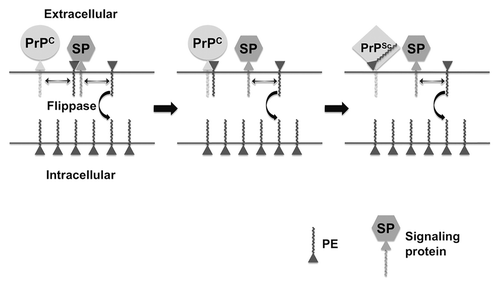
It is also interesting to speculate that the distribution of phospholipids in the membrane of infected neurons might also become disrupted if specific surface phospholipid molecules such as PE were irreversibly bound to PrPSc during the prion conversion process (). In this scenario, the pathogenic process (i.e., the mechanism by which PrPSc formation leads to cell death) would be tied to the infectious process (i.e., the mechanism by which PrPSc molecules are formed from PrPC molecules in cells) at a molecular level through the common involvement of an essential membrane phospholipid cofactor. Indeed, even minor changes in the distribution of PE across the plasma membrane are known to cause cellular dysfunction and degeneration, presumably by changing the biophysical properties of the membrane itself and by making surface PE less accessible to various signaling proteins that require it for activity (). This hypothetical model of prion pathogenesis provides an alternative mechanism that does not need to invoke the production of an unidentified toxic PrP species distinct from PrPSc during the process of prion formation.Citation21 The depletion of surface PE (or other phospholipids) would require membrane anchoring of PrPC during the conversion process, in agreement with the observation that transgenic mice expressing only PrPC molecules not anchored to the plasma membrane display attenuated pathological changes.Citation22 Also, the rate of depletion of surface phospholipid cofactors would be proportional to PrPC expression levels, in agreement with the observation that the rate of pathogenic progression in prion-infected animals (and their incubation period) is proportional to PrPC protein levels.Citation23
In conclusion, we have recently isolated PE as an endogenous cofactor for mammalian prion formation in vitro. Although new systems will be needed to study whether PE or other phospholipids play a similar role during prion replication in vivo, their identification allows us to envision a new unified model of prion propagation and pathogenesis.
NOTE: Author please cite references Citation4 and Citation5.
References
- Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, et al. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A 2012; 109:8546 - 51; http://dx.doi.org/10.1073/pnas.1204498109; PMID: 22586108
- Prusiner SB, Groth D, Serban A, Koehler R, Foster D, Torchia M, et al. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A 1993; 90:10608 - 12; http://dx.doi.org/10.1073/pnas.90.22.10608; PMID: 7902565
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991; 30:7672 - 80; http://dx.doi.org/10.1021/bi00245a003; PMID: 1678278
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; http://dx.doi.org/10.1073/pnas.90.23.10962; PMID: 7902575
- Caughey B, Kocisko DA, Raymond GJ, Lansbury PT Jr.. Aggregates of scrapie-associated prion protein induce the cell-free conversion of protease-sensitive prion protein to the protease-resistant state. Chem Biol 1995; 2:807 - 17; http://dx.doi.org/10.1016/1074-5521(95)90087-X; PMID: 8807814
- Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 1995; 375:698 - 700; http://dx.doi.org/10.1038/375698a0; PMID: 7791905
- Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT Jr., Caughey B. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci U S A 1995; 92:3923 - 7; http://dx.doi.org/10.1073/pnas.92.9.3923; PMID: 7732006
- Saborío GP, Soto C, Kascsak RJ, Levy E, Kascsak R, Harris DA, et al. Cell-lysate conversion of prion protein into its protease-resistant isoform suggests the participation of a cellular chaperone. Biochem Biophys Res Commun 1999; 258:470 - 5; http://dx.doi.org/10.1006/bbrc.1999.0660; PMID: 10329411
- Lucassen R, Nishina K, Supattapone S. In vitro amplification of protease-resistant prion protein requires free sulfhydryl groups. Biochemistry 2003; 42:4127 - 35; http://dx.doi.org/10.1021/bi027218d; PMID: 12680767
- Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell 2005; 121:195 - 206; http://dx.doi.org/10.1016/j.cell.2005.02.011; PMID: 15851027
- Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, et al. Synthetic mammalian prions. Science 2004; 305:673 - 6; http://dx.doi.org/10.1126/science.1100195; PMID: 15286374
- Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen HO, et al. Protease-sensitive synthetic prions. PLoS Pathog 2010; 6:e1000736; http://dx.doi.org/10.1371/journal.ppat.1000736; PMID: 20107515
- Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 2010; 285:14083 - 7; http://dx.doi.org/10.1074/jbc.C110.113464; PMID: 20304915
- Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 2010; 119:177 - 87; http://dx.doi.org/10.1007/s00401-009-0633-x; PMID: 20052481
- Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A 2007; 104:9741 - 6; http://dx.doi.org/10.1073/pnas.0702662104; PMID: 17535913
- Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 2010; 49:3928 - 34; http://dx.doi.org/10.1021/bi100370b; PMID: 20377181
- Taraboulos A, Raeber AJ, Borchelt DR, Serban D, Prusiner SB. Synthesis and trafficking of prion proteins in cultured cells. Mol Biol Cell 1992; 3:851 - 63; PMID: 1356522
- Zachowski A. Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem J 1993; 294:1 - 14; PMID: 8363559
- Devaux PF, Zachowski A, Morrot G, Cribier S, Fellmann P, Geldwerth D, et al. Control of the transmembrane phospholipid distribution in eukaryotic cells by aminophospholipid translocase. Biotechnol Appl Biochem 1990; 12:517 - 22; PMID: 2288706
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318:930 - 6; http://dx.doi.org/10.1126/science.1138718; PMID: 17991853
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435 - 9; http://dx.doi.org/10.1126/science.1110837; PMID: 15933194
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540 - 2; http://dx.doi.org/10.1038/nature09768; PMID: 21350487