Abstract
Shadoo (Sho) is a brain glycoprotein with similarities to the unstructured region of PrPC. Frameshift alleles of the Sho gene, Sprn, are reported in variant Creutzfeldt-Jakob disease (vCJD) patients while Sprn mRNA knockdown in PrP-null (Prnp0/0) embryos produces lethality, advancing Sho as the hypothetical PrP-like “pi” protein. Also, Sho levels are reduced as misfolded PrP accumulates during prion infections. To penetrate these issues we created Sprn null alleles (Daude et al., Proc. Natl. Acad. Sci USA 2012; 109(23): 9035–40). Results from the challenge of Sprn null and TgSprn transgenic mice with rodent-adapted prions coalesce to define downregulation of Sho as a “tracer” for the formation of misfolded PrP. However, classical BSE and rodent-adapted BSE isolates may behave differently, as they do for other facets of the pathogenic process, and this intriguing variation warrants closer scrutiny. With regards to physiological function, double knockout mice (Sprn0/0/Prnp0/0) mice survived to over 600 d of age. This suggests that Sho is not pi, or, given the accumulating data for many activities for PrPC, that the pi hypothesis invoking a discrete signaling pathway to maintain neuronal viability is no longer tenable.
Genotype/Phenotype Relationships for PrP and Sho
Genetic analysis can provide profound insights into complex biological systems at the whole organism level. However, few knockouts in systematic studies in Arabidopsis thaliana, C. Elegans and S. Cerevisiae produce a demonstrable phenotype - less than 2%, 14%, and 40–60% respectively.Citation1-Citation3 For mice, the functional genomic pipeline has generated thousands of null alleles, but – perhaps compounded by under-reporting of “negative results” – this is inferred to have yielded similar low percentages. Broadly speaking, these results can be rationalized by genetic redundancy in the case of gene families, and by genetic robustness for the case where parallel pathways perform similar functions.Citation3 With regards to the specifics of mammalian prion biology, the cellular prion protein (PrPC), a precursor to the infectious prion protein isoform, is encoded by a host locus, the Prnp gene. At one stage it was hoped that phenotypes in homozygous null mice would provide a simple penetrating insight into the riddle of PrPC function, but unfortunately this was not the case. Prnp0/0 mice are resistant to prion infection but have an ostensibly normal development. Here we review recent results on the knockout of the PrP-like Sho protein, the creation of PrP/Sho double-knockout animals and the implications of these findings for the so-called pi hypothesis.
PrPC
For context, we will first consider properties of these two GPI-linked neuronal glycoproteins (). For PrPC, the precursor to misfolded PrPSc protein in prion infection, the situation is quite complex and for the sake of brevity, the reader is directed to recent reviews.Citation4-Citation6 Nonetheless, it is worth mentioning that PrP has an anti-apoptotic role in vitroCitation7 and that in vivo it is involved in neuroprotection, as Prnp0/0 mice are more sensitive to ischemic insults and to seizure, whereas overexpression of PrP in a rat stroke model is protective. In terms of signaling, PrPC is coupled to Fyn kinase in 1C11 cells. PrPC is also involved in copper binding and modified response to oxidative stress, in cell adhesion and in the glutamatergic system by binding to NMDA receptors. Another way to discern function follows “guilt-by-association,” by identifying protein partners with (hopefully) clear-cut functions. However, this hasn’t proven particularly useful due to the wealth of interactors and the failure of different labs to routinely identify the same proteins.Citation4,Citation8,Citation9 Two routes to resolution of the PrPC puzzle invoke theory. One posits PrPC as a generalized scaffold and signaling node for many membrane proteins,Citation6 and a second posits two hypothetical “missing” partners. Thus, since PrPC deficiency is not lethal and since Prnp0/0 mice lack a strong phenotype, it has been inferred from these data, and from transgenetic studies with internally deleted forms of PrPC, that another protein takes over PrPC function in knockout mice, a hypothetical functional analog termed “pi” (π),Citation10 and that both PrPC and pi dock an entity termed “LPrP” to transmit signals needed for neuronal viability.
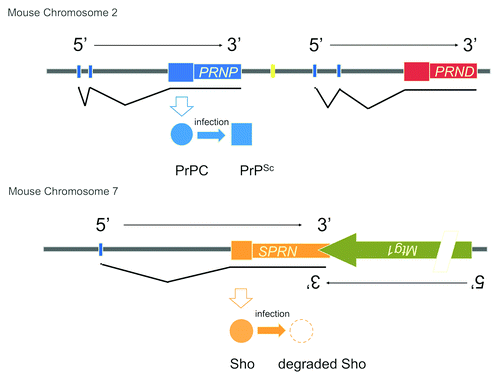
Figure 1. CNS-expressed prion proteins in mice. PrPC encoded by Prnp and Sho encoded by Sprn are located on different chromosomes. Their adjacent flanking genes (Prnd, Mtg1) are shown and these encode the doppel protein and mitochondrial GTPase 1, respectively. Protein products of Prnp and Sprn are shown but are omitted for Prnd and Mtg1 for the sake of simplicity. Sizes of the transcription units are not to scale and a complex intron/exon structure for Mtg1 has been simplified for the purpose of this figure (slash mark).
Sho
Could Sho be pi? The Sho glycoprotein encoded by Sprn has partial homology to the PrPC hydrophobic domain, a series of N-terminal repeats, a C-terminal N-linked glycosylation and a GPI anchor.Citation11,Citation12 In terms of physiological action, Sho, like PrPC, can exhibit neuroprotective properties and also shares a number of protein binding partners with PrPC.Citation12, Citation13 Unlike PrPC, this lab failed to define expression of Sho outside the central nervous system (CNS) by immunoblotting, although expression in reproductive tissues has been inferred from the analysis of GFP reporter mice driven by Sprn promoter elements (http://www.gensat.org). Also akin to PrPC, we have found that Sho is naturally endoproteolysed into N- and C-terminal fragments, with abundant N1 and C1 fragments but also including species of 12 and 14kDa.Citation12,Citation14 Given increasing interest in the protective bioactivity of PrP N1 fragments,Citation15 analogous studies of Sho N1 in the brain, a neuropeptide of sufficient abundance to be detected in wildtype (wt) mice would appear in order. An important task will be to define the protease that naturally cleaves Sho, which could be the same as the one that cleaves prior to the hydrophobic domain in PrPC—the latter activity has sometimes been equated with ADAM proteases but in response to complications in this storyCitation16,Citation17 has also been accorded the more cautious moniker of “alpha-PrPase”.Citation18
In light of a potential Sho/pi connection, we looked at neuroanatomical expression in the brain and compared it to PrP. Interestingly, expression of the two proteins was not fully superimposable. In the hippocampus, Sho is expressed in the molecular layer of the dentate gyrus whereas PrP expression is more prominent in the molecular layer adjacent to CA1 neurons,Citation14 and future versions of these experiments will benefit from monoclonal antibodies specific to N- and C-terminal fragments of Sho. Since the hippocampus is involved in modification of behavior, attention, and spatial memory, and since expression of Sho and PrP are abutted, memory-based behavioral tests to study the differences between Sprn0/0 and Prnp0/0 mice might be in order to pinpoint specific contributions of the two proteins.
The concept of biological redundancy forms a strand in this review and an implicit assumption that follows from this is that individual knockout stocks are without notable phenotypes in the resting state. This has been mentioned for Prnp0/0 mice and also proves to be the case for Sprn0/0 mice, as both male and female mice are fertile and do not show overt malformations. However, a subtle alteration in body weight was discerned at the p < 0.05 level in Sprn0/0 mice maintained on two genetic backgrounds, with knockout animals a little lighter than their wt counterparts. Furthermore, using Sprn0/0 mice as a negative control we also defined Sho expression, by immunostaining, in hypothalamic neurons in wt mice.Citation14 Of note (1) this neuroanatomical structure contains nuclei that control feeding behavior,Citation19 (2) weight loss can be observed in prion-infected animals,Citation20 suggestive to some of an endocrinopathy,Citation21 and (3) Sho is downregulated in prion infections. While these three observations are tantalizing, a mechanistic relationship featuring Sho as a determinant of body mass alteration subsequent to PrPSc accumulation remains speculative at this time. In terms of chemical phenotypes, PrPC levels in Sprn0/0 mice are comparable to wt mice of the same genotype, arguing against compensatory cross-regulated expression of the two genes.Citation14
Action of Sho in Prion Infections: Witness or Accessory?
A next phenotypic issue at hand is the outcome of prion challenge in Sprn0/0 mice. From a frameshift mutation observed in two vCJD cases, and a polymorphism in the signal sequence associated with risk of sporadic CJDCitation22 one can infer a potential role in human prion disease. In our studies so far we have studied prion infection in Sprn0/0 mice, challenging the animals by intracerebral, intraperitoneal and oral routes with the mouse-adapted RML isolate of scrapie prions. These experiments failed to reveal distinctions - either in the symptoms or the duration of the disease—from parallel infections of wt mouse controls. Future studies will need to encompass vCJD challenge of Sprn0/0 x Tg(HuPrP) “humanized” mice and 301V (mouse-adapted BSE) challenge of Sprn0/0 x Prnpb mice. However, while an active role of Sho in prion replication remains conjectural at this juncture, it is established beyond cavil that the levels of protein are markedly reduced in prion infections with several prion isolates (“strains”) producing slightly different end-stage pathologies.Citation23-Citation25 Provocatively, BSE, a prion isolate with unusual hyperglycosylated PrPScCitation26 and with a vast host-range including human and non-human primates, bovidae, and felidae,Citation27 may be an exception to this emerging rule. With regards to reduction in Sho being a non-specific response to tissue damage, this phenomenon was not observed for four other neurodegenerative syndromes. Our laboratory and other workers have generally observed a correlation between the accumulation of protease-resistant PrPSc in the brain and the disappearance of Sho, both in wt animals and in TgSprn mice overexpressing wt mouse Sho.Citation23,Citation24 How might this disappearance of Sho be explained? Sprn transcript levels are unperturbed by prion infection, so proteolysis is an obvious candidate and we favor the interpretation that Sho levels are passively monitoring an in vivo clearance mechanism directed against PrPSc (rather than Sho actively perturbing this process or, for that matter, perturbing prion replication). Chronically infected prion cultures do not usually exhibit morphological signs of cellular pathology (for review seeCitation28) yet can exhibit robust Sho downregulation (C. E. Mays, in preparation). Besides underscoring the conclusion that the Sho downregulation effect is not related to cellular damage, these systems should provide opportunities for profound mechanistic insight by use of antibiotics and protease inhibitors. Furthermore, the early preclinical disappearance of Sho—or perhaps the appearance of telltale proteolytic fragments—could provide a new diagnostic angle on prion infections.
Sho, PrPC and Embryonic Development
Returning to physiology there is an implicit question as to the viability of “double” knockout mouse embryos. The first attempt at addressing this issue was taken in Prnp0/0 embryos, where Sprn knockdown using lentiviral vectors was reported to result in embryonic lethality,Citation29 potentially validating the pi hypothesis with pi equated to Sho. Curiously, in contrast to this robust effect, using penetrant null alleles of Sprn and Prnp to generate double knockouts (Sprn0/0/Prnp0/0) we found the adult mice to be viable and fertile.Citation14 Moreover, we performed inbreeding crosses over several generations to ensure that this phenomenon is not due to maternal effects (e.g., carryover of maternally encoded Sho mRNAs into Sprn homozygous null embryos). So, it seems that although PrP and Sho have overlaps in their chemical properties, neither alone nor in combination are they required for the completion of embryogenesis when using fully penetrant, constitutive null alleles. How then did the lentiviral studies in Prnp0/0 embryos yield a seemingly contradictory result?
Besides a theoretical compensation phenomenon by other genes (discussed below), technical explanations also need consideration. The possibility that Sprn phenotypic effects are extremely sensitive to conditions of husbandry cannot be excluded. In terms of genetic effects, inbred strain background, a standard argument invoked in the case of divergent allelic phenotypes, is not so different between the two experiments (FVB/N for the knockdown studies vs. FVB/NCr x 129Pas for our study). Perhaps more pivotal is the adopted strategy, genetic deletion of all Sho ORF codons in the germline vs. virally-vectored shRNAs that target the 3' untranslated region (UTR) of Sprn mRNA within cells of infected embryos. It is known that the Sprn transcription unit overlaps with an adjacent gene encoding a mitochondrial protein, Mtg1, and it is possible that shRNAs against the Sprn 3′UTR might affect Mtg1 expression if there is transcriptional interference between the two loci. It is of interest to recall that an early interpretation of the impact of PrP knockout on cerebellar degenerationCitation30 had to be revisited subsequent to a demonstration of an artifactual effect of certain Prnp null alleles on the adjacent Prnd transcription unit.Citation31,Citation32 However, Mtg1 transcript levels are reported as being unaltered by shRNA vectors in a follow-up study,Citation33 tipping the balance to a consideration of off-axis effects of shRNAs against irrelevant genes. Further validation of shRNA knockdown strategies could involve (1) confirming successful knockdown of the Sho protein and (2) genetic rescue of the knockdown effect on protein levels and embryonic development by co-administration of a second vector with the shRNA vector: this second vector would encode a wt Sho protein open reading frame upstream of a heterologous 3'UTR (i.e., a 3' UTR not targeted by the current anti-Sprn shRNAs). Data to ascertain and distinguish (3) the phenotypic impact of lentiviral Mtg1 knockdown would also comprise a useful, additional point of reference.
Activities of Sho and PrPC in the Adult CNS
The viability of Sprn0/0 and Prnp0/0 knockout mice leads next to a consideration of action and synergy beyond embryos and, certainly, within the CNS of adult mice that has the most fulminant expression of these two proteins. While aging Prnp0/0 mice have a polyneuropathyCitation34,Citation35 that is not apparently enhanced by the absence of Sho,Citation14 it will be important to pursue other parameters in Sprn0/0 mice (e.g., susceptibility to stroke and seizure) and to compare the results with Prnp0/0 animals.
A theoretical concern with the constitutive null alleles is that phenotypic impacts might be masked by a counterbalancing expression of a functional homolog (or homologs) induced as a result of the gene deficiency. For PrPC, the possibility of other CNS proteins with functional homology is not inconceivable.Citation36 One way to address this is to perform “-omic” analyses (for example, but not limited to, microarray analyses), to compare Sprn0/0, Prnp0/0 and Sprn0/0/Prnp0/0 mice. Another tack is to use conditional null alleles. This is based on the assumption that compensatory proteins, induced transiently in embryogenesis, might not be available once adulthood is attained: hence a single conditional knockout in adults or activation of conditional null PrP alleles in adult Sprn0/0 mice might reveal the “true” phenotype of protein deficiency. This could lie in the behavioral domain and be revealed by forced swim and open field tests, for example, or it might comprise a neurodegenerative syndrome. If a neurologic phenotype was still absent under standard housing conditions, then focus would turn to conditions of CNS duress or adaptation. Since both Sho and PrP are putatively neuroprotective proteins, lesioning or neurotoxic challenge of double knockout mice might yield impairments that transcend the individual knockouts.
Pi, on the Face of It
While the equation “Sho = pi” might have been a foregone conclusion a few years backCitation29 this is not so clear now. One viewpoint is that Sho is as plausible a candidate for pi as can be mustered from biochemical data, and that the pi hypothesis may be erroneous. Observations that underpin this position are (1) largely overlapping patterns of Sho/PrP gene expression, (2) failure to pass a critical genetic test and (3) a vacant candidacy for a transmembrane protein (LPrP) with two distinct PrP binding sites. The fact that much of PrP is endoproteolyzed such that the N-and C-termini are separated might comprise a further caveat for the hypothesis (which posits simultaneous docking by N- and C-terminal PrP sites to the LPrP partner). One may also note that another hypothesis that seeks to explain the properties of internally-deleted PrP does not concern signaling for neuronal survival under basal conditions, but instead as a response to stress events.Citation37 Alternatively, if one believes in the pi hypothesis (which includes an assumption of genetic redundancy), then Sho cannot be pi because of the health of the Prnp0/0/Sprn0/0 mice.
In reaching a final position, there are other questions and subtleties that apply to the failure to discern a phenotype in a knockout strain. First, there is always a question as to whether the “right” phenotype is being tested, and, beyond redundancy from members of a gene family, there is another concept to explain a lack of a phenotype in knockouts, this being “genetic robustness.” Even if two proteins function in the same pathway, a parallel pathway might exist such that functionality and/or viability is maintained.Citation3 For PrPC there is already evidence against high affinity interaction with just single protein, evidence for interactions with diverse proteins and evidence for a diversity of actions including functional modulation of distinct ion channels and protection against different neurotoxic insults. Since these complex activities must inevitably involve a variety of accessory proteins (e.g., ligand and voltage gated channels are typically multi subunit complexes), and, since parallel systems from neuroprotection go beyond just biochemical pathways in neurons to include contributions from glia and microglia,Citation38 then the complex cell-surface landscape of PrPC and Sho seems far more reconcilable with the concept of genetic robustness than genetic redundancy. Thus, rather than double knockouts excluding that Sho is pi, we are more inclined to conclude that the pi hypothesis is closed as an avenue in prion biology research. Hopefully, however, the new knockout mouse lines will contribute to a better understanding of these small, enigmatic glycoproteins.
Related Research Data
References
- Bouché N, Bouchez D. Arabidopsis gene knockout: phenotypes wanted. Curr Opin Plant Biol 2001; 4:111 - 7; http://dx.doi.org/10.1016/S1369-5266(00)00145-X; PMID: 11228432
- Kim SK. Http://C. elegans: mining the functional genomic landscape. Nat Rev Genet 2001; 2:681 - 9; http://dx.doi.org/10.1038/35088523; PMID: 11533717
- Barbaric I, Miller G, Dear TN. Appearances can be deceiving: phenotypes of knockout mice. Brief Funct Genomic Proteomic 2007; 6:91 - 103; http://dx.doi.org/10.1093/bfgp/elm008; PMID: 17584761
- Watts JC, Westaway D. The prion protein family: diversity, rivalry, and dysfunction. Biochim Biophys Acta 2007; 1772:654 - 72; http://dx.doi.org/10.1016/j.bbadis.2007.05.001; PMID: 17562432
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 2008; 3:11 - 40; http://dx.doi.org/10.1146/annurev.pathmechdis.3.121806.154326; PMID: 18233951
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev 2008; 88:673 - 728; http://dx.doi.org/10.1152/physrev.00007.2007; PMID: 18391177
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 2001; 276:39145 - 9; http://dx.doi.org/10.1074/jbc.C100443200; PMID: 11522774
- Mehrpour M, Codogno P. Prion protein: From physiology to cancer biology. Cancer Lett 2010; 290:1 - 23; http://dx.doi.org/10.1016/j.canlet.2009.07.009; PMID: 19674833
- Aguzzi A, Baumann F, Bremer J. The prion’s elusive reason for being. Annu Rev Neurosci 2008; 31:439 - 77; http://dx.doi.org/10.1146/annurev.neuro.31.060407.125620; PMID: 18558863
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998; 93:203 - 14; http://dx.doi.org/10.1016/S0092-8674(00)81572-X; PMID: 9568713
- Premzl M, Sangiorgio L, Strumbo B, Marshall Graves JA, Simonic T, Gready JE. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene 2003; 314:89 - 102; http://dx.doi.org/10.1016/S0378-1119(03)00707-8; PMID: 14527721
- Watts JC, Drisaldi B, Ng V, Yang J, Strome B, Horne P, et al. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J 2007; 26:4038 - 50; http://dx.doi.org/10.1038/sj.emboj.7601830; PMID: 17703189
- Watts JC, Huo H, Bai Y, Ehsani S, Jeon AH, Shi T, et al. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog 2009; 5:e1000608; http://dx.doi.org/10.1371/journal.ppat.1000608; PMID: 19798432
- Daude N, Wohlgemuth S, Brown R, Pitstick R, Gapeshina H, Yang J, et al. Knockout of the prion protein (PrP)-like Sprn gene does not produce embryonic lethality in combination with PrP(C)-deficiency. Proc Natl Acad Sci U S A 2012; 109:9035 - 40; http://dx.doi.org/10.1073/pnas.1202130109; PMID: 22619325
- Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 2009; 284:35973 - 86; http://dx.doi.org/10.1074/jbc.M109.051086; PMID: 19850936
- Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, et al. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem 2009; 284:22590 - 600; http://dx.doi.org/10.1074/jbc.M109.032599; PMID: 19564338
- Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, et al. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem 2001; 276:37743 - 6; PMID: 11477090
- Oliveira-Martins JB, Yusa S, Calella AM, Bridel C, Baumann F, Dametto P, et al. Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS One 2010; 5:e9107; http://dx.doi.org/10.1371/journal.pone.0009107; PMID: 20161712
- Vianna CR, Coppari R. A treasure trove of hypothalamic neurocircuitries governing body weight homeostasis. Endocrinology 2011; 152:11 - 8; http://dx.doi.org/10.1210/en.2010-0778; PMID: 21068159
- Outram GW. Changes in drinking and feeding habits of mice with experimental scrapie. J Comp Pathol 1972; 82:415 - 27; http://dx.doi.org/10.1016/0021-9975(72)90041-2; PMID: 4630696
- Bailey JD, Berardinelli JG, Rocke TE, Bessen RA. Prominent pancreatic endocrinopathy and altered control of food intake disrupt energy homeostasis in prion diseases. J Endocrinol 2008; 197:251 - 63; http://dx.doi.org/10.1677/JOE-07-0516; PMID: 18434355
- Beck JA, Campbell TA, Adamson G, Poulter M, Uphill JB, Molou E, et al. Association of a null allele of SPRN with variant Creutzfeldt-Jakob disease. J Med Genet 2008; 45:813 - 7; http://dx.doi.org/10.1136/jmg.2008.061804; PMID: 18805828
- Westaway D, Genovesi S, Daude N, Brown R, Lau A, Lee I, et al. Down-regulation of Shadoo in prion infections traces a pre-clinical event inversely related to PrP(Sc) accumulation. PLoS Pathog 2011; 7:e1002391; http://dx.doi.org/10.1371/journal.ppat.1002391; PMID: 22114562
- Watts JC, Stöhr J, Bhardwaj S, Wille H, Oehler A, Dearmond SJ, et al. Protease-resistant prions selectively decrease Shadoo protein. PLoS Pathog 2011; 7:e1002382; http://dx.doi.org/10.1371/journal.ppat.1002382; PMID: 22163178
- Miyazawa K, Manuelidis L. Agent-specific Shadoo responses in transmissible encephalopathies. J Neuroimmune Pharmacol 2010; 5:155 - 63; http://dx.doi.org/10.1007/s11481-010-9191-1; PMID: 20112073
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996; 383:685 - 90; http://dx.doi.org/10.1038/383685a0; PMID: 8878476
- Imran M, Mahmood S. An overview of animal prion diseases. Virol J 2011; 8:493; http://dx.doi.org/10.1186/1743-422X-8-493; PMID: 22044871
- Vilette D. Cell models of prion infection. Vet Res 2008; 39:10; http://dx.doi.org/10.1051/vetres:2007049; PMID: 18073097
- Young R, Passet B, Vilotte M, Cribiu EP, Béringue V, Le Provost F, et al. The prion or the related Shadoo protein is required for early mouse embryogenesis. FEBS Lett 2009; 583:3296 - 300; http://dx.doi.org/10.1016/j.febslet.2009.09.027; PMID: 19766638
- Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, et al. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 1996; 380:528 - 31; http://dx.doi.org/10.1038/380528a0; PMID: 8606772
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 1999; 292:797 - 817; http://dx.doi.org/10.1006/jmbi.1999.3108; PMID: 10525406
- Silverman GL, Qin K, Moore RC, Yang Y, Mastrangelo P, Tremblay P, et al. Doppel is an N-glycosylated, glycosylphosphatidylinositol-anchored protein. Expression in testis and ectopic production in the brains of Prnp(0/0) mice predisposed to Purkinje cell loss. J Biol Chem 2000; 275:26834 - 41; PMID: 10842180
- Passet B, Young R, Makhzami S, Vilotte M, Jaffrezic F, Halliez S, et al. Prion protein and shadoo are involved in overlapping embryonic pathways and trophoblastic development. PLoS One 2012; 7:e41959; http://dx.doi.org/10.1371/journal.pone.0041959; PMID: 22860039
- Katamine S, Nishida N, Sugimoto T, Noda T, Sakaguchi S, Shigematsu K, et al. Impaired motor coordination in mice lacking prion protein. Cell Mol Neurobiol 1998; 18:731 - 42; http://dx.doi.org/10.1023/A:1020234321879; PMID: 9876879
- Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 2010; 13:310 - 8; http://dx.doi.org/10.1038/nn.2483; PMID: 20098419
- Schmitt-Ulms G, Ehsani S, Watts JC, Westaway D, Wille H. Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS One 2009; 4:e7208; http://dx.doi.org/10.1371/journal.pone.0007208; PMID: 19784368
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. EMBO J 2007; 26:548 - 58; http://dx.doi.org/10.1038/sj.emboj.7601507; PMID: 17245437
- Polazzi E, Monti B. Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol 2010; 92:293 - 315; http://dx.doi.org/10.1016/j.pneurobio.2010.06.009; PMID: 20609379