Abstract
Alzheimer disease (AD) is the most prevalent cause of dementia. Amyloid-β (Aβ) oligomers are potent synaptotoxins thought to mediate AD-related phenotypes. Cellular prion protein (PrPC) has been identified as a high-affinity receptor for Aβ oligomers. Herein, we review the functional consequences of Aβ oligomer binding to PrPC on the neuronal surface. We highlight recent evidence that Fyn kinase mediates signal transduction downstream of the PrPC-Aβ oligomer complex. These studies suggest that PrPC has a central role in AD pathogenesis and may provide a target for therapeutic intervention in AD.
Alzheimer disease (AD) is the most common cause of age-related dementia, affecting more than 35 million people worldwide,Citation1 and potentially 115 million people by 2050. The histopathological hallmarks of AD are extracellular amyloid β (Aβ) peptide aggregates or ‘amyloid plaques’ and neurofibrillary tangles (NFTs) containing the microtubule-associated protein, tau. The histological features are associated with neurodegeneration and cognitive decline, leading eventually to death.
The leading hypothesis for AD postulates causation by Aβ peptide (especially Aβ42), which is derived proteolytically from amyloid precursor protein (APP) by the action of β- and γ-secretases.Citation2 Aβ peptide can exist as soluble monomers, soluble oligomers, intermediate protofibrils and insoluble fibrillar aggregates.Citation3 Among these Aβ species, cognitive impairment and the degree of synaptic deficit in the AD brain are most strongly correlated with soluble oligomeric assemblies of Aβ, suggesting that soluble Aβ oligomers trigger neuronal toxicity to cause dementia.Citation3-Citation7
Identification of PrPC as a High-Affinity Receptor for Aβ Oligomers
Cellular prion protein (PrPC) was identified as a high affinity binding site of Aβ by a genome-wide unbiased expression cloning.Citation8 After the binding affinity is corrected for the extent of oligomerization, the estimated affinity of oligomeric Aβ is 1–10 nM. In vitro and in vivo studies have confirmed a direct and oligomer-specific high affinity interaction of soluble Aβ oligomer with PrPC.Citation9-Citation12
PrPC is a membrane-anchored neuronal glycoprotein whose normal function is uncertain. For a class of fatal neurodegenerative disorders affecting human and animals, termed prion diseases or transmissible spongiform encephalopathies (TSEs), PrPC is required. In these conditions, normally folded endogenous PrPC undergoes a transformation to a conformationally altered scrapie prion protein (PrPSc) that accumulates in the brain as insoluble aggregates.Citation13,Citation14 This process leads to neuronal dysfunction and progressive neurodegeneration, for which there is no clinically effective treatment. PrPC mediates toxic signaling of PrPSc by its binding to β-sheet rich conformers of PrPSc.Citation15 The oligomeric state of Aβ recognized by PrPC may share conformational properties of other misfolded proteins such as PrPSc bound to PrPC.
Definition of the molecular and cellular consequences of Aβ-PrPC complex formation provides the opportunity to advance understanding of AD pathophysiology.
Inhibition of LTP by Aβ Oligomer is Mediated by PrPC
Hippocampal long-term potentiation (LTP), a leading experimental model for the synaptic changes underlying learning and memory, is strongly inhibited by Aβ oligomer application, as reported by numerous laboratories.Citation4-Citation7 Aβ oligomer-induced LTP blockade is absent in the hippocampal slices from Prnp–/– mice, and those anti-PrPC antibodies preventing Aβ oligomer binding to PrPC rescue synaptic plasticity in hippocampal slices from oligomeric Aβ.Citation8 Thus, disruption of synaptic plasticity by soluble Aβ oligomers is mediated through PrPC under these conditions.
However, Kessels et al. observed Aβ42 suppression of hippocampal CA1 LTP in slices from Prnp−/− mice.Citation16 Furthermore, the impairment of hippocampal synaptic plasticity in one AD mouse model was not altered by ablation or overexpression of PrPC.Citation17 These findings challenged the role of PrPC as a mediator of Aβ oligomer toxicity. In this context, it is important to recognize that Aβ preparations vary greatly in composition, and this variability may explain variable outcomes in functional experiments.Citation3 For this reason, a careful characterization of Aβ oligomers is required by size exclusion chromatography, electron microscopy and atomic force microscopy, as well as SDS-PAGE. Without such characterization, there is a potential for Aβ to elicit nonspecific toxicity, bypassing the need for any specific neuronal receptor, including PrPC. It is critical to evaluate the action of well-characterized Aβ species, including those derived from human AD brains, as being most relevant for AD pathophysiology. With synthetic Aβ oligomers resembling those of the original study,Citation8 the requirement for PrPC to mediate LTP inhibition has been verified.Citation12 In addition, two studies showed that Tris-soluble extracts from human AD brain, which contain disease-relevant Aβ oligomers, require PrPC for inhibition of LTP in mouse brain slices.Citation12,Citation18 One of these studies included in vivo electrophysiological outcomes. Aβ oligomers derived from human AD brain were injected into the rat brain and LTP was measured. Unlike the synthetic Aβ oligomer preparation used by studies from Kessel et al., which caused a marked reduction in baseline excitatory synaptic transmission,Citation16 the Aβ-containing AD brain extract selectively inhibits LTP in vivo. The in vivo inhibition of LTP by Aβ oligomer is fully alleviated by preinjection into the hippocampus of anti-PrPC antibodies that recognize the Aβ binding domain in PrPC. In addition to supporting the earlier finding that PrPC functions as a receptor for mediating toxicity of Aβ oligomer, these data provide evidence to support a biological, antibody-based approach to PrPC as a target for AD.
Role of PrPC in Synaptic Loss
A hallmark of AD is the massive synaptic loss that occurs at an early stage of the disease.Citation19 Synapse loss can also be detected in APP/PS1 mice by staining for synaptic marker proteins, and the loss of synaptic markers is fully dependent on PrPC.Citation20 Treatment of aged APP/PS1 mice with anti-PrPC antibodies allows a recovery of depleted synaptic density in the dentate gyrus.Citation21 In vitro studies have described dendritic spine loss after acute Aβ oligomer exposure. Imaging of spines continuously over 6h shows that Aβ oligomer treatment of dissociated hippocampal neurons induces a 10–15% loss of spines, but Prnp–/– cultures are fully protected.Citation22
It has been known that Aβ oligomer binding sites localize to the postsynaptic density,Citation8,Citation23,Citation24 consistent with their inducing synaptic loss. To the extent that PrPC mediates Aβ oligomer effects on spine composition, morphology and density, the protein is predicted to be concentrated at synapses and present in the postsynaptic density. Biochemical subcellular fractionation, immunohistological studies and unbiased proteomic studiesCitation22,Citation25 show that PrPC is located at synapses and enriched at postsynaptic densities.
Role of PrPC in Rodent Memory Impairment
The experimental paradigm most closely related to human AD is analysis of rodent memory. Mouse genetic models expressing familial AD mutant APP with or without familial AD mutant PS1 are useful tools for the study of AD. Transgenic mice expressing APP mutations exhibit age-dependent deficits of spatial learning and memory in water maze studies.Citation26,Citation27 Injection of Aβ oligomer isolated from transgenic mice, human- or cell-derived Aβ oligomers into naive mice produces learning and memory impairment.Citation7,Citation28,Citation29 Balducci and colleagues injected synthetic Aβ oligomer into the hippocampus and assessed novel object recognition with and without PrPC.Citation11 This model is simple, but relies on synthetic peptide and on acute effects. Aβ oligomer injection prevented novel object recognition memory by wild type mice. In Aβ-injected mice lacking PrPC, the recognition of novel vs. familiar objects was preserved, but mice preferred familiar objects rather than novel objects.Citation11 This reversal complicates any assessment of the role for PrPC in Aβ action using this paradigm.
The most direct assessment of PrPC in AD-related deficits entails measuring spatial learning and memory in mice carrying familial AD genes but null for PrPC expression. Mice lacking PrPC, but containing Aβ plaque derived from APPswe/PS1ΔE9 transgenes, show no detectable impairment of spatial learning and memory.Citation20 Furthermore, the treatment of aged APPswe/PS1-M146L mice with anti-PrPC antibodies reverses memory impairments.Citation21 These data suggest that memory deficits in AD transgenic mice require the presence of PrPC. However, Cisse et al. reported that PrPC is not essential for this phenotype in the J20 line.Citation30 Discrepancies between different animal AD models with respect to a possible role for PrPC might be anticipated. The many reported animal models of AD are each likely to recapitulate only part of the human syndrome. While PrPC appears to be an essential receptor for certain toxic Aβ species, it would not be expected that PrPC ablation to rescue all aspects of pathology in each model. The most critical difference between these studies is the AD transgenic line investigated, and the age of onset for behavioral deficits. Critically, the J20 mice are known to be memory-impaired as young adults, perhaps due to the high juvenile Aβ levels in this strain.Citation31,Citation32 There is no evidence for progressive memory loss in these mice after normal development, as occurs in AD. In contrast, the APP/PS1 lines, which show PrPC dependence of memory dysfunction, possess normal spatial memory at 3–6 months of age.Citation20,Citation33 The most plausible conclusion is that PrPC is required for adult-onset AD transgene-driven progressive disease, whereas developmental-onset deficits in the J20 occur by a different mechanism. The developmental disorder appears to be PrPC-independent while the late-onset progressive disorder is PrPC-dependent.
Role of PrPC in Seizures
Patients with AD have an increased incidence of unprovoked seizures.Citation34,Citation35 Epileptiform EEG discharges, including spikes and sharp waves in temporal lobe epilepsy patients can generate AD-like memory dysfunction.Citation36 Accordingly, transgenic AD model mice such as J20 and APPswe/PS1 ΔE9 mice exhibit altered network activity and epileptiform discharges.Citation22,Citation37 Interestingly, the APP/PS1 transgenic mice lacking PrPC does not exhibit a convulsive seizure phenotype, which suggests that this electrographic abnormality of AD model mice requires PrPC.Citation22
Fyn Kinase in AD
The discovery of Aβ oligomer binding to PrPC in AD pathogenesis implies a neuronal signaling pathway downstream of the Aβ-PrPC complex. Our recent study published shows that Fyn kinase functions to mediate signal transduction from Aβ-PrPC complexes.Citation22 We found that Aβ-PrPC activates Fyn pathway, and subsequently dysregulates NMDA receptor function.
Fyn is a member of the Src family of intracellular non-receptor tyrosine kinases family.Citation38 There are nine members of Src family kinases. Five of them (Src, Fyn, Lck, Lyn, Yes) are expressed in central nervous system, but Src and Fyn are most highly expressed in the brain. Fyn activity, like that of other Src family kinases, is regulated by intramolecular interactions that depend on an equilibrium between tyrosine phosphorylation and dephosphorylation. In the basal state, catalytic activity is constrained by intramolecular interactions, such as engagement of the SH2 domain by a phosphorylated C-terminal tyrosine 527. Disruption of these interactions by phosphorylation at Tyr 416 in the activation loop of the kinase domain and/or by dephosphorylation of Tyr 527 results in Fyn activation.Citation39
Src family kinases have been implicated in neurodegenerative disease, including AD. In one study, PP2, a selective inhibitor of Src family kinases, was shown to be neuroprotective against Aβ.Citation40 For AD transgenic models, the toxic effect of Aβ is blocked by the genetic ablation of Fyn, whereas overexpression of Fyn enhances Aβ-induced toxicity and AD-related phenotypes.Citation41 Fyn regulates NMDA receptor trafficking and synaptic plasticity,Citation42 and Fyn interacts with Tau to modulate phenotypes in AD mouse models.Citation43,Citation44
There are multiple lines of evidence linking Fyn kinase to PrPC. Fyn is localized to lipid rafts as is PrPC, and clustering of PrPC activates Fyn kinase in certain cell lines.Citation45,Citation46 Fyn mediates mutant PrPC phenotypes in fish and worms.Citation47,Citation48 Based on these reports, we focused on Fyn as a candidate transducer of Aβ-PrPC signaling.Citation22
Fyn Activation by Aβ-PrPC
We found that Fyn kinase is specifically activated in response to synthetic Aβ oligomer in cell lines overexpressing PrPC22 (). In addition, Aβ oligomer-induced Fyn activation is observed in wild type cortical neurons, but not in Prnp–/– neurons. More importantly, AD brain extract containing PrPC-interacting Aβ species stimulates neuronal Fyn activation, but age-matched control brain extracts do not.Citation22 The ability of AD brain extract to induce Fyn activation is absent from Prnp–/– neurons. Thus, AD-relevant preparations of Aβ oligomers activate neuronal Fyn via PrPC.
Figure 1. Molecular model for Aβ oligomer induced synaptic dysfunction. The amyloid precursor protein (APP) is processed by β- and γ-secretase to yield Aβ monomer. The Aβ monomer can undergo a conformational change coincident with assembly into toxic oligomers. Aβ oligomer binding to cellular prion protein (PrPC) at the neuronal post-synaptic density activates Fyn tyrosine kinase. PrPC may couple to Fyn through an as yet unidentified transmembrane protein. Aβ oligomer stimulation of Fyn signaling drives the tyrosine phosphorylation of the NR2B subunit of NMDA receptors. NMDA receptor phosphorylation in turn produces altered surface expression, dysregulation of receptor function, excitoxicity and dendritic spine retraction.
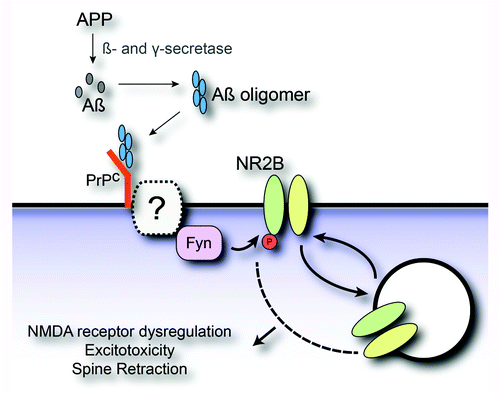
Fyn is known to phosphorylate the NMDA receptor subunit, NR2B.Citation49 This phosphorylation regulates NMDA receptor trafficking and synaptic plasticity.Citation42 NR2B phosphorylation and subsequent cell surface level of NR2B are enhanced during first 15 min of Aβ oligomer exposure but after 1–3 h, phosphorylation is suppressed and surface level of NR2B returns to the basal level.Citation22 The biphasic effect of Aβ oligomer through PrPC/Fyn on surface NR2B is paralleled by changes in NMDA-induced calcium signaling. The NMDA receptor dysregulation induced by Aβ is eliminated in Prnp–/– and Fyn-/– neurons. Furthermore, we found that the Fyn activation by Aβ-PrPC complexes induces excitotoxicity and destabilizes dendritic spines.Citation22 Aβ oligomer-induced cell death has now been shown to be PrPC-dependent in a range of experiments.Citation15,Citation22,Citation50,Citation51
Our studies define an Aβ oligomer signal transduction pathway that requires PrPC and Fyn to alter synaptic function, and that is likely to be directly relevant to AD.Citation22 These observations pose the issue of how PrPC might be connected to Fyn kinase, since two proteins are localized in the opposite sides of the plasma membrane. One or more transmembrane proteins might serve as a co-receptor(s) for Aβ oligomer to couple PrPC with Fyn (). Further study should identify a co-receptor(s) connecting PrPC to Fyn. It will be of interest to examine whether other Aβ signaling pathways, such as calcineurin, insulin receptors, autophagy, and Tau-directed kinases, are also PrPC-dependent. Investigation of the role of Aβ-PrPC-Fyn pathway in an even broader set of preclinical AD models will further define its relevance for human AD pathophysiology.
Conclusion
In summary, we have reviewed a series of studies showing that Aβ oligomer binding to PrPC mediates deleterious effects on neurons. Both well-characterized synthetic Aβ oligomers and human TBS-soluble AD extracts impair neuronal synapse function in a PrPC-dependent manner. The role of the Aβ/PrP complex in AD pathophysiology is further supported by the identification of a downstream Fyn pathway linking Aβ-PrPC to NMDA receptor and Tau dysfunction. Being distinct from regulating the levels of Aβ, the PrPC-Fyn pathway represents a novel target for AD therapy. This pathway is based specifically on neuronal toxicity of Aβ oligomers, and provides a link between Aβ and Tau pathologies in the disease. Of importance, the limited or absent phenotype of Prnp−/− mice suggests no serious side effect from modulating PrPC function. Thus, these findings open attractive new avenues for AD therapy development.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest have been disclosed.
Acknowledgments
We acknowledge support from the National Institutes of Health, the Falk Medical Research Trust and the Alzheimer Association to S.M.S. S.M.S. is a member of the Kavli Institute for Neuroscience at Yale University. S.M.S. is a co-founder of Axerion Therapeutics, seeking to develop NgR- and PrP-based therapeutics.
References
- Wimo A, Prince M. World Alzheimer Report 2010: The Global Economic Impact of Dementia (London: Alzheimer's Disease International). 2010: p. 1-56.
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297:353 - 6; http://dx.doi.org/10.1126/science.1072994; PMID: 12130773
- Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 2012; 15:349 - 57; http://dx.doi.org/10.1038/nn.3028; PMID: 22286176
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 1998; 95:6448 - 53; http://dx.doi.org/10.1073/pnas.95.11.6448; PMID: 9600986
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416:535 - 9; http://dx.doi.org/10.1038/416535a; PMID: 11932745
- Wang H-W, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, et al. Soluble oligomers of β amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res 2002; 924:133 - 40; http://dx.doi.org/10.1016/S0006-8993(01)03058-X; PMID: 11750898
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 2008; 14:837 - 42; http://dx.doi.org/10.1038/nm1782; PMID: 18568035
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Zou W-Q, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, et al. Amyloid-β42 interacts mainly with insoluble prion protein in the Alzheimer brain. J Biol Chem 2011; 286:15095 - 105; http://dx.doi.org/10.1074/jbc.M110.199356; PMID: 21393248
- Chen S, Yadav SP, Surewicz WK. Interaction between Human Prion Protein and Amyloid-β (Aβ) Oligomers. J Biol Chem 2010; 285:26377 - 83; http://dx.doi.org/10.1074/jbc.M110.145516; PMID: 20576610
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A 2010; 107:2295 - 300; http://dx.doi.org/10.1073/pnas.0911829107; PMID: 20133875
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, et al. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat Commun 2011; 2:336; http://dx.doi.org/10.1038/ncomms1341; PMID: 21654636
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93:337 - 48; http://dx.doi.org/10.1016/S0092-8674(00)81163-0; PMID: 9590169
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J 2011; 30:2057 - 70; http://dx.doi.org/10.1038/emboj.2011.86; PMID: 21441896
- Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature 2010; 466:E3 - 4, discussion E4-5; http://dx.doi.org/10.1038/nature09217; PMID: 20703260
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, et al. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2010; 2:306 - 14; http://dx.doi.org/10.1002/emmm.201000082; PMID: 20665634
- Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, et al. Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci 2011; 31:7259 - 63; http://dx.doi.org/10.1523/JNEUROSCI.6500-10.2011; PMID: 21593310
- Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging 1990; 11:29 - 37; http://dx.doi.org/10.1016/0197-4580(90)90059-9; PMID: 2325814
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, et al. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 2010; 30:6367 - 74; http://dx.doi.org/10.1523/JNEUROSCI.0395-10.2010; PMID: 20445063
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, et al. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci 2010; 11:130; http://dx.doi.org/10.1186/1471-2202-11-130; PMID: 20946660
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, et al. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 2012; 15:1227 - 35; http://dx.doi.org/10.1038/nn.3178; PMID: 22820466
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci 2007; 27:796 - 807; http://dx.doi.org/10.1523/JNEUROSCI.3501-06.2007; PMID: 17251419
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. [beta] Nat Neurosci 2005; 8:1051 - 8; http://dx.doi.org/10.1038/nn1503; PMID: 16025111
- Collins MO, Husi H, Yu L, Brandon JM, Anderson CN, Blackstock WP, et al. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J Neurochem 2006; 97:Suppl 1 16 - 23; http://dx.doi.org/10.1111/j.1471-4159.2005.03507.x; PMID: 16635246
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, et al. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Nature 2000; 408:975 - 9; http://dx.doi.org/10.1038/35046031; PMID: 11140684
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996; 274:99 - 102; http://dx.doi.org/10.1126/science.274.5284.99; PMID: 8810256
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006; 440:352 - 7; http://dx.doi.org/10.1038/nature04533; PMID: 16541076
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 2005; 8:79 - 84; http://dx.doi.org/10.1038/nn1372; PMID: 15608634
- Cissé M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci 2011; 31:10427 - 31; http://dx.doi.org/10.1523/JNEUROSCI.1459-11.2011; PMID: 21775587
- Harris JA, Devidze N, Halabisky B, Lo I, Thwin MT, Yu GQ, et al. Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer’s disease are independent of caspase cleavage of the amyloid precursor protein. J Neurosci 2010; 30:372 - 81; http://dx.doi.org/10.1523/JNEUROSCI.5341-09.2010; PMID: 20053918
- Deipolyi AR, Fang S, Palop JJ, Yu GQ, Wang X, Mucke L. Altered navigational strategy use and visuospatial deficits in hAPP transgenic mice. Neurobiol Aging 2008; 29:253 - 66; http://dx.doi.org/10.1016/j.neurobiolaging.2006.10.021; PMID: 17126954
- Park JH, Widi GA, Gimbel DA, Harel NY, Lee DH, Strittmatter SM. Subcutaneous Nogo receptor removes brain amyloid-beta and improves spatial memory in Alzheimer’s transgenic mice. J Neurosci 2006; 26:13279 - 86; http://dx.doi.org/10.1523/JNEUROSCI.4504-06.2006; PMID: 17182778
- Cabrejo L, Guyant-Maréchal L, Laquerrière A, Vercelletto M, De la Fournière F, Thomas-Antérion C, et al. Phenotype associated with APP duplication in five families. Brain 2006; 129:2966 - 76; http://dx.doi.org/10.1093/brain/awl237; PMID: 16959815
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, et al. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 2006; 47:867 - 72; http://dx.doi.org/10.1111/j.1528-1167.2006.00554.x; PMID: 16686651
- Sinforiani E, Manni R, Bernasconi L, Banchieri LM, Zucchella C. Memory disturbances and temporal lobe epilepsy simulating Alzheimer’s disease: a case report. Funct Neurol 2003; 18:39 - 41; PMID: 12760413
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007; 55:697 - 711; http://dx.doi.org/10.1016/j.neuron.2007.07.025; PMID: 17785178
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol 1997; 13:513 - 609; http://dx.doi.org/10.1146/annurev.cellbio.13.1.513; PMID: 9442882
- Hunter T. A tail of two src’s: mutatis mutandis. Cell 1987; 49:1 - 4; http://dx.doi.org/10.1016/0092-8674(87)90745-8; PMID: 3030562
- Wu G-M, Hou X-Y. Oligomerized Abeta25-35 induces increased tyrosine phosphorylation of NMDA receptor subunit 2A in rat hippocampal CA1 subfield. Brain Res 2010; 1343:186 - 93; http://dx.doi.org/10.1016/j.brainres.2010.04.055; PMID: 20441772
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, et al. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci 2005; 25:9694 - 703; http://dx.doi.org/10.1523/JNEUROSCI.2980-05.2005; PMID: 16237174
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 2005; 47:845 - 57; http://dx.doi.org/10.1016/j.neuron.2005.08.016; PMID: 16157279
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010; 142:387 - 97; http://dx.doi.org/10.1016/j.cell.2010.06.036; PMID: 20655099
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci 2004; 7:221 - 8; http://dx.doi.org/10.1038/nn1188; PMID: 14966521
- Pantera B, Bini C, Cirri P, Paoli P, Camici G, Manao G, et al. PrPc activation induces neurite outgrowth and differentiation in PC12 cells: role for caveolin-1 in the signal transduction pathway. J Neurochem 2009; 110:194 - 207; http://dx.doi.org/10.1111/j.1471-4159.2009.06123.x; PMID: 19457127
- Stuermer CA, Langhorst MF, Wiechers MF, Legler DF, Von Hanwehr SH, Guse AH, et al. PrPc capping in T cells promotes its association with the lipid raft proteins reggie-1 and reggie-2 and leads to signal transduction. FASEB J 2004; 18:1731 - 3; PMID: 15345693
- Bizat N, Peyrin JM, Haïk S, Cochois V, Beaudry P, Laplanche JL, et al. Neuron dysfunction is induced by prion protein with an insertional mutation via a Fyn kinase and reversed by sirtuin activation in Caenorhabditis elegans. J Neurosci 2010; 30:5394 - 403; http://dx.doi.org/10.1523/JNEUROSCI.5831-09.2010; PMID: 20392961
- Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, et al. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol 2009; 7:e55; http://dx.doi.org/10.1371/journal.pbio.1000055; PMID: 19278297
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 2004; 5:317 - 28; http://dx.doi.org/10.1038/nrn1368; PMID: 15034556
- Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, et al. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum Mol Genet 2012; 21:1138 - 44; http://dx.doi.org/10.1093/hmg/ddr542; PMID: 22100763
- You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, et al. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A 2012; 109:1737 - 42; http://dx.doi.org/10.1073/pnas.1110789109; PMID: 22307640