Abstract
In prion diseases, the normal prion protein is transformed by an unknown mechanism from a mainly α-helical structure to a β-sheet-rich, disease-related isomer. In this study, we surprisingly found that a slow, spontaneous α-to-coil-to-β transition could be monitored by circular dichroism spectroscopy in one full-length mouse recombinant prion mutant protein, denoted S132C/N181C, in which the endogenous cysteines C179 and C214 were replaced by Ala and S132 and N181 were replaced by Cys, during incubation in a non-denaturing neutral buffer. No denaturant was required to destabilize the native state for the conversion. The product after this structural conversion is toxic β-oligomers with high fluorescence intensity when binding with thioflavin T. Site-directed spin-labeling ESR data suggested that the structural conversion involves the unfolding of helix 2. After examining more protein mutants, it was found that the spontaneous structural conversion is due to the disulfide-deletion (C to A mutations). The recombinant wild-type mouse prion protein could also be transformed into β-oligomers and amyloid fibrils simply by dissolving and incubating the protein in 0.5 mM NaOAc (pH 7) and 1 mM DTT at 25°C with no need of adding any denaturant to destabilize the prion protein. Our findings indicate the important role of disulfide bond reduction on the structural conversion of the recombinant prion protein, and highlight the special “intrinsically disordered” conformational character of the recombinant prion protein.
Introduction
Prion disease is a transmissible neurodegenerative disease in mammals caused by the multimeric misfolded form of the mammalian host-encoded PrP (prion protein) named “prion.”Citation1 Prion, also known as PrPSc, has a high β-structure content of 43%, as estimated by circular dichroism spectroscopy and Fourier transformed infrared spectroscopyCitation2 compared with the 4% β-sheet content of the native PrP (PrPC), estimated from its NMR structure.Citation3 The structural conversion process has been conducted in a test tube using recombinant prion proteins (rPrPs) of various lengths, brain homogenates, or purified cellular PrPC.Citation4 In vitro-produced amyloid fibrils can be produced from rPrP by vigorous shaking under denaturing conditions.Citation5,Citation6 Proteinase K-resistant PrPres can be generated from rPrPs, brain homogenates, cell lysates, or purified PrPC simply by seedingCitation7,Citation8 and the process can be accelerated by sonication, a process known as protein misfolding cyclic amplification (PMCA),Citation9-Citation12 or by shaking, a process known as quaking-induced conversion (QUIC).Citation13
Recombinant PrP(90–231) in partially denaturing condition can adopt two kinds of abnormal β-sheet-rich quaternary structures, the soluble β-oligomer (preferred form in acidic pH) and insoluble amyloid fibril (preferred form at neutral pH).Citation5,Citation14 The soluble β-oligomer, also designated β-PrP, can be produced from rPrP(90–231) by reduction in the presence of a denaturant under acidic conditions.Citation15-Citation18 β-PrP has been reported to show slight protease resistance,Citation15 is more toxic to cells than amyloid fibrils,Citation19,Citation20 and has been suspected of being a precursor or intermediate in the pathway of prion protein folding or PrPSc formation.Citation16,Citation20 In the present study, we initially intended to use recombinant full-length mouse prion protein mPrPwt and site-directed spin-labeling electron spin resonance spectroscopy (SDSL-ESR)Citation21 to monitor the conformational change during the formation of β-PrP and amyloid fibrils. To perform spin labeling, the two Cys in mPrPwt were mutated to Ala, generating mPrP-CtoA, then both Ser-132 (in the flexible loop region before the first helix) and Asn-181 (in the second helix and one of the two N-linked glycosylation sites) were mutated to Cys and the resulting mutant protein, named S132C/N181C, was labeled with a commonly used nitroxide-based spin probe MTSSL. However, we surprisingly found that the CD spectrum of S132C/N181C, dissolved in NaOAc buffer (pH 7), was very different from that of mPrPwt in the same buffer and very similar to the CD spectrum of the previously reported β-PrP.Citation15 Here, we report the slow, spontaneous formation of β-PrP under non-denaturing neutral pH conditions and its relationship to the removal of the endogenous disulfide bond of prion protein.
Results and Discussion
β-PrP oligomer formation at neutral pH
The two Cys residues (C179, C214) of mPrPwt were replaced by Ala by site-directed mutagenesis to generate mutant mPrP-CtoA, then S132 and N181 of mPrP-CtoA were further replaced by Cys to generate the mutant S132C/N181C. As shown in , in contrast to mPrPwt, which had a CD spectrum of characteristic α-helical structure (designated α-PrP conformer, black line), the CD spectrum of S132C/N181C (green line) dissolved in 10 mM NaOAc, pH 7, showed a strong β-sheet signal. After spectral deconvolution, S132C/N181C was found to have an α-helical content of 6% and a β-sheet content of 35%, in contrast to the 30% α-helix and 20% β-sheet content of mPrPwt (). Since formation of β-PrP in a neutral buffer has not been reported previously, analytical ultracentrifugation (AUC) was used to investigate whether S132C/N181C formed oligomers, and the results showed two major peaks of 194 kDa and 420 kDa, corresponding, respectively, to an 8–9 mer and an 18–19 mer, with only a very small amount of monomer (), confirming that β-PrP oligomers could be generated simply by dissolving the protein in 10 mM NaOAc, pH 7. To determine whether this phenomenon resulted from the mutations introduced, the CD spectra of the disulfide-deletion mutant mPrP-CtoA and the two single Cys mutants of mPrP-CtoA, S132C and N181C, in the same buffer were also measured (). All three spectra and their deconvolution results were very similar to those for S132C/N181C, suggesting that the disulfide deletion promoted β-PrP formation. In the presence of DTT, S132C/N181C could still form β-PrP, demonstrating that the β-PrP formation is not due to intermolecular disulfide bond formation (Fig. S1)
Figure 1. (A) CD spectra of mPrPwt, mPrP-CtoA, S132C, N181C, and S132C/N181C. The proteins were dissolved at a concentration of 0.15 mg/mL in 10 mM NaOAc buffer, pH 7, and their CD spectra immediately recorded. (B) Secondary structure content of each protein determined by CD spectral deconvolution using CDPro software.
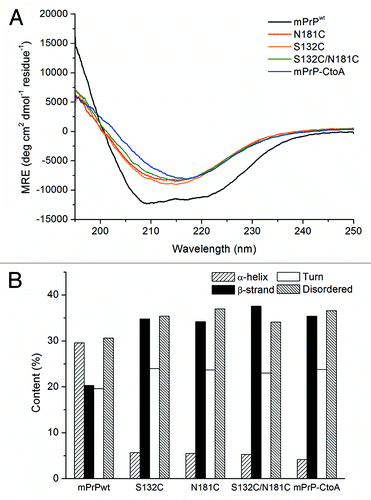
Figure 2. The SV-AUC spectrum of S132C/N181C (0.1 mg/mL) in 10 mM NaOAc (pH 7).
Short prion peptides can go through a coil-to-β transition simply by incubation in buffer.Citation22-Citation25 These peptides are random coil when dissolved in buffer and, since random coil is not a stabilized structural state, it is relatively easy for the peptides to cross the transition barrier in the structural transformation from random coil to the amyloid precursor structure, which can finally associate into amyloid fibrils. In contrast, prion protein has a stable structural domain with 3 α-helices and 2 short β-strands. Since the energy barrier of the structural transition from random coil to β-sheet is much lower than that from α-helical to β-sheet, in experimental studies on prion protein, denaturants or detergents are commonly used to destabilize the native α-helical structure to facilitate the structural conversion. In previous reports, β-PrP oligomers could only be formed under acidic conditions in the presence of denaturants with or without reduction.Citation5,Citation14-Citation16,Citation19,Citation20 In this study, we found that simply dissolving our disulfide-deletion mutants in 10 mM NaOAc at neutral pH (pH 7) resulted in the formation of soluble β-PrP oligomers, instead of an α-helical structure, like mPrPwt. The association process was irreversible, since, once the β-PrP oligomers were formed, diluting the sample with water to lower the protein concentration did not restore the structure to its native α-helical structure (data not shown).
The structural conversion process could be monitored at low salt condition by CD spectroscopy
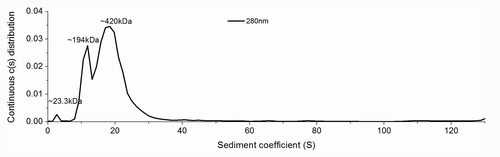
Interestingly, the kinetics of β-PrP formation was strongly dependent on the salt concentration. When the salt concentration was decreased to 0.5 mM NaOAc, the whole process was slowed down, which allowed us to monitor the time-dependent structural conversion process by CD spectroscopy. S132C/N181C oligomerization could be analyzed as a three-state process consisting of a “α-to-coil-to-β “ transition. When S132C/N181C was first dissolved in 0.5 mM NaOAc, pH 7, the initial CD spectrum clearly indicated that it formed an α-PrP structure, like mPrPwt. S132C/N181C retained the same α-PrP structure for about 12 hr, then, on standing in the CD quartz cuvette, the structure underwent the first transition to a coil-rich structure (spectrum blue-shifting) spontaneously at 25 °C within 2 days. The spectral deconvolution results in show a dramatic decrease in α-helical content and an increase in β-sheet, turn, and disordered structure over the period from 12 to 32 hr. Over the following days, the protein then underwent a slow oligomerization process in which the unstructured component slowly decreased and the β-sheet content slowly increased, finally displaying the characteristic β-PrP CD spectrum on day 13. The first “α-to-coil” transition and the second “coil-to-β” transition are shown separately in for clarity. No reagent was added to destabilize the native state of the protein. This phenomenon conflicts with the protein folding theory - “one sequence under one condition, one structure” and suggests that the energy barrier between the native state of the reduced prion protein and its β-conformer is low.
Figure 3. (A and B) CD spectra of S132C/N181C (0.15 mg/mL) in 0.5 mM NaOAc buffer, pH 7, recorded after incubated for the indicated time at 25°C. The protein solution was kept inside the quartz cuvette throughout the experiment. Two structural transitions during the whole time course could be seen. The first transition from an α-helical state to a coil-rich state is shown in (A) and the second transition from a coil-rich state to β-oligomers is shown in (B). The direction of the spectral change is indicated by an arrow. The spectral deconvolution results are shown in (C).
For the protein S132C, the structural conversion process was similar to that of S132C/N181C, but its spectral blue-shifting was not so apparent (Fig. S2). On the other hand, N181C and mPrP-CtoA did not show a coil-rich state. N181C showed a predominant structure change between 96 and 168 h, whereas mPrP-CtoA retained its α-PrP structure for a longer period (~192 h) and took longer to form β-PrP than the other three mutants (Fig. S2). Our data indicated that the mutation of S132 or N181 to Cys destabilized the native state of the protein and facilitated the structural conversion. Moreover, the co-existence of these two mutations shifts the equilibrium of the protein conformational ensemble toward the disordered conformation. The formation of β-PrP was very sensitive to the salt concentration, suggesting that the association might be driven by hydrophobic interaction (Fig. S3).
One may argue that the buffer used in this experiment (0.5 and 10 mM NaOAc) is non-physiological. It should be noted that PrPC concentration in brain is very low (0.07 μg/mg brain homogenate)Citation26 and cellular prion protein is glycosylated and anchored on membrane using GPI anchor. Its physiological protein concentration is too low for studying protein conformation using spectroscopic methods. Moreover, protein solubility of unglycosylated, recombinant prion protein is not as good as glycosylated natural PrPC, hence most of the studies used denaturant to dissolve recombinant prion protein. We are lucky to find that low concentration of sodium acetate at neutral pH can dissolve recombinant prion protein at a protein concentration suitable for conformational study.
Conformational characterization and structural properties of S132C/N181C β-PrP produced at neutral pH
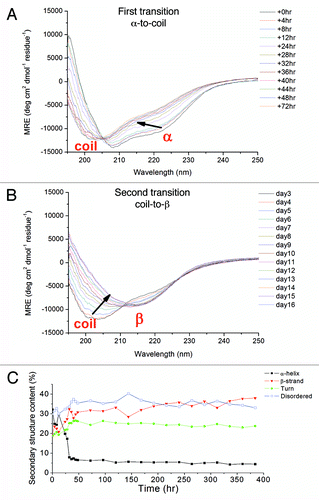
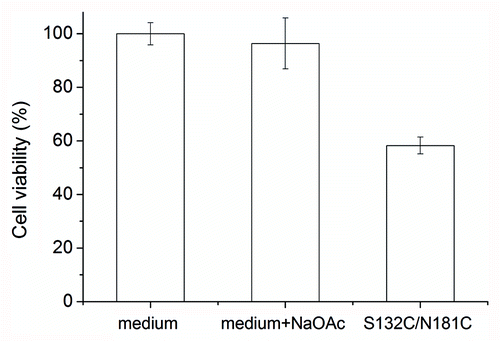
The AUC results showed that β-PrP formed from S132C/N181C in 10 mM NaOAc, pH 7, was an oligomer and this was corroborated by its transmission electron microscopy (TEM) image. In , bead-like oligomers can be easily seen and these could further associate one-dimensionally as protofibril-like morphology. Similar to amyloid fibrils, these β-PrP oligomers showed strong fluorescence in the ThT fluorescence assay (), suggesting that they contain hydrophobic ThT binding sites, like amyloids. Our data supported the previous finding that ThT fluorescence does not necessarily correlate with amyloid formation.Citation27 However, the previously reported β-PrP formed from mouse PrP(89–231) under acidic conditions shows no ThT fluorescence.Citation14 The difference of these two β-PrPs suggests that our β-PrP produced in non-denaturing neutral condition might be different from previously reported β-PrP. A cytotoxicity assay showed that S132C/N181C β-PrP is toxic for cultured mouse N2a cells (). Whether the ThT-positive β-PrP is infectious or not remains to be examined.
Figure 4. (A) TEM images of S132C/N181C dissolved in 10 mM NaOAc, pH 7. The sample was stained with 2% uranyl acetate and images taken using a Hitachi H-7000 electron microscope at 75 kV. The bars represent 500 nm. (B) ThT fluorescence assay of mPrPwt fibrils (dash line) and S132C/N181C β-PrP oligomers (solid line). S132C/N181C was dissolved at a concentration of 20 μM in 10 mM NaOAc, pH 7, while mPrPwt was incubated at a concentration of 22 μM in 1 M GdnHCl, 3 M urea in PBS at 37°C with shaking at 200 rpm for 5 d. The two protein solutions were mixed with the same volume of ThT working solution and incubated for 1 min, then the fluorescence spectra were recorded from 450 to 600 nm with excitation at 420 nm.
Figure 5. Cytotoxicity of S132C/N181C β-PrP. The β-PrP prepared from S132C/N181C (30 μM) in 10 mM NaOAc, pH 7 was added to the cultured mouse N2a cells. The cytotoxicity was measured by using the MTT assay. The addition of 10 mM NaOAc (pH 7) into the cultured cells was also used as one control. The values are the mean ± standard deviation (n = 6 for S132C/N181C and n = 14 for the others).
After prolonged incubation, except oligomers and protofibrils, amorphous aggregate and amyloid fibrils could also be observed under electron microscopy (Fig. S4).
Examining the conformational change upon oligomerization by SDSL-ESR
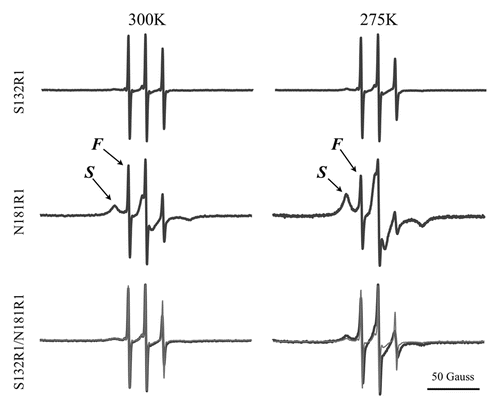
To explore the structural change upon oligomerization, site-directed spin labeling (SDSL) ESR was employed. The mPrP single-Cys mutants, S132C and N181C, were labeled with one MTSSL molecule and the spin-labeled proteins, denoted as S132R1 and N181R1, were purified by HPLC to remove unlabeled protein and free spin molecules. The double Cys mutant S132C/N181C was labeled with two MTSSL molecules and the resulting product, named S132R1/N181R1, was also purified by HPLC. shows the ESR spectra of these spin-labeled PrP mutants obtained at temperatures of 300 and 275 K. The S132R1 spectra showed a typical spectral lineshape in the fast-motional regime, indicating that the local environment of site 132 is characterized by a high mobility, consistent with the fact that site 132 is on a loop of the prion protein. The spectra suggested that the local environment of S132R1 remained highly mobile and disordered upon induction of oligomerization. In the case of N181R1, the spectra clearly showed two distinctly different coexisting spectral components, indicated as slow (S) and fast (F) on the figure. In a prion protein in a native monomer state, site 181 is in the α-helical H2 region, in which the nitroxide R1 side chain would typically show a slow-motional lineshape (i.e., a lineshape resembling the S component) due to the local environment within a structured helical region. Several sites in the H2 region of prion protein have been previously studied by ESR and shown to display a slow-motional-like ESR lineshape.Citation28,Citation29 Thus, the existence of the F component in the N181R1 spectra provides evidence that the local environment of the H2 region is partially unstructured/unfolded under the experimental conditions that induce oligomerization. This observation was consistent with the decrease in the α-helical content of this protein upon oligomerization seen by CD spectroscopy. The spectral plots at the bottom of are the superimposed spectra of the double-labeled S132R1/N181R1 (bold lines) and the average of the spectra for the two single-labeled mutants (thin lines) and clearly show that the two spectra superimpose very well at 300 K, but differ slightly at 275 K. This indicates that the dipolar interaction between the two spin probes at sites 132 and 181 is small and insignificant; suggesting that the inter-spin distance is greater than the upper limit of the cw-ESR measurement (ca. 2 nm). The ESR spectra in provide supporting evidence that, upon oligomerization of these mPrP mutants, the local environment of site 132 remains unstructured, whereas the local environment of site 181 is not as rigid as in the native structure but is in an equilibrium between two states, i.e., unstructured vs. structured.
Figure 6. ESR spectra of the spin-labeled mPrP mutants S132R1, N181R1, and S132R1/N181R1. Protein samples were dissolved in 10 mM NaOAc containing 40% sucrose, pH 7.0. The average of the spectra for S132R1 and N181R1 (thin lines) and the spectra of S132R1/N181R1 (bold lines) are shown at the bottom of the figure. ESR spectra were measured either at 275K or 300K.
Therefore, the large drop in α-helical content during oligomerization was probably due to the unfolding of helix 2, which is consistent with the NMR data published by Hosszu et al.Citation30
Relation of disulfide bond and β-PrP formation
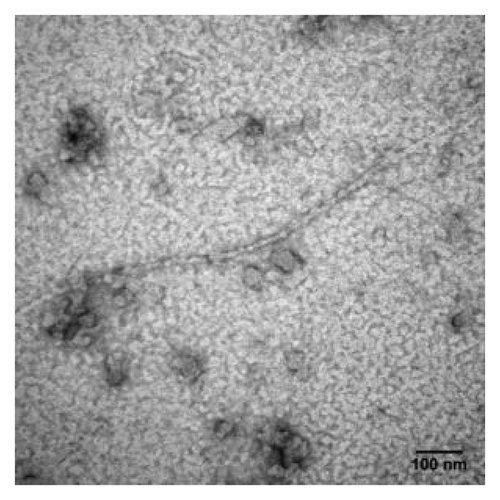
Helices 2 and 3 of the prion protein are linked by a single disulfide bond between Cys-179 and Cys-214. The role of this disulfide bond in the α-to-β structural transition has been studied. This intramolecular disulfide bond has been found to be crucial for the folding of the α-helical conformer.Citation25 Maiti and Surewicz have studied the role of disulfide bridge in the folding and stability of prion protein and reported that the recombinant human PrP as well as its Cys→Ala mutant formed insoluble aggregate at neutral pH and underwent a transition from α-helix to a β-sheet-rich structure under acidic, mildly denaturing condition and the presence of NaCl.Citation31 Similar to our findings, it has been reported that the reduced form of hamster rPrP(90–231) has a lower solubility and high β-sheet content at pH values above 7.Citation32 In our study, oligomerization and fibrillization was seen with all disulfide-deletion mutants, suggesting the importance of this disulfide bond in the stabilization of the native state. Due to the protein solubility problem, previous studies were all done in acidic condition (pH 4). To verify whether loss of this disulfide bond was the key factor for this structural conversion in a neutral condition, the CD spectra of mPrPwt following addition of the reducing agent dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP) were recorded (). In , when the oxidized form of mPrPwt was treated with DTT, a similar structural transition was observed by CD spectroscopy and, again, the kinetics of this transition was salt concentration-dependent, as mPrPwt was converted much faster in 10 mM NaOAc than in 0.5 mM NaOAc (Fig. S5). Interestingly, TCEP was able to promote rapid structural conversion of mPrPwt, even at a low NaOAc concentration (). The difference in the time scale of β-PrP formation after disulfide bond reduction by TCEP and DTT is probably due to the different reduction mechanisms for these two agents. The mechanism by which DTT reduces disulfide bonds is disulfide exchange, which gradually shifts the equilibrium from an oxidized to a reduced protein, whereas TCEP causes disulfide reduction by a SN2 mechanism involving a phosphorous nucleophile.Citation33 Thus, the reducing power of TCEP is much greater than that of DTT. The amyloid fibrils and β-PrP oligomers formed by dissolving and incubating mPrPwt in 0.5 mM NaOAc (pH 7) and 1 mM DTT were clearly seen under TEM ().
Figure 7. Spontaneous conformational transition of mPrPwt after addition of disulfide-bond reducing agents. (A) The CD spectra of mPrPwt (0.14 mg/mL) in 0.5 mM NaOAc, pH 7 with 1 mM DTT, recorded after incubated for the indicated time at 25°C. The protein was kept inside the quartz cuvette throughout the experiment. The direction of the spectral change is indicated by an arrow. (B) The CD spectrum of mPrPwt (0.14 mg/mL) in 0.5 mM NaOAc, pH 7 with 1 mM TCEP was recorded immediately after sample preparation.
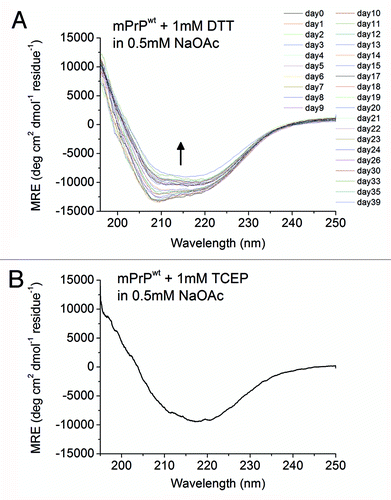
Figure 8. TEM image of mPrPwt dissolved in 0.5 mM NaOAc (pH 7), 1 mM DTT and incubated at 25°C for two days. The sample was stained with 2% uranyl acetate and images taken using a Hitachi H-7000 electron microscope at 75 kV. The bar represents 100 nm.
The structural conversion of mPrPwt promoted by adding reducing agents suggests the potential role of the redox state on initiation of prion conversion. Previous studies regarding whether the disulfide bridge reduction is necessary in prion conversion are controversial. Prusiner and his colleagues have reported that both PrPC and PrPSc have an intact intramolecular disulfide bondCitation34 and the Cys-179→Ala mutation inhibits PrPSc conversion in ScN2a cells.Citation35 Caughey and his colleagues have reported that PrPSc can initiate the conversion of the oxidized, disulfide-intact PrPC to its protease-resistant form without the temporary breakage and subsequent re-formation of the disulfide bond in cell-free reactionsCitation36 and reducing agents can inhibit the in vitro PrPC → PrPres conversion.Citation37 However, in contrast, Imamura et al. recently reported that high concentrations of DTT do not inhibit either the binding or conversion reactions of recombinant mouse prion protein to PrPSc and even accelerate the mouse-adapted BSE-seeded conversion.Citation38 Similarly, Maeda et al. recently reported that reducing the disulfide bond of insulin using protein disulfide isomerase (PDI) promotes the formation of β-sheet-rich aggregates by insulin.Citation39 Based our results, we suggest that breaking the disulfide bridge increases the structural flexibility of the recombinant prion protein at neutral pH and in turn facilitates the association of certain conformers into oligomer. The intrinsically disordered property of the reduced prion protein might be important for its nucleation step in sporadic prion diseases. Here, we propose a model for the structural conversion of the prion protein initiated by reduction (). The disulfide bond might serve as a “lock” to trap the prion protein in a thermodynamic metastable stateCitation40 and, once the lock is removed, prion protein is intrinsically disordered and starts searching for the conformation with the thermodynamic minimum. The S132C/N181C mutation might favor a more disordered conformer, so the coil-rich state can be seen in the CD spectrum of the conformational ensemble. Finally the β-PrP conformer is stabilized by association into β-oligomers, protofibrils or fibrils. Our work implies a potential role of redox state, probably affected by the glutathione concentration or PDI, in initiating the structural conversion of the prion protein. PDI is mainly present in the endoplasmic reticulum (ER), but some members of the PDI family have been reported to have a non-ER location (e.g., cytosol, nucleus, plasma membrane rafts, cell surface, or secreted).Citation41 Interestingly, overexpression of PDI ER-60 (also called ERp60, GRp58) has been found in brains of patients with sporadic Creutzfeldt-Jacob diseaseCitation42 and scrapie-infected mice.Citation43 The effect of disulfide bond reduction on facilitating the α-to-β conversion of the prion protein implies that PDI might be the “protein X” involved in prion formation. If so, screening for inhibitors of prion-related PDI might uncover agents that could be used in therapy for prion disease.Citation44 However, it has been reported that inhibition of GRp58 with siRNA enhances PrPSc toxicity.Citation43 The roles of PDI in sporadic and infected prion diseases deserve further investigation.
Figure 9. Proposed model for the structural conversion of the mouse prion protein after disulfide bond reduction in neutral condition. The flexible N-terminal domain is not displayed. Helices 1 and 3 are colored red, helix 2 is colored blue, the two β-strands are colored green, and the disulfide bond between Cys-179 and Cys-214 is shown in yellow. The mutation sites used in our study, Ser-132 and Asn-181, are indicated. After the disulfide bond is reduced, helix 2 is no longer stable and is partially unfolded. The disordered part associates and the protein structure is stabilized by the formation of oligomers, which show a characteristic β-sheet signal in the CD spectrum. These β-oligomers have a bead-like morphology under TEM and can associate one-dimensionally into protofibrils.
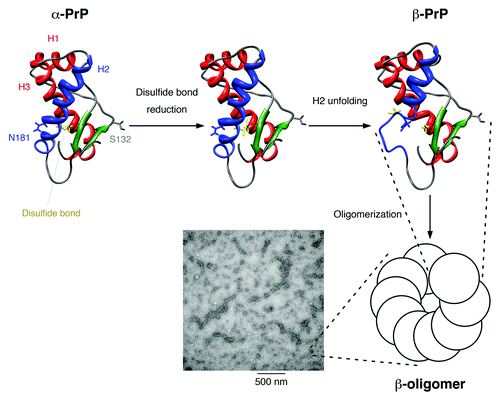
In summary, we report, for the first time, that full-length mPrP disulfide-deletion mutants is intrinsically disordered and can spontaneously undergo a structural conversion from the native-like α-PrP structure to soluble β-PrP oligomers or fibrils under non-denaturing neutral conditions. This conversion is due to the disulfide-deletion, and full-length wild-type mPrP can be converted from α-PrP into β-PrP simply by adding reducing agent.
Materials and Methods
Protein expression and purification
The mouse PrP gene cloned in the pET101/D-TOPO vector was kindly provided by Dr. Ilia V. Baskakov (Center for Biomedical Engineering and Technology, University of Maryland Biotechnology Institute, USA). The expressed protein, mPrPwt, contains mouse PrP residues 23–230 with an extra Met at the N-terminus. Mutations were constructed by site-directed mutagenesis. The proteins were expressed in BL21 Star™ (DE3) cells (Invitrogen) and were purified using a protocol modified from a published method.Citation45 An overnight culture was used to inoculate fresh TB medium containing ampicillin (100 μg/mL) at a ratio of 2:100 and the cells were grown at 37°C with vigorous shaking (250 rpm) for 3 h (A600 nm = ~0.6) Protein expression was then induced by adding IPTG to a final concentration of 1 mM and the culture grown for an additional 5 h, then the cells were harvested by centrifugation at 1,900 g at 4°C for 10 min. The cell pellet (about 34 g from 2.4 L of culture) was resuspended in 300 mL of cell lysis buffer (50 mM TRIS-HCl, 100 mM NaCl, pH 8.0) and the cells lysed by addition of 0.5 X CelLyticB (Sigma), 0.15 mg/mL of lysozyme, 50 μg/mL of DNaseI, 7 mM MgCl2, and 1 mM PMSF. The lysate was then centrifuged at 6,000 g at 4°C for 30 min and the pellet was resuspended in freshly prepared IMAC A buffer (8 M urea, 100 mM Na2HPO4, 10 mM TRIS-HCl, 2 mM TCEP, pH 8.0). The suspension was incubated at room temperature for 2 h, then centrifuged at 18,000 g at 4°C for 20 min. The supernatant was applied to a column packed with chelating Fast Flow Sepharose resin (GE Healthcare) precharged with Ni-ions at a flow rate of 150 cm/hr, which was then washed with 5 column volumes of IMAC A buffer. The prion protein was eluted with 4 column volumes of IMAC A buffer containing 20 mM EDTA and further purified by reversed-phase chromatography on a C5 HPLC column (Discovery BIO Wide Pore C5, 10 μm, 25 cm × 10.0 mm, Supelco, Sigma-Aldrich) using a linear gradient of 27–45% buffer B in 30 min (Buffer A: 94.9% water, 5% acetonitrile, 0.1% TFA; buffer B: 99.9% acetonitrile, 0.1% TFA) at a flow rate of 3 mL/min. The mPrP mutant proteins were typically eluted at 31–33% buffer B. The proteins were identified by SDS-PAGE and ESI-TOF mass spectrometry and lyophilized and stored at -30°C.
In contrast to the mPrP mutant proteins, the wild-type mPrPwt has an intramolecular disulfide bond and an oxidation step was required before the final HPLC purification step. In the IMAC purification step, TCEP was replaced by 10 mM reduced glutathione and the eluent was desalted on a HiPrep 26/10 desalting column (GE Healthcare) using 6 M urea, 0.1 M TRIS-HCl, pH 7.5, as desalting buffer. For oxidation to form the disulfide bond, 5 mM EDTA and 0.2 mM oxidized glutathione were then added to the desalted eluent and the mixture shaken at room temperature overnight.
Circular dichroism (CD) spectroscopy
The sample (0.15 mg/mL) was placed in a 1-mm path-length quartz cuvette and incubated at 25°C. The far-UV CD spectrum between 195 and 250 nm was recorded on a JASCO J-715 spectrometer (JASCO) with the band width set to 2 nm and the step resolution to 0.05 nm. Two scans were averaged for each sample. For time-course studies, the samples were kept in the cuvette throughout the experiment to avoid any possible contamination during sample transfer. CD deconvolution was performed using CDPro software. The fitting program was cdsstr and the basis set SP43.
Analytical ultracentrifugation (AUC)
Sedimentation velocity analytical ultracentrifugation (SV-AUC) was performed using a Beckman XL-A analytical ultracentrifuge (Beckman). S132C/N181C (0.1 mg/mL) in 10 mM NaOAc, pH 7, was loaded into an AUC sample cell with a 12-mm optical path two-channel centerpiece, with 10 mM NaOAc in the reference sector and the cells were loaded into a AnTi-60 rotor and spun at 60,000 rpm and scans of the absorbance at 280 nm were acquired at 1 min intervals over a period of 5 h. The sedimentation profiles were analyzed using the software SEDFIT (version 12.1b). The sedimentation velocity data were analyzed using the c(s) method of distribution to characterize the sedimentation coefficient distribution of all species in solution. The partial specific volume (v) for S132C/N181C and the buffer density and viscosity were calculated using SEDNTERP software.
Thioflavin T (ThT) binding assay
A stock solution of 2 mM ThT was prepared by dissolving ThT in 140 mM KCl, 100 mM sodium phosphate buffer, pH 7.85, and filtering the solution through a 0.22 μm Millipore syringe filter. The working solution was freshly prepared before use by adjusting the final concentration to 200 μM in the same buffer, then 30 μL of the solution was mixed with an equal volume of sample and the mixture left for 1 min. Fluorescence measurement was performed on a Jasco FP-750 spectrophotometer using a 3-mm path-length rectangular cuvette with excitation at 420 nm. Fluorescence spectra were recorded over the range of 450–600 nm at a scan speed of 125 nm/min with a data pitch of 1 nm. The slits for excitation and emission were set to 10 nm and 5 nm, respectively.
Transmission electron microscopy
The samples were deposited on carbon-coated 300-mesh copper grids and incubated for 3 min for absorption. Negative staining was performed by staining with 2% uranyl acetate for 3 min. After drying, the samples were viewed using a Hitachi H-7000 electron microscope (Hitachi, Tokyo, Japan).
Cytotoxicity assay
Mouse N2a cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Invitrogen) containing 10% fetal bovine serum (FBS; HyClone) at 37°C in 5% CO2. To evaluate cytotoxicity, the cells were harvested, suspended at a density of 350,000 cells/mL in DMEM and 100 μL was plated into each well of a 96-well CellBIND polystyrene microplate (Corning). The plates were then incubated at 37°C in 5% CO2 for 24 h to allow the cells to attach. At the same time, S132C/N181C was dissolved in 10 mM NaOAc buffer, pH 7.0, as a 300 μM stock solution. The S132C/N181C stock solution was then diluted 10-fold in DMEM to a final concentration of 30 μM and incubated for 24 h with gentle shaking. On the second day, the cell medium was replaced with the DMEM containing S132C/N181C and incubated for 2 d, then cell viability was measured using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) toxicity assayCitation46; 10 μL of a 5 mg/mL solution of MTT in PBS was added to each well, then, after incubation for 4 h, the medium was removed and the MTT crystals dissolved in 100 μL of DMSO and the absorption at 570 nm measured.
ESR spectroscopy
1.5 mg of S132C, N181C, or S132C/N181C was dissolved in 478.4 μL of DMSO and mixed with 500 μL of 50 mM TRIS-HCl, pH 7.5, 20 μL of TCEP (50 mM in DMSO), and 1.6 μL of 126 mM (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl) methanethiosulfonate spin label (MTSSL) (Alexis Biochemicals) at a protein to MTSSL molar ratio of 1 to 3. The pH of the final solution was about 7.0. The mixture was left overnight at room temperature in the dark, then free MTSSL and unlabeled protein were removed by HPLC and the purified labeled protein lyophilized and stored at -30°C. The amino acid labeled with MTSSL was designated R1. To record the ESR spectra, the spin-labeled protein sample was dissolved at a concentration of 0.25 mM in 10 mM NaOAc containing 40% sucrose, pH 7.0. The spectra samples were recorded in a Bruker ELEXSYS E580 CW/Pulse spectrometer (Bruker, Billerica) equipped with an ER 4131VT variable nitrogen temperature accessory. The samples were loaded into two capillaries (O.D. 0.8 mm), each about 20 μL, and the ends were sealed with Parafilm, then the capillaries were introduced into a quartz tube (O.D. Four mm), which was then sealed with Parafilm. The experiments were performed at X-band (9.44 GHz) frequency with a 1.5 mW incident microwave. The scan width was 200 Gauss (G) and the field modulation amplitude was 0.8 G.
| Abbreviations: | ||
| PrP | = | prion protein |
| mPrPwt | = | recombinant mouse full-length PrP containing residues 23-230 |
| mPrP-CtoA | = | a mPrPwt mutant in which Cys-179 and Cys-214 of mPrPwt are replaced by Ala |
| S132C | = | an mPrP-CtoA mutant in which Ser-132 is replaced by Cys |
| N181C | = | an mPrP-CtoA mutant in which Asn-181 is replaced by Cys |
| S132C/N181C | = | an mPrP-CtoA mutant in which Ser-132 and Asn-181 are replaced by Cys |
| S132R1 | = | S132C with the side-chain of Cys-132 labeled with MTSSL |
| N181R1 | = | N181C with the side-chain of Cys-181 labeled with MTSSL |
| S132R1/N181R1 | = | S132C/N181C with the side-chains of Cys-132 and Cys-181 labeled with MTSSL. Residues are numbered based on hamster prion sequence in this work |
Additional material
Download Zip (8.8 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the Academia Sinica and the National Science Council, Taiwan, R. O. C (grant Nos. NSC 97–2321-B-001–032, NSC 98–2311-B-001–019, NSC 99–2321-B-001–038, and NSC 100–2321-B-001–026). We thank Dr. Ilia Baskakov for kindly providing the plasmid containing the mouse PrP gene. The ESI-TOF mass identification of proteins was performed by the core facilities at the Institute of Biological Chemistry, Academia Sinica, supported by the National Science Council and the Academia Sinica. We thank Mr. Tai-Lang Lin and the Core Facility of the Institute of Cellular and Organismic Biology, Academia Sinica, Taiwan for assistance in transmission electron microscopy. We thank Prof. Kuan Wang for his useful comments in manuscript writing.
Related Research Data
References
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, et al. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 1993; 90:10962 - 6; http://dx.doi.org/10.1073/pnas.90.23.10962; PMID: 7902575
- Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, et al. Structure of the recombinant full-length hamster prion protein PrP(29-231): the N terminus is highly flexible. Proc Natl Acad Sci U S A 1997; 94:13452 - 7; http://dx.doi.org/10.1073/pnas.94.25.13452; PMID: 9391046
- Ryou C, Mays CE. Prion propagation in vitro: are we there yet?. Int J Med Sci 2008; 5:347 - 53; http://dx.doi.org/10.7150/ijms.5.347; PMID: 19015743
- Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J Mol Biol 2005; 346:645 - 59; http://dx.doi.org/10.1016/j.jmb.2004.11.068; PMID: 15670611
- Baskakov IV. Autocatalytic conversion of recombinant prion proteins displays a species barrier. J Biol Chem 2004; 279:7671 - 7; http://dx.doi.org/10.1074/jbc.M310594200; PMID: 14668351
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, et al. Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 4; http://dx.doi.org/10.1038/370471a0; PMID: 7913989
- Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 1995; 375:698 - 700; http://dx.doi.org/10.1038/375698a0; PMID: 7791905
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411:810 - 3; http://dx.doi.org/10.1038/35081095; PMID: 11459061
- Soto C, Saborio GP, Anderes L. Cyclic amplification of protein misfolding: application to prion-related disorders and beyond. Trends Neurosci 2002; 25:390 - 4; http://dx.doi.org/10.1016/S0166-2236(02)02195-1; PMID: 12127750
- Gonzalez-Montalban N, Makarava N, Ostapchenko VG, Savtchenk R, Alexeeva I, Rohwer RG, et al. Highly efficient protein misfolding cyclic amplification. PLoS Pathog 2011; 7:e1001277; http://dx.doi.org/10.1371/journal.ppat.1001277; PMID: 21347353
- Mays CE, Yeom J, Kang HE, Bian J, Khaychuk V, Kim Y, et al. In vitro amplification of misfolded prion protein using lysate of cultured cells. PLoS One 2011; 6:e18047; http://dx.doi.org/10.1371/journal.pone.0018047; PMID: 21464935
- Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, et al. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods 2008; 5:211 - 2; http://dx.doi.org/10.1038/nmeth0308-211; PMID: 18309304
- Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem 2002; 277:21140 - 8; http://dx.doi.org/10.1074/jbc.M111402200; PMID: 11912192
- Jackson GS, Hosszu LL, Power A, Hill AF, Kenney J, Saibil H, et al. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science 1999; 283:1935 - 7; http://dx.doi.org/10.1126/science.283.5409.1935; PMID: 10082469
- Sokolowski F, Modler AJ, Masuch R, Zirwer D, Baier M, Lutsch G, et al. Formation of critical oligomers is a key event during conformational transition of recombinant syrian hamster prion protein. J Biol Chem 2003; 278:40481 - 92; http://dx.doi.org/10.1074/jbc.M304391200; PMID: 12917432
- Bocharova OV, Breydo L, Salnikov VV, Gill AC, Baskakov IV. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob Disease. Protein Sci 2005; 14:1222 - 32; http://dx.doi.org/10.1110/ps.041186605; PMID: 15802644
- Bjorndahl TC, Zhou GP, Liu X, Perez-Pineiro R, Semenchenko V, Saleem F, et al. Detailed biophysical characterization of the acid-induced PrP(c) to PrP(β) conversion process. Biochemistry 2011; 50:1162 - 73; http://dx.doi.org/10.1021/bi101435c; PMID: 21189021
- Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J Biol Chem 2006; 281:13828 - 36; http://dx.doi.org/10.1074/jbc.M511174200; PMID: 16554307
- Khan MQ, Sweeting B, Mulligan VK, Arslan PE, Cashman NR, Pai EF, et al. Prion disease susceptibility is affected by beta-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci U S A 2010; 107:19808 - 13; http://dx.doi.org/10.1073/pnas.1005267107; PMID: 21041683
- Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol 2006; 16:644 - 53; http://dx.doi.org/10.1016/j.sbi.2006.08.008; PMID: 16949813
- Chen PY, Lin CC, Chang YT, Lin SC, Chan SI. One O-linked sugar can affect the coil-to-β structural transition of the prion peptide. Proc Natl Acad Sci U S A 2002; 99:12633 - 8; http://dx.doi.org/10.1073/pnas.192137799; PMID: 12235358
- Lee LY, Chen RP. Quantifying the sequence-dependent species barrier between hamster and mouse prions. J Am Chem Soc 2007; 129:1644 - 52; http://dx.doi.org/10.1021/ja0667413; PMID: 17243682
- Ho CC, Lee LY, Huang KT, Lin CC, Ku MY, Yang CC, et al. Tuning the conformational properties of the prion peptide. Proteins 2009; 76:213 - 25; http://dx.doi.org/10.1002/prot.22341; PMID: 19137620
- Zhang H, Stockel J, Mehlhorn I, Groth D, Baldwin MA, Prusiner SB, et al. Physical studies of conformational plasticity in a recombinant prion protein. Biochemistry 1997; 36:3543 - 53; http://dx.doi.org/10.1021/bi961965r; PMID: 9132005
- Pan KM, Stahl N, Prusiner SB. Purification and properties of the cellular prion protein from Syrian hamster brain. Protein Sci 1992; 1:1343 - 52; http://dx.doi.org/10.1002/pro.5560011014; PMID: 1363897
- Chang ES, Liao TY, Lim TS, Fann W, Chen RP. A new amyloid-like β-aggregate with amyloid characteristics, except fibril morphology. J Mol Biol 2009; 385:1257 - 65; http://dx.doi.org/10.1016/j.jmb.2008.11.009; PMID: 19041877
- Watanabe Y, Hiraoka W, Shimoyama Y, Horiuchi M, Kuwabara M, Inanami O. Instability of familial spongiform encephalopathy-related prion mutants. Biochem Biophys Res Commun 2008; 366:244 - 9; http://dx.doi.org/10.1016/j.bbrc.2007.11.145; PMID: 18062918
- Chiang YW, Otoshima Y, Watanabe Y, Inanami O, Shimoyama Y. Dynamics and local ordering of spin-labeled prion protein: an ESR simulation study of a highly PH-sensitive site. J Biomol Struct Dyn 2008; 26:355 - 66; http://dx.doi.org/10.1080/07391102.2008.10507250; PMID: 18808201
- Hosszu LL, Trevitt CR, Jones S, Batchelor M, Scott DJ, Jackson GS, et al. Conformational properties of β-PrP. J Biol Chem 2009; 284:21981 - 90; http://dx.doi.org/10.1074/jbc.M809173200; PMID: 19369250
- Bateman DA, Tycko R, Wickner RB. Experimentally derived structural constraints for amyloid fibrils of wild-type transthyretin. Biophys J 2011; 101:2485 - 92; http://dx.doi.org/10.1016/j.bpj.2011.10.009; PMID: 22098747
- Mehlhorn I, Groth D, Stöckel J, Moffat B, Reilly D, Yansura D, et al. High-level expression and characterization of a purified 142-residue polypeptide of the prion protein. Biochemistry 1996; 35:5528 - 37; http://dx.doi.org/10.1021/bi952965e; PMID: 8611544
- Dmitrenko O, Thorpe C, Bach RD. Mechanism of SN2 disulfide bond cleavage by phosphorus nucleophiles. Implications for biochemical disulfide reducing agents. J Org Chem 2007; 72:8298 - 307; http://dx.doi.org/10.1021/jo071271w; PMID: 17914842
- Turk E, Teplow DB, Hood LE, Prusiner SB. Purification and properties of the cellular and scrapie hamster prion proteins. Eur J Biochem 1988; 176:21 - 30; http://dx.doi.org/10.1111/j.1432-1033.1988.tb14246.x; PMID: 3138115
- Muramoto T, Scott M, Cohen FE, Prusiner SB. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc Natl Acad Sci U S A 1996; 93:15457 - 62; http://dx.doi.org/10.1073/pnas.93.26.15457; PMID: 8986833
- Welker E, Raymond LD, Scheraga HA, Caughey B. Intramolecular versus intermolecular disulfide bonds in prion proteins. J Biol Chem 2002; 277:33477 - 81; http://dx.doi.org/10.1074/jbc.M204273200; PMID: 12082114
- Herrmann LM, Caughey B. The importance of the disulfide bond in prion protein conversion. Neuroreport 1998; 9:2457 - 61; http://dx.doi.org/10.1097/00001756-199808030-00006; PMID: 9721914
- Imamura M, Kato N, Okada H, Iwamaru Y, Shimizu Y, Mohri S, et al. Strain-specific effects of reducing agents on the cell-free conversion of recombinant prion protein into a protease-resistant form. Microbiol Immunol 2011; 55:633 - 40; http://dx.doi.org/10.1111/j.1348-0421.2011.00357.x; PMID: 21645053
- Maeda R, Ado K, Takeda N, Taniguchi Y. Promotion of insulin aggregation by protein disulfide isomerase. Biochim Biophys Acta 2007; 1774:1619 - 27; http://dx.doi.org/10.1016/j.bbapap.2007.08.016; PMID: 17920002
- Baldwin AJ, Knowles TP, Tartaglia GG, Fitzpatrick AW, Devlin GL, Shammas SL, et al. Metastability of native proteins and the phenomenon of amyloid formation. J Am Chem Soc 2011; 133:14160 - 3; http://dx.doi.org/10.1021/ja2017703; PMID: 21650202
- Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol 2002; 193:154 - 63; http://dx.doi.org/10.1002/jcp.10172; PMID: 12384992
- Yoo BC, Krapfenbauer K, Cairns N, Belay G, Bajo M, Lubec G. Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt-Jakob disease. Neurosci Lett 2002; 334:196 - 200; http://dx.doi.org/10.1016/S0304-3940(02)01071-6; PMID: 12453628
- Hetz C, Russelakis-Carneiro M, Wälchli S, Carboni S, Vial-Knecht E, Maundrell K, et al. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci 2005; 25:2793 - 802; http://dx.doi.org/10.1523/JNEUROSCI.4090-04.2005; PMID: 15772339
- Smith AM, Chan J, Oksenberg D, Urfer R, Wexler DS, Ow A, et al. A high-throughput turbidometric assay for screening inhibitors of protein disulfide isomerase activity. J Biomol Screen 2004; 9:614 - 20; http://dx.doi.org/10.1177/1087057104265292; PMID: 15475481
- Makarava N, Baskakov IV. Expression and purification of full-length recombinant PrP of high purity. Methods Mol Biol 2008; 459:131 - 43; http://dx.doi.org/10.1007/978-1-59745-234-2_10; PMID: 18576153
- Shearman MS, Hawtin SR, Tailor VJ. The intracellular component of cellular 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) reduction is specifically inhibited by beta-amyloid peptides. J Neurochem 1995; 65:218 - 27; http://dx.doi.org/10.1046/j.1471-4159.1995.65010218.x; PMID: 7790863