Abstract
Growing evidence indicates that astrocytes cannot be just considered as passive supportive cells deputed to preserve neuronal activity and survival, but rather they are involved in a striking number of active functions that are critical to the performance of the central nervous system (CNS). As a consequence, it is becoming more and more evident that the peculiar properties of these cells can actively contribute to the extraordinary functional complexity of the brain and spinal cord.
This new perception of the functioning of the CNS opens up a wide range of new possibilities to interpret various physiological and pathological events, and moves the focus beyond the neuronal compartment toward astrocyte-neuron interactions. With this in mind, here we provide a synopsis of the activities astrocytes perform in normal conditions, and we try to discuss what goes wrong with these cells in specific pathological conditions, such as Alzheimer Disease, prion diseases and amyotrophic lateral sclerosis.
Introduction
The functioning of the central nervous system (CNS) has been long explained by a neuron-centric vision of the CNS, attributing all the main information processing activities to neuronal cells. Yet, more recent work has challenged this concept, revealing that the non-neuronal cell component of the nervous system, particularly glial cells, also perform a dynamic range of functions that are essential for the development and physiology of the brain and spinal cord.Citation1-Citation3 Thus, there is growing consensus that a more integrated level of neuroglial interactions needs to be considered in order to have a comprehensive overview of brain functions and dysfunctions.
The most abundant glial cell type in the mammalian brain is represented by the astrocytes, which constitute up to 50% of its volume.Citation4 These cells have been traditionally regarded just as passive housekeepers apt to preserve the optimal microenvironment for neuronal function and survival. However, the recent acknowledgment of a broader range of previously unrecognized properties and activities has dramatically changed this view, introducing the idea that astrocytes contribute to the performance of the CNS.Citation1,Citation5 The best-characterized astroglial cells, in terms of morphology and organization, are protoplasmic astrocytes, located in the parenchyma of the gray matter. Intracellular injection of fluorescent dyes revealed that protoplasmic astrocytes present a peculiar anatomical organization, based on the occupancy of exclusive, non-overlapping territories.Citation6-Citation8 Within its own territory, each astrocyte extends highly ramified processes that establish numerous connections with the adjacent cellular populations, i.e., neurons, other glial cells and the blood vessels. This intimate spatial relationship with both neurons and the blood vessel cells puts astrocytes in an ideal position to integrate the neural circuitry with the local microcirculation.Citation9-Citation11 As a consequence, astrocytes emerge as the main effectors of the CNS homeostasis. They supply energy metabolites to neurons,Citation11 regulate the blood flow and the blood-brain barrier,Citation12 and control the levels of extracellular ions, neurotransmitters and fluids.Citation13-Citation15 In addition, the expression of functional receptors on their plasma membrane allows astrocytes to sense neurotransmitters spilled over from nearby synaptic sites. In turn, astrocytes can respond to neurons by Ca2+-dependent release of various gliotransmitters (i.e., transmitters of glial origin, as opposite to neurotransmitters), which can influence neuronal and synaptic functions.Citation1-Citation3 This close anatomical and functional interactions between astrocytes and neurons prompted the formulation of a new concept of synaptic physiology, i.e., the tripartite synapse, wherein the flow of information involves the processes of perisynaptic astrocytes in addition to presynaptic and postsynaptic nerve terminals.Citation2,Citation16,Citation17
The multiplicity and complexity of these activities clearly indicate that the correct performance of the astrocytes is crucial for the physiological functioning of the CNS, and its derangement may affect both neuronal activity and survival.
Astrocyte Signaling in Physiology
Because of their inability to generate action potentials and communicate via electrical signals, astrocytes have been long considered as non-excitable cells. Actually, they display a peculiar form of excitability that is based on variations in the intracellular concentration of Ca2+ ions ([Ca2+]i). Originally described in the early nineties in cell cultures,Citation18 astroglial [Ca2+]i changes have been more recently characterized in acute brain slices and in vivo. Thus, it has been determined that astrocytic [Ca2+]i elevations can occur spontaneouslyCitation19-Citation23 or can be evoked in response to various physiological and pharmacological stimulations.Citation24-Citation35
Although the physiological significance of such intracellular Ca2+ rises has not been fully clarified, it appears that astroglial [Ca2+]i signaling represents a composite mechanism by which astrocytes control a broad variety of cellular processes, including the exocytotic release of various gliotransmitters. The discharge of such mediators seems to be generally triggered by activation of inositol 1,4,5 triphosphate (Ins(1,4,5)P3)-generating G-protein-coupled receptors (GPCRs), followed by Ins(1,4,5)P3-mediated release of Ca2+ from the endoplasmic reticulum (ER) stores.Citation36-Citation40
Remarkably, such regulated release of gliotransmitters appears to be relevant for the astrocyte-to-neuron communication as well as for the performance of the CNS, as indicated by several studies performed in various experimental paradigms. Thus, the release of the gliotransmitter glutamate from astrocytes was described to exert various forms of neuromodulatory activity in different brain regions.Citation19,Citation31-Citation33,Citation41-Citation46 Furthermore, the discharge of other glial mediators, particularly d-serine and adenosine, was reported to control the induction of long-term potentiation (LTP) at nearby excitatory synapsesCitation47 as well as to modulate a number of animal behaviors,Citation48-Citation50 respectively. Among the GPCRs competent to initiate the astrocytic release of gliotransmitters, there are group I metabotropic glutamate receptors (mGluRs), CXCR4 chemokine receptors and P2Y1 purinergic receptors (P2Y1Rs).Citation38,Citation51-Citation54 Notably, Ca2+-dependent release of glutamate from cultured astrocytes, in response to stimulation of the CXCR4 chemokine receptors or the purinergic P2Y1 receptors, was reported to be controlled by pro-inflammatory mediators, such as prostaglandins and cytokines, particularly the Tumor Necrosis Factor α (TNFα).Citation32,Citation38,Citation51,Citation52 Since the levels of these mediators are subjected to dramatic increases in several neurodegenerative diseases, it is reasonable to postulate that the molecular pathway controlling the glial release of glutamate can become over stimulated in pathological conditions, and this may perturb the astrocyte-to-neuron signaling and, possibly, trigger neurodamaging events.
Altogether, this scenario suggests that understanding in depth the versatility of both the astrocytes and the glial-neuronal interactions may be critical to gain new insights into the molecular basis of various neurodegenerative processes.
Astrocyte Signaling in Pathology
Several lines of evidence indicate that astrocytes do not react in a stereotyped fashion to all forms of injury and disease, but the mode and the extent of the astrocytic reaction can vary in dependence of both the severity of the insult and the context of the injury site. The most evident reaction of the astrocytes to several neurodegenerative diseases is represented by a vigorous activation, a condition commonly known as “reactive astrocytosis.”Citation55,Citation56
While the specific factors and cellular interactions that govern the activation of astroglial cells remain mostly elusive, it is clear that the shift from the resting to the activated phenotype is associated with morphological and biochemical changes of the astrocytes.Citation55,Citation56 Little is known about the impact that these alterations have on the astrocyte signaling. However, evidence indicates that reactive astrocytes are endowed with a plethora of molecules that are undetectable or present at lower levels in quiescent astroglia.Citation55,Citation56 Among these molecules, there are several cytokines and enzymes involved in the metabolic pathways of arachidonic acid, and thus in the generation of eicosanoids.Citation55,Citation56 This finding is particularly interesting considering that some of these molecules, such as TNFα and prostaglandins, control the Ca2+-dependent release of glutamate from astrocytes,Citation32,Citation38,Citation51,Citation52 which has been implicated in the excitotoxic death of neuronal cells in vitro, in neuron-glia co-culture systems.Citation38 Additional alterations of reactive astrocyte physiology have been described in a pioneer study by Aguado and colleagues. These authors reported that while spontaneous [Ca2+]i transients are a common feature of resting astrocytes, they are lost in reactive astrocytes in situ.Citation21 Since [Ca2+]i oscillations control astrocytic gliotransmitter release and may synchronize neuronal network activity, such defect appears relevant to CNS function, and should be also taken into consideration when studying neurodegenerative mechanisms.
Another aspect that should be considered carefully is that, in specific pathological circumstances, the reaction of astrocytes to injury or disease may take the form of atrophy or degeneration. This phenomenon was interpreted by some as a defensive mechanism to stem the overwhelming reaction of reactive astrocytes.Citation57 However, different causative hypotheses can be postulated, including the possibility that diseased astrocytes may become susceptible to physiological stimuli, and this progressively leads to the deterioration of their health conditions.Citation58,Citation59
On a mechanistic standpoint, the degenerative process of the astrocytes was described to be fine-tuned by a delicate balance between pro-survival and pro-apoptotic factors promoting or preventing astrocyte cell death, respectively.Citation57,Citation59 In this context, cytosolic Ca2+ may gain importance in view of its role in controlling the cell fate under stress conditions. It is generally well established that intracellular Ca2+ levels are critical to modulate a variety of cellular responses that are fundamental for a significant number of vital functions. However, cellular Ca2+ overload, or perturbation of intracellular Ca2+ compartmentalization, can cause cytotoxicity and trigger various forms of cell death.Citation60-Citation63 Thus, it is reasonable to assume that, in the astrocytes, intracellular Ca2+ concentrations may represent a critical switch between life and death. As a consequence, studying astrocytic Ca2+ signaling and Ca2+-dependent gliotransmitter release in pathological conditions emerges as a high priority to elucidate the contribution of this glial cell population to neurodegeneration and neurodegenerative diseases.
In this review, we tackle the abnormalities that the astrocytic signaling encounters in specific pathologies of the CNS, such as Alzheimer disease (AD), prion diseases and Amyotrophic Lateral Sclerosis (ALS). While these disorders are often seen as disparate pathologies, evidence indicates that they share prominent common features, including dramatic changes of the astrocytes. According to several observations, these glial alterations are unlikely to play a causal role in the pathogenesis of neurodegenerative disorders. Nevertheless, in transgenic models, glia appear to contribute to disease progression.Citation64 Thus, clarifying both how the glial responses are triggered under pathological conditions as well as their specific contribution to critical neurodamaging mechanisms may highlight novel cellular and molecular targets for therapeutic intervention.
Alzheimer Disease
Alzheimer disease (AD) is the most common form of progressive dementia in the elderly. Most cases of AD are sporadic, but approximately 1–2% of instances are genetically linked and can be distinguished by the early onset of dementia. The most critical risk factor for the development of AD is age, with the prevalence of cases increasing exponentially after 65 y.Citation65 On a clinical standpoint, the disease is characterized by the progressive impairment of higher cognitive function, loss of memory and altered behavior, which follow a gradual progression. The major histopathological hallmarks of AD are the deposition of extracellular amyloid plaques and the formation of intracellular neurofibrillary tangles in the brain.Citation65 These events are accompanied by a progressive neuroinflammatory reaction that involves the activation of glial cells around amyloid plaques.Citation66,Citation67 In the brain of both AD subjects and animal models, reactive microglia and astrocytes produce high levels of pro-inflammatory cytokines that were proposed to exacerbate neuronal damage.Citation68-Citation71 As the disease progresses, synapse loss and neuronal cell death become prominent, with the consequent shrinkage of the affected areas, i.e., the entorhinal cortex and the hippocampus. Amyloid plaques mostly consist of aggregated amyloid β (Aβ) peptide generated by the sequential proteolitic cleavages of the amyloid precursor protein (APP) via β- and γ-secretase enzymes.Citation72 The β-site APP-cleaving enzyme 1 (BACE1) has been identified as the β-secretase that iniziates the production of Aβ peptide.Citation73 Since neurons express higher levels of BACE1 than astrocytes,Citation74-Citation76 it was initially proposed that they are the major source of Aβ in brain.Citation74 However, in both autoptic AD cases and transgenic mice, BACE1 expression and Aβ deposits are not only restricted to neurons, but they co-localize also with astroglial markers.Citation77-Citation80 This suggests that astrocytes may contribute to the generation of pathological protein aggregates in vivo. In keeping, recent studies in vitro confirmed that, under normal conditions, astrocytes possess the APP-processing machinery for generating Aβ peptide, i.e., APP itself, BACE1 and/or its close homolog BACE2.Citation76,Citation80,Citation81 Furthermore, Aβ peptide was described to transcriptionally regulate the expression of the astrocytic BACE1 via the calcineurin/nuclear factor of activated T-cells 4 (NFAT4) or nuclear factor-kB (NF-kB) signaling pathways,Citation82,Citation83 suggesting a feed-forward mechanism to maintain or amplify Aβ deposits in astrocytes. More controversial is the issue of reactive astroglia as some studies indicated that cultured astrocytes, activated with cytokine combinations, positively regulate β-secretase activity and Aβ secretion,Citation84 whereas others found opposite results.Citation76
A correlation between the deposition of Aβ protein in the extracellular space of forebrain regions and the progressive decline in cognitive functions was originally established in morphological and biochemical studies in AD patients.Citation85-Citation87 Subsequent results obtained in various transgenic AD models provided additional evidence that cognitive deficits are the consequence of the synaptic failure induced by Aβ.Citation88 Synaptic transmission and plasticity is strongly based on Ca2+ signals, and these latter appear to be disrupted in AD neurons.Citation89-Citation91
Altogether, these observations can be explained by the “calcium hypothesis” of AD according to which the amyloidogenic APP processing may perturb the neuronal Ca2+ signaling pathways that are responsible for cognitive functions. Eventually, this can cause the learning and memory deficits that characterize the early stages of AD.Citation92 Alterations of neuronal Ca2+ levels may also influence the metabolism and production of Aβ, a peptide whose accumulation in the brain may further exacerbate Ca2+ dyshomeostasis and neurodegeneration.Citation93
Besides neurons, important alterations of Ca2+ signaling were also described in the astrocytes by two-photon Ca2+ imaging in vivo in the cerebral cortex of a mouse model of AD.Citation94
While the specific implications of astrocytic Ca2+ signaling alterations are not yet known, the existence of such phenomenon suggests that, in AD, the circuital dysfunction may be generalized, involving not only synaptic transmission but also neuron-glia communication.
Additional evidence in favor of this idea was provided in vitro, in cell culture experiments.
Administration of Aβ peptides to mixed cultures of hippocampal neurons and astrocytes was in fact described to cause abnormal [Ca2+]i transients and mitochondrial depolarization in astrocytes, long before any impairment was visible in neurons. Blocking the astrocytic Ca2+ protected neurons from delayed cell death,Citation95,Citation96 thereby establishing a direct correlation between Aβ−dependent alterations of the astrocytic Ca2+ signals and neuronal loss in vitro. Furthermore, astrocytic Ca2+ variations appear to be critical also for the release of neurotoxic concentrations of the gliotransmitter glutamate initiated by high levels of TNFα.Citation38 Because this cytokine is highly represented in the CNS of AD patients and transgenic mice,Citation67,Citation68,Citation70,Citation71 it is plausible to hypothesize that such process may be relevant also for AD-linked neurodamaging events. Our group tested this hypothesis using the PDAPP mice, a transgenic model of AD.Citation97 In particular, we utilized aged animals (12 mo-old), presenting abundant amyloid plaque deposition and reactive gliosis in the forebrain, and pre-symptomatic animals (4 mo-old), with little or no amyloid deposits and no apparent glial alteration. Ca2+-dependent glutamate release from astrocytes was stimulated in brain slices from PDAPP animals and controls by direct application of high concentrations of exogenous TNFα. The amount of glutamate secreted by hippocampal slices from aged PDAPP animals was significantly lower compared with pre-symptomatic mice and age-matched controls.Citation97 The defect was region-selective as the glutamate release response from cerebellar slices of aged PDAPP mice was identical to that of controls.Citation97 Altogether, these observations were quite unexpected, particularly in view of previous data in cultures where, in an acute experiment, production of high TNFα levels by reactive microglia strongly potentiated the astrocytic release of glutamate.Citation38 However, in AD, glial inflammation is a chronic phenomenon, particularly around protein aggregates, where endogenous TNFα levels are presumably constantly high and may over-stimulate receptors, thus causing functional uncoupling.Citation68,Citation70,Citation71
At present, the functional significance of this defect is not established. However, one can speculate that a reduced astrocytic glutamate input to neurons may result in weakened connectivity of excitatory hippocampal synapses.Citation31,Citation32 Remarkably, TNFα was also reported to critically control glutamatergic gliotransmission in the hippocampus,Citation32 and astrocytic TNFα-dependent signaling was shown to favor synaptic strengthCitation98 and induce homeostatic synaptic up-scalingCitation98-Citation100 by promoting insertion of post-synaptic AMPA receptor subunits. Therefore, disruption of TNFα signal-transduction in astrocytes could lead to several alterations converging in a progressive reduction of synaptic efficacy and, possibly, underlie behavioral deficits in AD.
Prion Diseases
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases, are a group of invariably fatal neurodegenerative disorders affecting humans and a wide range of mammals. An important feature of these disorders is the accumulation in the CNS of PrPSc, a protease-resistant conformer of the host-encoded cellular prion protein (PrPC), believed to be the main or only constituent of the transmissible agent (‘prion’).Citation101-Citation103 Despite considerable attention resulting from its involvement in these disorders, the physiological function of PrPC remains elusive.
Thus, an unsolved issue is whether disease progression is mainly affected by the accumulation of PrPSc in brain and/or by the loss of PrPC function.Citation104-Citation106
Although prion diseases present some common neuropathological traits with AD, the Ca2+ pathophysiology has been much less characterized in the context of these disorders, and knowledge appears to be limited to neuronal cells. Thus, receptor-mediated Ins(1,4,5)P3 and [Ca2+]i responses resulted markedly reduced in scrapie-infected mouse neuroblastoma cells.Citation107,Citation108
Furthermore, the ER Ca2+ content was shown to be decreased in cells chronically infected with scrapie prions, and this correlated with a higher sensitivity to ER stress-induced cell death.Citation109 Since a similar reduction in the ER Ca2+ levels were recently reported in primary neurons lacking the cellular prion protein, it was postulated that PrPC may be part of the machinery deputed to maintain the intracellular Ca2+ homeostasis. As a consequence, the loss of its function, during disease progression, may contribute to Ca2+ derangement and synaptic failure.Citation110 Even though astrocytes were described to be the earliest site of PrPSc accumulation in brain,Citation111 no information is currently available on the Ca2+ signaling of scrapie-infected astroglia.
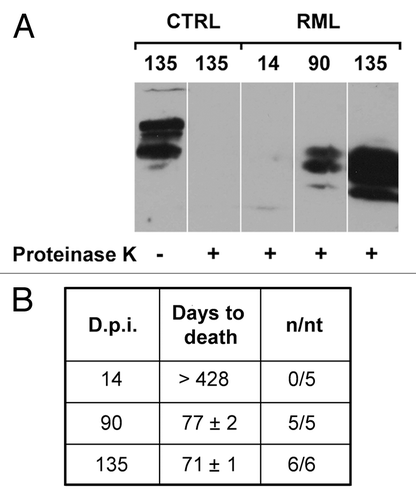
The presence of chronically activated glial cells around PrPSc depositions is a well documented feature in prion diseases.Citation112-Citation114 While it is not clear whether they play a causative role in the disease, recent data from animal models suggest that activated glia might at least contribute to accelerate its progression. Since high levels of pro-inflammatory cytokines, produced by reactive microglia and astrocytes, are detected in the brain of patients and animal models for these disorders,Citation115-Citation117 we investigated the TNFα- and Ca2+-dependent release of glutamate from astrocytes on scrapie-infected tissues. Wild-type C57Bl/6J mice were intracerebrally (i.c.) inoculated with the mouse-adapted scrapie strain Rocky Mountain Laboratory (RML). Brains from RML-infected and saline-treated (control) mice were taken at 14, 90 and 135 d post-inoculation (d.p.i.) and used to prepare acute slices. Stimulation of scrapie-infected hippocampal slices with exogenous TNFα resulted in a significant reduction of the release of glutamate from astrocytes at the early symptomatic stage of 135 d.p.i. when compared with the pre-symptomatic phases and to slices from age-matched control animals (). Impairment in the astrocytic release of glutamate correlated with a prominent astrocytosis () and the accumulation of both the proteinase K-resistant PrPSc and infectivity in the hippocampus (). Interestingly, such result could not be ascribed to a local neuroinflammatory reaction triggered by intracerebral manipulation and injection of scrapie prions, because we could confirm similar results in hippocampal slices from mice intraperitoneally (i.p.) challenged with RML at the early symptomatic stage of 180 d.p.i. when compared with slices from younger or control animals () (unpublished observations).
Figure 1. Impaired TNFα-dependent glutamate release from hippocampal astrocytes of scrapie-infected mice. Histograms indicate the glutamate release induced by TNFα (30ng/ml) in acute hippocampal slices from C57Bl/6J mice intracerebrally (30 μl, (A) or intraperitoneally (100 μl, (B) inoculated with 1% homogenate from saline-treated (CTRL) or RML-infected, terminally ill CD1 mouse brains. Brains were taken at 14, 90 and 135 d post intracerebral inoculation (d.p.i.) and at 14 and 180 d post intraperitoneal inoculation (Days to terminal disease: i.c.: 164 ± 3; i.p.: 209 ± 5). Acute hippocampal slices were prepared and the amount of glutamate released upon stimulation with TNFα was measured using a previously described enzymatic assay.Citation97 Data are expressed as mean ± s.e.m. (n = 3–6 animals for each experimental group). *p < 0.05 vs. CTRL, two-way ANOVA followed by Scheffe’s F-test.
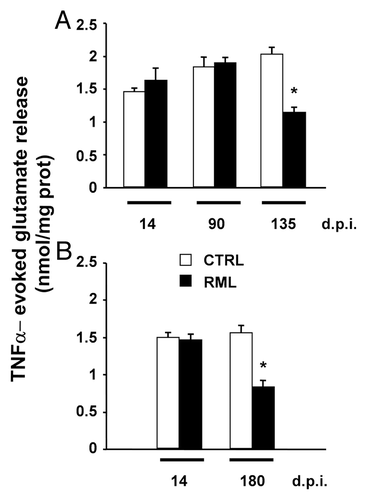
Figure 2. Progressive astrocytosis and spongiosis in the hippocampus of scrapie-infected mice. Coronal sections from saline-treated (CTRL) and RML-infected animals (i.c.) at 90 and 135 d.p.i were stained with hematoxylin alone or hematoxylin with the astrocytic marker Glial Fibrillary Acidic Protein (GFAP). Stainings highlight spongiosis and pronounced astrocytosis in the hippocampus of RML-infected mice at 135 d.p.i. Images are representative of 3 animals. Scale bar, 50μm.
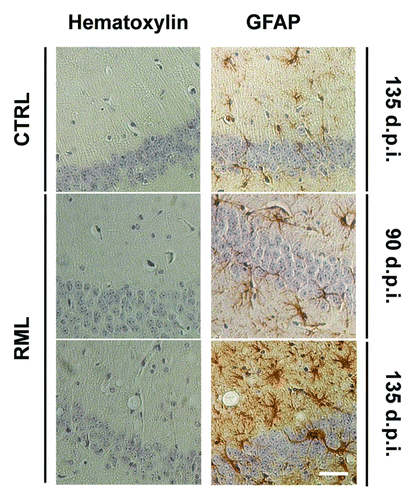
Figure 3. PrPSc accumulation and prion titers in the hippocampus of RML-infected mice. (A) Hippocampal homogenates from saline-treated (CTRL) or RML-infected mice (i.c.) at 14, 90 and 135 d.p.i. were treated in the presence (+) or in the absence (-) of proteinase K to distinguish native PrPC from the proteinase K-resistant PrPSc and analyzed by western blotting (n = 3). (B) Prion titers in the hippocampus of infected animals at 14, 90 and 135 d.p.i. were determined by inoculating homogenates i.c. into CD-1 indicator mice. Data highlight a progressive increase in the amount of PrPSc and infectivity in the hippocampi of RML-infected mice at 135 d.p.i. n/nt = Number of mice with scrapie/ total number of mice inoculated.
Because these results are fully consistent with the data obtained on the PDAPP model of Alzheimer Disease, one may infer that the alterations of the TNFα- and Ca2+-dependent glutamate release from astrocytes cannot be a distinctive feature of a specific pathology, but it is rather a peculiarity of chronically activated astrocytes.
Amyotrophic Lateral Sclerosis
Another disorder of the nervous system that results particularly interesting because of the overt involvement of glial cells in the development and progression of the disease is Amyotrophic Lateral Sclerosis (ALS), a pathological condition characterized by the progressive loss of corticospinal and spinal motor neurons. Although multiple genes and genetic loci have been recently linked to this disorder,Citation118-Citation120 most of the current knowledge on ALS pathogenesis is based on the discovery of mutations in the enzyme Cu-Zn superoxide dismutase (SOD1)Citation121 and the subsequent generation of transgenic animal models.Citation122-Citation127
While the primary toxic property of mutant SOD1s remains unresolved, a hint of the cascade of events implicated in motor neuron degeneration came by the landmark observation that death of the motor cells is a non-cell-autonomous process, but instead involves interaction with neighboring non-neuronal cells, particularly microglia and astrocytes.Citation128 Massive activation of microglia and astrocytes in areas of motor neuron loss was reported in both sporadic and familial human cases, as well as in transgenic animal models.Citation129-Citation131 Though microglial alterations were shown to be directly implicated in favoring disease progression in vivo,Citation130,Citation132 silencing mutant SOD1 expression in astrocytes was reported to affect disease onset and progression in transgenic mice.Citation131,Citation133 Furthermore, transplantation of mutant SOD1-expressing glial progenitors, capable of differentiating into astrocytes, into the spinal cord of wild-type rats induced motor neuron degeneration and disease symptoms in vivo, definitively confirming a causative role for these cells in ALS.Citation134
A peculiar histopathological abnormality in tissues of ALS patients is the presence of ubiquitin inclusions within astrocytes.Citation135 Noteworthy, similar features are shared by mutant SOD1 mice, which show protein aggregates made of SOD1, ubiquitin and/or activated caspase-3 in astroglial cells.Citation124,Citation136 To clarify the impact of such inclusions on the astrocyte performance in ALS, our group performed a thorough histopathological analysis of the lumbar tract of the spinal cord from transgenic mice carrying the Gly93→Ala substitution in the human SOD1 amino acid sequence (hSOD1G93A). We localized a subpopulation of astrocytes harboring protein inclusions specifically in the neighborhood of motor cells. These astrocytes displayed morphological and biochemical features reminiscent of degenerating cells,Citation58 including a spheroid cell body with increased diameter and a reduced number or even absence of GFAP-positive cell processes.
When present, such processes appeared short and abnormally thick compared with those of the normal astrocytes. Degenerating astroglial cells were first observed at the pre-symptomatic stage, when motor neurons show axonal damage but are still alive.Citation137 Their number significantly increased concomitant with the onset of neuronal degeneration and the appearance of ALS symptoms. We then studied the mechanism underlying astrocyte degeneration in cultured spinal cord astrocytes and surprisingly found that mutant SOD1s, either G93A or G85R, do not exert a direct pro-apoptotic action, but rather make the astrocytes vulnerable to glutamate, even at physiological concentrations. The glutamatergic mechanism responsible for the deleterious effect was found to involve metabotropic glutamate receptor 5 (mGluR5) signaling. Thus, blockage of this receptor reduced apoptosis of mutant SOD1-expressing astrocytes in response to glutamate challenges. Moreover, administration of a mGluR5 antagonist in vivo reduced astrocyte degeneration in the lumbar spinal cord, delayed the appearance of ALS symptoms and extended survival in hSOD1G93A transgenic mice.Citation58
In normal astrocytes, the activation of mGluR5 triggers the formation of Ins(1,4,5)P3 and the consequent release of Ca2+ from the ER, resulting in intracellular Ca2+ oscillations.Citation138,Citation139 Thus, we investigated the astrocytic Ca2+ response downstream mGluR5 in mutant SOD1- expressing astrocytes. We found that the mutant cells responded to the receptor stimulation with an aberrant and persistent Ca2+ release from the intracellular stores that correlated with cytochrome c release from mitochondria and astrocyte degeneration.Citation59 These results are fully consistent with the description of mitochondrial dysfunction in mutant SOD1-expressing astrocytes by other authors.Citation140 Since the Bcl-2 family members were reported to exert their anti-apoptotic activity by fine-tuning intracellular Ca2+ signaling through direct interaction with the Ins(1,4,5)P3 receptors (Ins(1,4,5)P3Rs), we then investigated the impact of the BH4 domain of Bcl-XL on astrocyte Ca2+ signaling by exploiting a biologically active BH4 peptide fused to the HIV-1 TAT protein (TAT-BH4). We realized that TAT-BH4 modulates the Ins(1,4,5)P3R-dependent Ca2+ release from the ER, and restores spontaneous Ca2+ oscillations in mutant SOD1-expressing astrocytes. This tight control of Ins(1,4,5)P3Rs by the peptide prevents the mGluR5-dependent aberrant release of Ca2+ from the intracellular stores, precludes the release of cytochrome c from mitochondria and protects the cells from excitotoxic damage. Furthermore, chronic administration of TAT-BH4 in vivo, to hSOD1G93A transgenic mice, reduces degeneration of spinal cord astrocytes and shows a positive impact on disease manifestations.Citation59
But what is the relevance of these findings in the context of ALS? And what are their repercussions on motor neurons? As mentioned above, astrocytes are intimately associated with synapses and can sense neurotransmitter released during synaptic activity.Citation1 Therefore, we infer that spinal cord astrocytes, endangered by the expression of ALS-linked mutant SOD1s, become vulnerable to physiological glutamate concentrations released at neighboring synapses and start to degenerate. This in turn may deprive the neighboring motor neurons of the optimal microenvironment and accelerate their degeneration in an interactive process of reciprocal damage.
Conclusions
The deposition of misfolded protein aggregates in regions of neuronal degeneration represents a typical feature of most brain disorders. Some of the findings briefly summarized in this review, indicate that, such protein aggregates not only accumulate in neurons, but they are present also within astroglial cells, possibly suggesting that astrocytes themselves may be functionally compromised in pathological conditions. In keeping with this hypothesis, some astroglial pathways, such as those controlling the astrocytic Ca2+ signaling and the Ca2+-dependent release of gliotransmitters, appear to be disrupted in various CNS disorders. Considering that these processes are critically involved in astrocyte survival as well as in the modulation of synaptic activity, it becomes clear that glial derangements in neurodegenerative conditions should not be considered only as epiphenomena or late reactions to neuronal injury, but rather as intrinsic components of the pathological process. Additional steps are certainly necessary to improve our understanding of the astrocyte pathophysiology. Nevertheless, the awareness that astrocytes are actively involved in the CNS functioning offers a wide range of new possibilities to unravel physiological and pathological mechanisms. This may open new perspectives for original therapeutic strategies.
Acknowledgments
The original studies were supported by grants from EMBO (ALTF 279–2002), MIUR-Italy (FIRB 2003 and PRIN 2006), European Community (QLK6–1999–02203) and Fondazione Telethon (GGP02052 and GGP05244). We are grateful to Prof. G. Poli (University of Milan) and Dr. E. Flechsig (University of Würzburg) for facilities and for technical and experimental support to the experiments with scrapie prions.
References
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 2005; 6:626 - 40; http://dx.doi.org/10.1038/nrn1722; PMID: 16025096
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 2009; 32:421 - 31; http://dx.doi.org/10.1016/j.tins.2009.05.001; PMID: 19615761
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol 2010; 72:335 - 55; http://dx.doi.org/10.1146/annurev-physiol-021909-135843; PMID: 20148679
- Tower DB, Young OM. The activities of butyrylcholinesterase and carbonic anhydrase, the rate of anaerobic glycolysis, and the question of a constant density of glial cells in cerebral cortices of various mammalian species from mouse to whale. J Neurochem 1973; 20:269 - 78; http://dx.doi.org/10.1111/j.1471-4159.1973.tb12126.x; PMID: 4633361
- Rossi D, Martorana F, Brambilla L. Implications of gliotransmission for the pharmacotherapy of CNS disorders. CNS Drugs 2011; 25:641 - 58; http://dx.doi.org/10.2165/11593090-000000000-00000; PMID: 21790208
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 2002; 22:183 - 92; PMID: 11756501
- Bushong EA, Martone ME, Ellisman MH. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J Dev Neurosci 2004; 22:73 - 86; http://dx.doi.org/10.1016/j.ijdevneu.2003.12.008; PMID: 15036382
- Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci 2007; 27:6473 - 7; http://dx.doi.org/10.1523/JNEUROSCI.1419-07.2007; PMID: 17567808
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature 2010; 468:232 - 43; http://dx.doi.org/10.1038/nature09613; PMID: 21068832
- Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron 2011; 71:782 - 97; http://dx.doi.org/10.1016/j.neuron.2011.08.009; PMID: 21903073
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 2011; 14:724 - 38; http://dx.doi.org/10.1016/j.cmet.2011.08.016; PMID: 22152301
- Quaegebeur A, Lange C, Carmeliet P. The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. Neuron 2011; 71:406 - 24; http://dx.doi.org/10.1016/j.neuron.2011.07.013; PMID: 21835339
- Danbolt NC. Glutamate uptake. Prog Neurobiol 2001; 65:1 - 105; http://dx.doi.org/10.1016/S0301-0082(00)00067-8; PMID: 11369436
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience 2004; 129:1045 - 56; http://dx.doi.org/10.1016/j.neuroscience.2004.06.008; PMID: 15561419
- Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004; 129:877 - 96; http://dx.doi.org/10.1016/j.neuroscience.2004.09.053; PMID: 15561405
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 1999; 22:208 - 15; http://dx.doi.org/10.1016/S0166-2236(98)01349-6; PMID: 10322493
- Volterra A, Magistretti PJ, Haydon PG, eds. The Tripartite Synapse: glia in synaptic transmission (Oxford University Press, Oxford, U.K., 2002).
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 1990; 247:470 - 3; http://dx.doi.org/10.1126/science.1967852; PMID: 1967852
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci 2001; 4:803 - 12; http://dx.doi.org/10.1038/90507; PMID: 11477426
- Nett WJ, Oloff SH, McCarthy KD. Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J Neurophysiol 2002; 87:528 - 37; PMID: 11784768
- Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E. Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J Neurosci 2002; 22:9430 - 44; PMID: 12417668
- Hirase H, Qian L, Barthó P, Buzsáki G. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol 2004; 2:E96; http://dx.doi.org/10.1371/journal.pbio.0020096; PMID: 15094801
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods 2004; 1:31 - 7; http://dx.doi.org/10.1038/nmeth706; PMID: 15782150
- Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron 1992; 8:429 - 40; http://dx.doi.org/10.1016/0896-6273(92)90271-E; PMID: 1347996
- Murphy TH, Blatter LA, Wier WG, Baraban JM. Rapid communication between neurons and astrocytes in primary cortical cultures. J Neurosci 1993; 13:2672 - 9; PMID: 8501531
- Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci 1997; 17:7817 - 30; PMID: 9315902
- Porter JT, McCarthy KD. GFAP-positive hippocampal astrocytes in situ respond to glutamatergic neuroligands with increases in [Ca2+]i. Glia 1995; 13:101 - 12; http://dx.doi.org/10.1002/glia.440130204; PMID: 7544323
- Porter JT, McCarthy KD. Adenosine receptors modulate [Ca2+]i in hippocampal astrocytes in situ. J Neurochem 1995; 65:1515 - 23; http://dx.doi.org/10.1046/j.1471-4159.1995.65041515.x; PMID: 7561845
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci 1996; 16:5073 - 81; PMID: 8756437
- Latour I, Gee CE, Robitaille R, Lacaille JC. Differential mechanisms of Ca2+ responses in glial cells evoked by exogenous and endogenous glutamate in rat hippocampus. Hippocampus 2001; 11:132 - 45; http://dx.doi.org/10.1002/hipo.1031; PMID: 11345120
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, et al. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci 2007; 10:331 - 9; http://dx.doi.org/10.1038/nn1849; PMID: 17310248
- Santello M, Bezzi P, Volterra A. TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 2011; 69:988 - 1001; http://dx.doi.org/10.1016/j.neuron.2011.02.003; PMID: 21382557
- Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, et al. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci 2011; 14:1276 - 84; http://dx.doi.org/10.1038/nn.2929; PMID: 21909085
- Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, et al. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci 2006; 9:816 - 23; http://dx.doi.org/10.1038/nn1703; PMID: 16699507
- Winship IR, Plaa N, Murphy TH. Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. J Neurosci 2007; 27:6268 - 72; http://dx.doi.org/10.1523/JNEUROSCI.4801-06.2007; PMID: 17554000
- Sanzgiri RP, Araque A, Haydon PG. Prostaglandin E(2) stimulates glutamate receptor-dependent astrocyte neuromodulation in cultured hippocampal cells. J Neurobiol 1999; 41:221 - 9; http://dx.doi.org/10.1002/(SICI)1097-4695(19991105)41:2<221::AID-NEU5>3.0.CO;2-A; PMID: 10512979
- Jeremic A, Jeftinija K, Stevanovic J, Glavaski A, Jeftinija S. ATP stimulates calcium-dependent glutamate release from cultured astrocytes. J Neurochem 2001; 77:664 - 75; http://dx.doi.org/10.1046/j.1471-4159.2001.00272.x; PMID: 11299329
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 2001; 4:702 - 10; http://dx.doi.org/10.1038/89490; PMID: 11426226
- Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, et al. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci U S A 2005; 102:16466 - 71; http://dx.doi.org/10.1073/pnas.0506382102; PMID: 16254051
- Kang N, Xu J, Xu Q, Nedergaard M, Kang J. Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J Neurophysiol 2005; 94:4121 - 30; http://dx.doi.org/10.1152/jn.00448.2005; PMID: 16162834
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 2004; 24:6920 - 7; http://dx.doi.org/10.1523/JNEUROSCI.0473-04.2004; PMID: 15295027
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 2004; 43:729 - 43; http://dx.doi.org/10.1016/j.neuron.2004.08.011; PMID: 15339653
- Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci 2005; 25:2192 - 203; http://dx.doi.org/10.1523/JNEUROSCI.3965-04.2005; PMID: 15745945
- D’Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, et al. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci U S A 2007; 104:1995 - 2000; http://dx.doi.org/10.1073/pnas.0609408104; PMID: 17259307
- Navarrete M, Araque A. Endocannabinoids mediate neuron-astrocyte communication. Neuron 2008; 57:883 - 93; http://dx.doi.org/10.1016/j.neuron.2008.01.029; PMID: 18367089
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci 2008; 28:6659 - 63; http://dx.doi.org/10.1523/JNEUROSCI.1717-08.2008; PMID: 18579739
- Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature 2010; 463:232 - 6; http://dx.doi.org/10.1038/nature08673; PMID: 20075918
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, et al. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 2009; 61:213 - 9; http://dx.doi.org/10.1016/j.neuron.2008.11.024; PMID: 19186164
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, et al. Astrocytes control breathing through pH-dependent release of ATP. Science 2010; 329:571 - 5; http://dx.doi.org/10.1126/science.1190721; PMID: 20647426
- Huxtable AG, Zwicker JD, Alvares TS, Ruangkittisakul A, Fang X, Hahn LB, et al. Glia contribute to the purinergic modulation of inspiratory rhythm-generating networks. J Neurosci 2010; 30:3947 - 58; http://dx.doi.org/10.1523/JNEUROSCI.6027-09.2010; PMID: 20237265
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, et al. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 1998; 391:281 - 5; http://dx.doi.org/10.1038/34651; PMID: 9440691
- Domercq M, Brambilla L, Pilati E, Marchaland J, Volterra A, Bezzi P. P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J Biol Chem 2006; 281:30684 - 96; http://dx.doi.org/10.1074/jbc.M606429200; PMID: 16882655
- Bowser DN, Khakh BS. Two forms of single-vesicle astrocyte exocytosis imaged with total internal reflection fluorescence microscopy. Proc Natl Acad Sci U S A 2007; 104:4212 - 7; http://dx.doi.org/10.1073/pnas.0607625104; PMID: 17360502
- Calì C, Marchaland J, Regazzi R, Bezzi P. SDF 1-alpha (CXCL12) triggers glutamate exocytosis from astrocytes on a millisecond time scale: imaging analysis at the single-vesicle level with TIRF microscopy. J Neuroimmunol 2008; 198:82 - 91; http://dx.doi.org/10.1016/j.jneuroim.2008.04.015; PMID: 18538866
- Eddleston M, Mucke L. Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience 1993; 54:15 - 36; http://dx.doi.org/10.1016/0306-4522(93)90380-X; PMID: 8515840
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 1997; 20:570 - 7; http://dx.doi.org/10.1016/S0166-2236(97)01139-9; PMID: 9416670
- Dietrich PY, Walker PR, Saas P. Death receptors on reactive astrocytes: a key role in the fine tuning of brain inflammation?. Neurology 2003; 60:548 - 54; http://dx.doi.org/10.1212/01.WNL.0000042049.74547.7F; PMID: 12607528
- Rossi D, Brambilla L, Valori CF, Roncoroni C, Crugnola A, Yokota T, et al. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ 2008; 15:1691 - 700; http://dx.doi.org/10.1038/cdd.2008.99; PMID: 18617894
- Martorana F, Brambilla L, Valori CF, Bergamaschi C, Roncoroni C, Aronica E, et al. The BH4 domain of Bcl-X(L) rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals. Hum Mol Genet 2012; 21:826 - 40; http://dx.doi.org/10.1093/hmg/ddr513; PMID: 22072391
- Giacomello M, Drago I, Pizzo P, Pozzan T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ 2007; 14:1267 - 74; http://dx.doi.org/10.1038/sj.cdd.4402147; PMID: 17431419
- Joseph SK, Hajnóczky G. IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 2007; 12:951 - 68; http://dx.doi.org/10.1007/s10495-007-0719-7; PMID: 17294082
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 2003; 4:552 - 65; http://dx.doi.org/10.1038/nrm1150; PMID: 12838338
- Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008; 27:6407 - 18; http://dx.doi.org/10.1038/onc.2008.308; PMID: 18955969
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 2009; 187:761 - 72; http://dx.doi.org/10.1083/jcb.200908164; PMID: 19951898
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet 2006; 368:387 - 403; http://dx.doi.org/10.1016/S0140-6736(06)69113-7; PMID: 16876668
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response?. Nat Med 2006; 12:1005 - 15; PMID: 16960575
- McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis 2010; 19:355 - 61; PMID: 20061650
- Janelsins MC, Mastrangelo MA, Oddo S, LaFerla FM, Federoff HJ, Bowers WJ. Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer’s disease mice. J Neuroinflammation 2005; 2:23; http://dx.doi.org/10.1186/1742-2094-2-23; PMID: 16232318
- McGeer PL, McGeer EG. Local neuroinflammation and the progression of Alzheimer’s disease. J Neurovirol 2002; 8:529 - 38; http://dx.doi.org/10.1080/13550280290100969; PMID: 12476347
- Tobinick E. Tumour necrosis factor modulation for treatment of Alzheimer’s disease: rationale and current evidence. CNS Drugs 2009; 23:713 - 25; http://dx.doi.org/10.2165/11310810-000000000-00000; PMID: 19689163
- Van Eldik LJ, Thompson WL, Ralay Ranaivo H, Behanna HA, Martin Watterson D. Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int Rev Neurobiol 2007; 82:277 - 96; http://dx.doi.org/10.1016/S0074-7742(07)82015-0; PMID: 17678967
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 2007; 8:101 - 12; http://dx.doi.org/10.1038/nrm2101; PMID: 17245412
- Vassar R, Kovacs DM, Yan R, Wong PC. The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci 2009; 29:12787 - 94; http://dx.doi.org/10.1523/JNEUROSCI.3657-09.2009; PMID: 19828790
- Zhao J, Paganini L, Mucke L, Gordon M, Refolo L, Carman M, et al. Beta-secretase processing of the beta-amyloid precursor protein in transgenic mice is efficient in neurons but inefficient in astrocytes. J Biol Chem 1996; 271:31407 - 11; http://dx.doi.org/10.1074/jbc.271.49.31407; PMID: 8940150
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 2005; 25:11693 - 709; http://dx.doi.org/10.1523/JNEUROSCI.2766-05.2005; PMID: 16354928
- Bettegazzi B, Mihailovich M, Di Cesare A, Consonni A, Macco R, Pelizzoni I, et al. β-Secretase activity in rat astrocytes: translational block of BACE1 and modulation of BACE2 expression. Eur J Neurosci 2011; 33:236 - 43; http://dx.doi.org/10.1111/j.1460-9568.2010.07482.x; PMID: 21073551
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer’s disease. J Neurosci 1996; 16:5795 - 811; PMID: 8795633
- Hartlage-Rübsamen M, Zeitschel U, Apelt J, Gärtner U, Franke H, Stahl T, et al. Astrocytic expression of the Alzheimer’s disease beta-secretase (BACE1) is stimulus-dependent. Glia 2003; 41:169 - 79; http://dx.doi.org/10.1002/glia.10178; PMID: 12509807
- Leuba G, Wernli G, Vernay A, Kraftsik R, Mohajeri MH, Saini KD. Neuronal and nonneuronal quantitative BACE immunocytochemical expression in the entorhinohippocampal and frontal regions in Alzheimer’s disease. Dement Geriatr Cogn Disord 2005; 19:171 - 83; http://dx.doi.org/10.1159/000083496; PMID: 15677864
- Rossner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR. Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J Neurochem 2005; 92:226 - 34; http://dx.doi.org/10.1111/j.1471-4159.2004.02857.x; PMID: 15663471
- Grolla AA, Fakhfouri G, Balzaretti G, Marcello E, Gardoni F, Canonico PL, et al. Aβ leads to Ca(2+) signaling alterations and transcriptional changes in glial cells. Neurobiol Aging 2012; In Press http://dx.doi.org/10.1016/j.neurobiolaging.2012.05.005; PMID: 22673114
- Bourne KZ, Ferrari DC, Lange-Dohna C, Rossner S, Wood TG, Perez-Polo JR. Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J Neurosci Res 2007; 85:1194 - 204; http://dx.doi.org/10.1002/jnr.21252; PMID: 17385716
- Jin SM, Cho HJ, Kim YW, Hwang JY, Mook-Jung I. Aβ-induced Ca(2+) influx regulates astrocytic BACE1 expression via calcineurin/NFAT4 signals. Biochem Biophys Res Commun 2012; 425:649 - 55; http://dx.doi.org/10.1016/j.bbrc.2012.07.123; PMID: 22846573
- Zhao J, O’Connor T, Vassar R. The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation 2011; 8:150; http://dx.doi.org/10.1186/1742-2094-8-150; PMID: 22047170
- Cummings BJ, Pike CJ, Shankle R, Cotman CW. Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging 1996; 17:921 - 33; http://dx.doi.org/10.1016/S0197-4580(96)00170-4; PMID: 9363804
- Näslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, et al. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 2000; 283:1571 - 7; http://dx.doi.org/10.1001/jama.283.12.1571; PMID: 10735393
- Bussière T, Friend PD, Sadeghi N, Wicinski B, Lin GI, Bouras C, et al. Stereologic assessment of the total cortical volume occupied by amyloid deposits and its relationship with cognitive status in aging and Alzheimer’s disease. Neuroscience 2002; 112:75 - 91; http://dx.doi.org/10.1016/S0306-4522(02)00056-8; PMID: 12044473
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science 2002; 298:789 - 91; http://dx.doi.org/10.1126/science.1074069; PMID: 12399581
- Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging Cell 2007; 6:307 - 17; http://dx.doi.org/10.1111/j.1474-9726.2007.00295.x; PMID: 17465978
- Green KN, LaFerla FM. Linking calcium to Abeta and Alzheimer’s disease. Neuron 2008; 59:190 - 4; http://dx.doi.org/10.1016/j.neuron.2008.07.013; PMID: 18667147
- Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010; 47:183 - 9; http://dx.doi.org/10.1016/j.ceca.2009.12.014; PMID: 20080301
- Berridge MJ. Calcium hypothesis of Alzheimer’s disease. Pflugers Arch 2010; 459:441 - 9; http://dx.doi.org/10.1007/s00424-009-0736-1; PMID: 19795132
- Green KN, Smith IF, Laferla FM. Role of calcium in the pathogenesis of Alzheimer’s disease and transgenic models. Subcell Biochem 2007; 45:507 - 21; http://dx.doi.org/10.1007/978-1-4020-6191-2_19; PMID: 18193650
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009; 323:1211 - 5; http://dx.doi.org/10.1126/science.1169096; PMID: 19251629
- Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 2003; 23:5088 - 95; PMID: 12832532
- Abramov AY, Canevari L, Duchen MR. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 2004; 24:565 - 75; http://dx.doi.org/10.1523/JNEUROSCI.4042-03.2004; PMID: 14724257
- Rossi D, Brambilla L, Valori CF, Crugnola A, Giaccone G, Capobianco R, et al. Defective tumor necrosis factor-alpha-dependent control of astrocyte glutamate release in a transgenic mouse model of Alzheimer disease. J Biol Chem 2005; 280:42088 - 96; http://dx.doi.org/10.1074/jbc.M504124200; PMID: 16253995
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science 2002; 295:2282 - 5; http://dx.doi.org/10.1126/science.1067859; PMID: 11910117
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature 2006; 440:1054 - 9; http://dx.doi.org/10.1038/nature04671; PMID: 16547515
- Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 2008; 58:673 - 80; http://dx.doi.org/10.1016/j.neuron.2008.04.023; PMID: 18549780
- Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SB, Aebersold R, et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985; 40:735 - 46; http://dx.doi.org/10.1016/0092-8674(85)90333-2; PMID: 2859120
- McKinley MP, Taraboulos A, Kenaga L, Serban D, Stieber A, DeArmond SJ, et al. Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab Invest 1991; 65:622 - 30; PMID: 1684401
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992; 356:577 - 82; http://dx.doi.org/10.1038/356577a0; PMID: 1373228
- Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003; 302:871 - 4; http://dx.doi.org/10.1126/science.1090187; PMID: 14593181
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 1994; 8:121 - 7; http://dx.doi.org/10.1007/BF02780662; PMID: 7999308
- Kristensson K, Feuerstein B, Taraboulos A, Hyun WC, Prusiner SB, DeArmond SJ. Scrapie prions alter receptor-mediated calcium responses in cultured cells. Neurology 1993; 43:2335 - 41; http://dx.doi.org/10.1212/WNL.43.11.2335; PMID: 8232952
- Wong K, Qiu Y, Hyun W, Nixon R, VanCleff J, Sanchez-Salazar J, et al. Decreased receptor-mediated calcium response in prion-infected cells correlates with decreased membrane fluidity and IP3 release. Neurology 1996; 47:741 - 50; http://dx.doi.org/10.1212/WNL.47.3.741; PMID: 8797473
- Torres M, Castillo K, Armisén R, Stutzin A, Soto C, Hetz C. Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS One 2010; 5:e15658; http://dx.doi.org/10.1371/journal.pone.0015658; PMID: 21209925
- Lazzari C, Peggion C, Stella R, Massimino ML, Lim D, Bertoli A, et al. Cellular prion protein is implicated in the regulation of local Ca2+ movements in cerebellar granule neurons. J Neurochem 2011; 116:881 - 90; http://dx.doi.org/10.1111/j.1471-4159.2010.07015.x; PMID: 21214552
- Diedrich JF, Bendheim PE, Kim YS, Carp RI, Haase AT. Scrapie-associated prion protein accumulates in astrocytes during scrapie infection. Proc Natl Acad Sci U S A 1991; 88:375 - 9; http://dx.doi.org/10.1073/pnas.88.2.375; PMID: 1671170
- DeArmond SJ, Mobley WC, DeMott DL, Barry RA, Beckstead JH, Prusiner SB. Changes in the localization of brain prion proteins during scrapie infection. Neurology 1987; 37:1271 - 80; http://dx.doi.org/10.1212/WNL.37.8.1271; PMID: 3112607
- DeArmond SJ, Yang SL, Lee A, Bowler R, Taraboulos A, Groth D, et al. Three scrapie prion isolates exhibit different accumulation patterns of the prion protein scrapie isoform. Proc Natl Acad Sci U S A 1993; 90:6449 - 53; http://dx.doi.org/10.1073/pnas.90.14.6449; PMID: 8101989
- Giese A, Brown DR, Groschup MH, Feldmann C, Haist I, Kretzschmar HA. Role of microglia in neuronal cell death in prion disease. Brain Pathol 1998; 8:449 - 57; http://dx.doi.org/10.1111/j.1750-3639.1998.tb00167.x; PMID: 9669696
- Campbell IL, Eddleston M, Kemper P, Oldstone MB, Hobbs MV. Activation of cerebral cytokine gene expression and its correlation with onset of reactive astrocyte and acute-phase response gene expression in scrapie. J Virol 1994; 68:2383 - 7; PMID: 8139024
- Kim JI, Ju WK, Choi JH, Choi E, Carp RI, Wisniewski HM, et al. Expression of cytokine genes and increased nuclear factor-kappa B activity in the brains of scrapie-infected mice. Brain Res Mol Brain Res 1999; 73:17 - 27; http://dx.doi.org/10.1016/S0169-328X(99)00229-6; PMID: 10581394
- Tribouillard-Tanvier D, Striebel JF, Peterson KE, Chesebro B. Analysis of protein levels of 24 cytokines in scrapie agent-infected brain and glial cell cultures from mice differing in prion protein expression levels. J Virol 2009; 83:11244 - 53; http://dx.doi.org/10.1128/JVI.01413-09; PMID: 19710140
- Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009; 323:1205 - 8; http://dx.doi.org/10.1126/science.1166066; PMID: 19251627
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323:1208 - 11; http://dx.doi.org/10.1126/science.1165942; PMID: 19251628
- Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis 2009; 4:3; http://dx.doi.org/10.1186/1750-1172-4-3; PMID: 19192301
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362:59 - 62; http://dx.doi.org/10.1038/362059a0; PMID: 8446170
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994; 264:1772 - 5; http://dx.doi.org/10.1126/science.8209258; PMID: 8209258
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995; 14:1105 - 16; http://dx.doi.org/10.1016/0896-6273(95)90259-7; PMID: 7605627
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997; 18:327 - 38; http://dx.doi.org/10.1016/S0896-6273(00)80272-X; PMID: 9052802
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 2007; 10:615 - 22; http://dx.doi.org/10.1038/nn1876; PMID: 17435755
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 2002; 99:1604 - 9; http://dx.doi.org/10.1073/pnas.032539299; PMID: 11818550
- Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol 2008; 85:94 - 134; http://dx.doi.org/10.1016/j.pneurobio.2008.01.001; PMID: 18282652
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillée S, Rule M, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003; 302:113 - 7; http://dx.doi.org/10.1126/science.1086071; PMID: 14526083
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002; 26:459 - 70; http://dx.doi.org/10.1002/mus.10191; PMID: 12362410
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006; 312:1389 - 92; http://dx.doi.org/10.1126/science.1123511; PMID: 16741123
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 2008; 11:251 - 3; http://dx.doi.org/10.1038/nn2047; PMID: 18246065
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2006; 103:16021 - 6; http://dx.doi.org/10.1073/pnas.0607423103; PMID: 17043238
- Wang L, Gutmann DH, Roos RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet 2011; 20:286 - 93; http://dx.doi.org/10.1093/hmg/ddq463; PMID: 20962037
- Papadeas ST, Kraig SE, O’Banion C, Lepore AC, Maragakis NJ. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc Natl Acad Sci U S A 2011; 108:17803 - 8; http://dx.doi.org/10.1073/pnas.1103141108; PMID: 21969586
- Mendonça DM, Chimelli L, Martinez AM. Expression of ubiquitin and proteasome in motorneurons and astrocytes of spinal cords from patients with amyotrophic lateral sclerosis. Neurosci Lett 2006; 404:315 - 9; http://dx.doi.org/10.1016/j.neulet.2006.06.009; PMID: 16806703
- Pasinelli P, Houseweart MK, Brown RH Jr., Cleveland DW. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2000; 97:13901 - 6; http://dx.doi.org/10.1073/pnas.240305897; PMID: 11095709
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 2006; 9:408 - 19; http://dx.doi.org/10.1038/nn1653; PMID: 16474388
- Gunnarson E, Song Y, Kowalewski JM, Brismar H, Brines M, Cerami A, et al. Erythropoietin modulation of astrocyte water permeability as a component of neuroprotection. Proc Natl Acad Sci U S A 2009; 106:1602 - 7; http://dx.doi.org/10.1073/pnas.0812708106; PMID: 19164545
- Zur Nieden R, Deitmer JW. The role of metabotropic glutamate receptors for the generation of calcium oscillations in rat hippocampal astrocytes in situ. Cereb Cortex 2006; 16:676 - 87; http://dx.doi.org/10.1093/cercor/bhj013; PMID: 16079243
- Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de León A, et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci 2008; 28:4115 - 22; http://dx.doi.org/10.1523/JNEUROSCI.5308-07.2008; PMID: 18417691