Abstract
Despite recent progress in the understanding of prion diseases, little is known about the host-defense mechanisms against prion. Although it has long been thought that type I interferon (IFN-I) has no protective effect on prion infection, certain key molecules in innate immunity such as toll-like receptor (TLR) 4 seemed to be involved in the host response. For this reason we decided to focus on TLRs and investigate the role of a transcription factor, interferon regulatory factor 3 (IRF3), because the absence of MyD88, a major adaptor signaling molecule of TLRs, has no effect on the survival of prion infected mice. Intriguingly, survival periods of prion inoculated IRF3-knockout mice became significantly shorter than those of wild-type mice. In addition, IRF3 stimulation inhibited PrPSc replication in prion persistently-infected cells, and a de novo prion infection assay revealed that IRF3-overexpression could make host cells resistant to prion infection. Our work suggests that IRF3 may play a key role in innate immune responses against invasion of prion pathogens. Activated IRF3 could upregulate several anti-pathogen factors, including IFN-I, and induce sequential responses. Although the mechanism for the anti-prion effects mediated by IRF3 has yet to be clarified, certain interferon responsive genes might be involved in the anti-prion host-defense mechanism.
The Hallmarks of Prion Disease
Transmissible spongiform encephalopathies (TSEs) are fatal progressive neurodegenerative disorders which feature three major histopathological findings: spongiform change, neuronal loss and gliosis. Although TSEs were originally thought to be caused by slow-virus infections, no exogenous viral genome has been identified. The infectious agent, now called prion, is thought not to possess its own genome and to be composed uniquely of prion proteins, which are encoded by the host gene.Citation1 The infectious particles are composed mainly of proteinase K (PK)-resistant and β-sheet rich amyloid isoforms of prion protein (PrPSc) which are generated by conformational conversion of PrPC via unknown post-translational modifications. Effective therapeutics have yet to be established, although several compounds are known to inhibit the conversion process. Virus-like interference between distinct prion strains has been reported, but little is known thus far about the host-defense mechanisms against prion. It has long been thought that the host immune system does not recognize prion, because the sequence of PrPSc is identical to that of host PrP and also because the agent lacks its own genome, but several recent reports including ours suggest that the host defense system does indeed play at least a partially protective role against prion infection.
Interference between Distinct Prion Infections
Biological diversity among prion strains is known to exist, with different strains producing distinct symptoms, histopathological lesion profiles and incubation periods. These phenotypic traits are handed down through serial transmission,Citation2 and strain characteristics are maintained through serial passage in a variety of experimental animals and cell cultures. Interference is known to exist among prion strains. The pre-infection of mice with an attenuated strain (SY) featuring a long-incubation period significantly suppressed the effect of superinfection with a strong strain (FK) possessing a short incubation period,Citation3 in an in vitro pure cell culture system in the absence of immunocompetent cells.Citation4 One of the best studied mechanisms of viral interference is the anti-viral effect of type I interferon (IFN-I) which is induced following recognition of virus-derived nucleic acids or proteins by the host. It was not known, however, whether such IFN-responses were also evoked in host cells in the case of prion infection. As early as the 1970s, it was reported that the administration of IFNs and anti-interferon globulin had no therapeutic effect against goat-derived scrapie infection in animal models.Citation5-Citation7 In another early study, IFNs were not detected in the serum, spleens, or brains of mice infected with scrapie.Citation8 More recently, IFN-β mRNA was shown not to be increased in the brains of CJD patientsCitation9 or in mice infected with ME7 prion strain.Citation10 On the other hand, IFN-stimulated genes (ISGs), such as Mx and 2’5′-OAS, were increased by 263K infection in hamstersCitation11,Citation12 and by 139A, ME7 or Rocky Mountain Laboratory (RML) strains in mice.Citation12,Citation13 In the microglia of CJD-affected human brains, increases in interferon regulatory factor (IRF) family gene expression were also documented.Citation9 These observations would suggest that although the initial activation of the innate immune system is slight, provoking only subtle IFN production, this may in turn stimulate more abundant IFN production. Further elucidation of the role of the innate immune system is needed to uncover the mechanisms behind this phenomenon.
Pattern-Recognition Receptor (PRR)-Mediated Innate Immune Responses to Prion Infection
Generally, the invasion of pathogens is recognized initially by the innate immune system with the switching on of the cellular defense system in the lymphoid cells, leading to the production of cytokines and IFNs. The innate immune responses are initiated through both TLRs and intracellular sensor molecules such as retinoic acid inducible gene-I (RIG-I) and melanoma differentiation associated gene-5 (MDA5).Citation14,Citation15 These molecules are termed PRRs as they can recognize characteristic structures, collectively known as pathogen-associated molecular patterns (PAMPs), in various types of foreign pathogens, such as bacterial cell wall components and viral envelope glycoproteins.Citation16 The various intracellular signaling cascades that follow PRR stimulation eventually converge to synthesize type I IFN (-α and -β), pro-inflammatory cytokines such as TNF-α and anti-inflammatory cytokines such as IL-10Citation17, that are mediated by transcription factors of the IRF family (IRF3 and/or IRF7). The secreted IFNs stimulate cells in both an autocrine and paracrine manner to upregulate various IFN responsive genes. Finally, chemoattractants induced by IFN render host cells resistant to further infection at sites of foreign antigen infection and/or by proteins that directly interfere with viral replication.Citation15
The role of conventional PAMPs in prion infection is puzzling. It has been reported that pretreatment with innate immune activators, such as complete Freund’s adjuvant (CFA)Citation18 and unmethylated CpG DNA,Citation19 both of which are known to activate immune response-mediated TLR2 and -9, delayed the onset of TSE in mice inoculated with RML strain. On the other hand, LPS post-treatment, despite strongly activating innate immunity mediated TLR4 in lymphocytes, exacerbated the pathology in mice following prion inoculation,Citation20 and Poly[I:C] post-treatment, selectivity acting on TLR3, RIG-I and MDA5, showed similar effects on prion infection.Citation10 Poly[I:C] pre-treatment also had no effect on survival times following scrapie agent infection.Citation8 Collectively, prion pathogenesis was modified by the innate immune response of the host by the stimulators under certain experimental conditions, but the molecular mechanism underlying these complicated results remains to be elucidated.
Deletion of MyD88 gene, a major intracellular signal transducer in most TLRs, with the exception of TLR3, did not significantly affect the incubation time in the same mouse RML prion model.Citation21 On the other hand, mice expressing a refractory mutation of TLR4 showed accelerated disease onset when they were infected with 139A and ME7 strains.Citation22 In addition, mice deficient in CD40L, which is also located upstream of IRF3, readily succumbed to prion disease.Citation23 As the signals following TLR4 stimulation will be transduced via both MyD88 and TRIF, one can speculate that signal transduction mediated by TRIF-IRF3 might play a crucial role in the host defense system against prion infection.
Although the innate immune response to infectious agents in the central nervous system (CNS) has not been well studied, neurons were found to express most innate immunity-related genes and produce IFN-I in response to viral infection.Citation24 IRF3 is constitutively expressed in many CNS tissues and cells, including lymphocytes, glial cells, and neuroblastoma cells, as well as neurons.Citation25-Citation27 Furthermore, it was recently reported that TLR3 and IRF3 have a role in herpes simplex encephalitisCitation28 and rabies.Citation29 Accordingly, we focused on IRF3, which is a key transcription factor in the MyD88-independent (i.e., TRIF-dependent) pathway, and induces IFN-I. In our study, IRF3 knockout (IRF3−/−) mice died significantly earlier than wild-type (WT) mice following intra-peritoneal inoculation with 22L, Fukuoka-1 (FK-1), or a mouse-adapted BSE (mBSE) strain. The accumulation of PrPSc in the spleens was detected earlier in the IRF3−/− mice compared with WT mice.Citation30 Although the pathological changes, such as the degree of degeneration and also the accumulation of PK resistant PrP in the brains of terminally ill mice were not obviously different between WT and IRF3−/−, innate immune responses mediated via IRF3 seemed to inhibit, in part at least, the disease progress. Using prion infected cell cultures, we were able to demonstrate that stimulation of IRF3 inhibits the production of PrPSc, and expression levels of IRF3 bore an inverse relation to resistance to prion infection.Citation30 These results, therefore, indicate that IRF3 in the MyD88-independent pathway signaling cascades is a key molecule in the host defense mechanism against prion pathogenesis.
How Does IRF3 Suppress Prion Pathogenesis?
The fact that activated IRF3 upregulates mainly IFN-I in most cell types raises the possibility that ISGs such as Mx and OAS, which are located downstream of IFN signaling, have some kind of protective role against prion infection. Indeed, these ISGs have been reported to be upregulated in the brains of prion-infected animalsCitation11-Citation13 and CJD.Citation9 Although evidence of the increased secretion of IFN in prion-infected tissue or cells remains elusive, it is possible that the IFN produced at low levels by infected cells sets up a positive feedback loop that results in enhanced signals to infected and adjacent cells.Citation31 Recently, it was reported that this constitutive weak IFN signaling is crucial for the immune responsiveness that subsequently produces a strong IFN signal at the time of invasion of foreign pathogens,Citation32 and also has a cell-intrinsic role that prevents cells from transformations leading to cancer.Citation33 Consequently, even subtle IFN secretion provoked by basal activity of IRF3 might have a role in the host defense machinery against prion invasion or propagation in the brain. In addition, evidence that the disease onset is accelerated in IL-10- or TNF-α gene-deficient miceCitation34,Citation35 support our hypothesis that signals via PRRs may have a protective role against prion infection. Moreover, expression of TNF-α and IL-6 was induced in macrophages of WT mice following exposure to PrPSc-mimicking peptides, but not in mice with TLR4 dysfunction.Citation22 It is likely that host cells respond to prion invasion through TLR4 signal transduction which induces not only IFN-I but also NF-κB, resulting in the production of both pro-inflammatory and anti-inflammatory cytokines (). It also remains to be determined whether IRF3-mediated signaling directly suppresses the production of PrPSc or facilitates its degradation. Moreover, we are currently investigating what types of host molecules induced by IRF3 can help protect cells from prion. Given these results, we believe that it would be of great value to reassess the effect of exogenous IFN-I treatment using purified recombinant interferons (-α-2a, -α-2b and -β-1a) on prion infection.
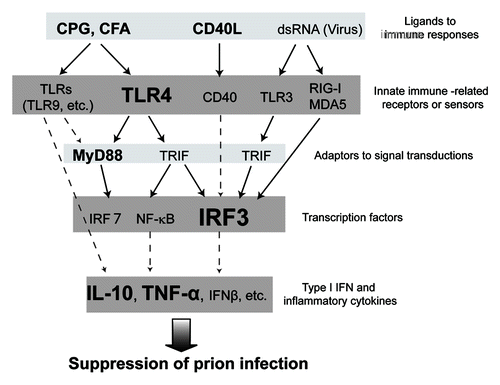
Figure 1. Schema of the host factors involved in innate immune responses against prion. The figure shows prion infection-related innate immune signal transductions from ligands to Type I IFN and inflammatory cytokines. Molecules relating closely to prion infection, as cited in previously published papers, are indicated in bold type. Well-defined pathways of signal transduction in innate immune responses are shown as solid lines, and probable pathways as dashed lines. We speculate that not only TLR4 but also TLR3 and RIG-I/MDA5 might be involved in prion infection. Additionally, it might be possible that type I IFN and inflammatory cytokines such as IL-10 might suppress prion infection, by an undetermined mechanism.
In Conclusion
We demonstrated that the transcription factor IRF3 has a protective role against prion infection. To further elucidate the host defense machinery against prion infection, the relationship between prion infection and IRF3 signaling should be studied, using, for example, conditional transgenic, neuron-specific IRF3-deficient, neuron-specific IRF3-expressing or IRF3-constitutively activated animals. It is our hope that IRF3 signaling-based prophylaxis and therapeutics against prion could one day dramatically help individuals suffering from this mysterious and deadly disease.
Acknowledgments
We thank Drs. Katsuya Satoh, Naohiro Yamaguchi, Takayuki Fuse, Hitoki Yamanaka, Takehiro Matsubara and Kazunori Sano for helpful discussions; graduate students Takehiro Nakagaki, Takujiro Homma, Hanae Takatsuki and Kaori Ono-Ubagai for assistance with experiments; and Mari Kudo, Ayumi Yamakawa and Atsuko Matsuo for technical assistance. This work was supported in part by the global COE Program (F12); a grant-in-aid for science research (C) (No. 24591482) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; a grant for BSE research, and a grant-in-aid of the Research Committee of Prion disease and Slow Virus Infection, from the Ministry of Health, Labor and Welfare of Japan.
Related Research Data
References
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 2007; 8:552 - 61; http://dx.doi.org/10.1038/nrm2204; PMID: 17585315
- Manuelidis L. Vaccination with an attenuated Creutzfeldt-Jakob disease strain prevents expression of a virulent agent. Proc Natl Acad Sci U S A 1998; 95:2520 - 5; http://dx.doi.org/10.1073/pnas.95.5.2520; PMID: 9482918
- Nishida N, Katamine S, Manuelidis L. Reciprocal interference between specific CJD and scrapie agents in neural cell cultures. Science 2005; 310:493 - 6; http://dx.doi.org/10.1126/science.1118155; PMID: 16239476
- Katz M, Koprowski H. Failure to demonstrate a relationship between scrapie and production of interferon in mice. Nature 1968; 219:639 - 40; http://dx.doi.org/10.1038/219639a0; PMID: 5691075
- Field EJ, Joyce G, Keith A. Failure of interferon to modify scrapie in the mouse. J Gen Virol 1969; 5:149 - 50; http://dx.doi.org/10.1099/0022-1317-5-1-149; PMID: 5387845
- Gresser I, Maury C, Chandler RL. Failure to modify scrapie in mice by administration of interferon or anti-interferon globulin. J Gen Virol 1983; 64:1387 - 9; http://dx.doi.org/10.1099/0022-1317-64-6-1387; PMID: 6189965
- Worthington M. Interferon system in mice infected with the scrapie agent. Infect Immun 1972; 6:643 - 5; PMID: 4628902
- Baker CA, Lu ZY, Manuelidis L. Early induction of interferon-responsive mRNAs in Creutzfeldt-Jakob disease. J Neurovirol 2004; 10:29 - 40; http://dx.doi.org/10.1080/13550280490261761; PMID: 14982726
- Field R, Campion S, Warren C, Murray C, Cunningham C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun 2010; 24:996 - 1007; http://dx.doi.org/10.1016/j.bbi.2010.04.004; PMID: 20399848
- Riemer C, Queck I, Simon D, Kurth R, Baier M. Identification of upregulated genes in scrapie-infected brain tissue. J Virol 2000; 74:10245 - 8; http://dx.doi.org/10.1128/JVI.74.21.10245-10248.2000; PMID: 11024157
- Stobart MJ, Parchaliuk D, Simon SL, Lemaistre J, Lazar J, Rubenstein R, et al. Differential expression of interferon responsive genes in rodent models of transmissible spongiform encephalopathy disease. Mol Neurodegener 2007; 2:5; http://dx.doi.org/10.1186/1750-1326-2-5; PMID: 17367538
- Xiang W, Windl O, Wünsch G, Dugas M, Kohlmann A, Dierkes N, et al. Identification of differentially expressed genes in scrapie-infected mouse brains by using global gene expression technology. J Virol 2004; 78:11051 - 60; http://dx.doi.org/10.1128/JVI.78.20.11051-11060.2004; PMID: 15452225
- Takeuchi O, Akira S. Genetic approaches to the study of Toll-like receptor function. Microbes Infect 2002; 4:887 - 95; http://dx.doi.org/10.1016/S1286-4579(02)01615-5; PMID: 12106781
- Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell Res 2006; 16:141 - 7; http://dx.doi.org/10.1038/sj.cr.7310019; PMID: 16474426
- Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 2006; 6:644 - 58; http://dx.doi.org/10.1038/nri1900; PMID: 16932750
- Samanta M, Iwakiri D, Takada K. Epstein-Barr virus-encoded small RNA induces IL-10 through RIG-I-mediated IRF-3 signaling. Oncogene 2008; 27:4150 - 60; http://dx.doi.org/10.1038/onc.2008.75; PMID: 18362887
- Tal Y, Souan L, Cohen IR, Meiner Z, Taraboulos A, Mor F. Complete Freund’s adjuvant immunization prolongs survival in experimental prion disease in mice. J Neurosci Res 2003; 71:286 - 90; http://dx.doi.org/10.1002/jnr.10474; PMID: 12503092
- Sethi S, Lipford G, Wagner H, Kretzschmar H. Postexposure prophylaxis against prion disease with a stimulator of innate immunity. Lancet 2002; 360:229 - 30; http://dx.doi.org/10.1016/S0140-6736(02)09513-2; PMID: 12133662
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 2005; 25:9275 - 84; http://dx.doi.org/10.1523/JNEUROSCI.2614-05.2005; PMID: 16207887
- Prinz M, Heikenwalder M, Schwarz P, Takeda K, Akira S, Aguzzi A. Prion pathogenesis in the absence of Toll-like receptor signalling. EMBO Rep 2003; 4:195 - 9; http://dx.doi.org/10.1038/sj.embor.embor731; PMID: 12612611
- Spinner DS, Cho IS, Park SY, Kim JI, Meeker HC, Ye X, et al. Accelerated prion disease pathogenesis in Toll-like receptor 4 signaling-mutant mice. J Virol 2008; 82:10701 - 8; http://dx.doi.org/10.1128/JVI.00522-08; PMID: 18715916
- Burwinkel M, Schwarz A, Riemer C, Schultz J, van Landeghem F, Baier M. Rapid disease development in scrapie-infected mice deficient for CD40 ligand. EMBO Rep 2004; 5:527 - 31; http://dx.doi.org/10.1038/sj.embor.7400125; PMID: 15071493
- Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, et al. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci U S A 2006; 103:7835 - 40; http://dx.doi.org/10.1073/pnas.0602460103; PMID: 16682623
- Au WC, Moore PA, Lowther W, Juang YT, Pitha PM. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc Natl Acad Sci U S A 1995; 92:11657 - 61; http://dx.doi.org/10.1073/pnas.92.25.11657; PMID: 8524823
- Karpova AY, Howley PM, Ronco LV. Dual utilization of an acceptor/donor splice site governs the alternative splicing of the IRF-3 gene. Genes Dev 2000; 14:2813 - 8; http://dx.doi.org/10.1101/gad.813800; PMID: 11090129
- Zhai J, Gao D, Liu W, Hong R, Qin Y, Ouyang H, et al. Characterization of a novel isoform of murine interferon regulatory factor 3. Biochem Biophys Res Commun 2008; 377:384 - 8; http://dx.doi.org/10.1016/j.bbrc.2008.09.147; PMID: 18851954
- Yokota S, Yokosawa N, Okabayashi T, Suzutani T, Miura S, Jimbow K, et al. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J Virol 2004; 78:6282 - 6; http://dx.doi.org/10.1128/JVI.78.12.6282-6286.2004; PMID: 15163721
- Ménager P, Roux P, Mégret F, Bourgeois JP, Le Sourd AM, Danckaert A, et al. Toll-like receptor 3 (TLR3) plays a major role in the formation of rabies virus Negri Bodies. PLoS Pathog 2009; 5:e1000315; http://dx.doi.org/10.1371/journal.ppat.1000315; PMID: 19247444
- Ishibashi D, Atarashi R, Fuse T, Nakagaki T, Yamaguchi N, Satoh K, et al. Protective role of interferon regulatory factor 3-mediated signaling against prion infection. J Virol 2012; 86:4947 - 55; http://dx.doi.org/10.1128/JVI.06326-11; PMID: 22379081
- Hata N, Sato M, Takaoka A, Asagiri M, Tanaka N, Taniguchi T. Constitutive IFN-alpha/beta signal for efficient IFN-alpha/beta gene induction by virus. Biochem Biophys Res Commun 2001; 285:518 - 25; http://dx.doi.org/10.1006/bbrc.2001.5159; PMID: 11444873
- Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol 2001; 2:378 - 86; http://dx.doi.org/10.1038/35073080; PMID: 11331912
- Chen HM, Tanaka N, Mitani Y, Oda E, Nozawa H, Chen JZ, et al. Critical role for constitutive type I interferon signaling in the prevention of cellular transformation. Cancer Sci 2009; 100:449 - 56; http://dx.doi.org/10.1111/j.1349-7006.2008.01051.x; PMID: 19076978
- Thackray AM, McKenzie AN, Klein MA, Lauder A, Bujdoso R. Accelerated prion disease in the absence of interleukin-10. J Virol 2004; 78:13697 - 707; http://dx.doi.org/10.1128/JVI.78.24.13697-13707.2004; PMID: 15564479
- Tamgüney G, Giles K, Glidden DV, Lessard P, Wille H, Tremblay P, et al. Genes contributing to prion pathogenesis. J Gen Virol 2008; 89:1777 - 88; http://dx.doi.org/10.1099/vir.0.2008/001255-0; PMID: 18559949