Abstract
The cellular prion protein (PrPC) is attached to the cell membrane via its glycosylphosphatidylinositol (GPI)-anchor and is constitutively shed into the extracellular space. Here, three different mechanisms are presented that concurrently shed PrPC from the cell. The fast α-cleavage released a N-terminal fragment (N1) into the medium and the extreme C-terminal cleavage shed soluble full-length (FL-S) PrP and C-terminally cleaved (C1-S) fragments outside the cell. Also, a slow exosomal release of full-length (FL) and C1-fragment (C1) was demonstrated. The three separate mechanisms acting simultaneously, but with different kinetics, have to be taken into consideration when elucidating functional roles of PrPC and also when processing of PrPC is considered as a target for intervention in prion diseases. Further, in this study it was shown that metalloprotease inhibitors affected the extreme C-terminal cleavage and shedding of PrPC. The metalloprotease inhibitors did not influence the α-cleavage or the exosomal release. Taken together, these results are important for understanding the different mechanisms acting in parallel in the shedding and cleavage of PrPC.
Introduction
The prion protein, PrPC, is expressed on the surface of many mammalian cells as a glycosylphosphatidylinositol (GPI) anchored glycoprotein, in particular on neural cells or cells of the lymphatic system.Citation1,Citation2 Following transport to the cell surface, PrPC is attached to the outer leaflet of the plasma membrane via its GPI moiety. Internalization via membrane microdomains is followed by constitutive cycling between the plasma membrane and a yet unidentified intracellular compartment.Citation3 The normal function of PrPC is still obscure but it is essential for the pathogenesis and transmission of prion diseases.Citation4 As part of its normal cellular processing, in the brain and in cultured cells, PrPC is N-terminally truncated producing a soluble N-terminal fragment (N1) and a GPI-anchored fragment denoted C1.Citation2,Citation5 The cleavage, referred to as the α-cleavage, takes place between the alternative residues K110/H111 or H111/M112 (human numbering).Citation6,Citation5 The α-cleavage occurs within a hydrophobic segment that is highly conserved which underlines the importance of this processing. The hydrophobic region is characterized as amyloidogenic and is thought to play a major role in the conformational change of PrPC to PrPSc.Citation7 The N- and the C-terminal fragments produced after the cleavage are critically involved in physiological and pathophysiological functions.Citation8-Citation11
Ectodomain shedding of PrPC into the cell culture medium has been reported and analysis has revealed that the C-terminal cleavage takes place between Gly228 and Arg229, three residues away from the GPI-anchor attachment site.Citation2,Citation5,Citation12 This cleavage results in the release of a GPI-anchorless full-length (FL-S) fragment and a C1-terminal fragment into the extracellular medium. In addition to this, exosomal release of PrPC and PrPSc has been suggested and it has been demonstrated that PrPSc-infected cell cultures discharge both PrPC and PrPSc into the extracellular medium in association with exosomes.Citation13-Citation15 However, the importance and functional role of cell-released PrP, both for the physiological function as PrPC, and in disease transmission via PrPSc is still not known.
Members of the ADAM (a disintegrin and metalloproteinase) family of proteases have been suggested to be involved both in the α-cleavage and in the shedding of PrPC. ADAM8, ADAM9, ADAM10 and ADAM17 have been shown to possess α-cleavage activities.Citation16-Citation18 Recently, studies have been done suggesting that ADAM10 instead is involved in the shedding of PrPC.Citation12,Citation19,Citation20
To investigate the α-cleavage and the shedding of PrPC, a specific deletion mutation in the α-cleavage site of PrPC was designed. Multiplex western immunoblot analysis was used to analyze the cleavage events taking place at the cell surface and the N1 fragment was used as a veridical measurement of the α-cleavage. Pulse-chase experiments were performed to analyze the influence of the deletion mutant on the shedding of PrPC. Also, the shedding of PrPC by an exosome-mediated mechanism and the role of ADAMs in the α-cleavage and ectodomain shedding of PrPC were investigated.
Results
Deletion of the α-cleavage site hindered shedding of the N1 fragment into the cell medium and the generation of C1 fragment in the cell lysate
In many cell culture systems, as well as in brain tissue, the full-length (FL) PrPC is subjected to a cleavage leaving a C-terminal fragment called C1 anchored in the cell membrane.Citation6,Citation21 The released N-terminal part called N1 is a measure of the N1/C1 cleavage, in the literature also named the α-cleavage. It has been reported that the α-cleavage is remarkably tolerant to sequence variations but that deletions in the site of the α-cleavage reduced the cleavage.Citation22,Citation23 In order to analyze the α-cleavage, the mutant PrPΔ121–123 was created by deleting the three amino acids encompassing the α-cleavage site, K121H122V123 (bovine numbering, corresponding to amino acid 110-112 in human PrP) (). After transient expression of wild type PrP (PrPwt) and PrPΔ121-123, the cell lysate and the cell medium were analyzed for the presence of N1 by using the N-terminal antibody R505. In the cell lysate after deglycosylation with PNGase F, the N-terminal antibody detected the FL fragment from both PrPwt and PrPΔ121-123 expressing cells (, lanes 2 and 3). Interestingly, although transfection conditions were identical, the amount of PrPC present in the lysate from the PrPΔ121-123 expressing cells was two to four times larger than in the PrPwt expressing cells. This might reflect differences in the protease sensitivity between PrPΔ121-123 and PrPwt.
Figure 1. Deletion of the α-cleavage site of PrPC hinders the α-cleavage. (A) Schematic representation of the α-cleavage site. The deleted amino acids are underlined. Numbers indicate amino acid positions in the bovine PrP. (B) Western immunoblot detection of PrP (N-terminal antibody R505) in cell lysate and in cell medium from cells expressing PrPwt (lanes 2 and 4, respectively), PrPΔ121–123 (lanes 3 and 5, respectively) and vector only (lane 1). The samples were treated with PNGase F to remove N-linked oligosaccharides. IRDye800 goat anti-rabbit (green) was used as secondary antibody. The positions of full length (FL), GPI-anchorless FL (FL-S) and N1 fragments are indicated. Approximate molecular masses are indicated on the left-hand side (kDa). (C) Inhibition of the α-cleavage was quantified by measuring the amount of accumulated N1 in the cell medium conditioned for 6 h from equivalent number of cells transiently expressing PrPwt (black column) and PrPΔ121-123 (gray column). The amount of N1-fragment from PrPwt expressing cells was set to 100%. Bars represent standard deviation based on two experiments. (D) Western immunoblot detection of PrP (C-terminal antibody L42) in cell lysate from cells expressing PrPwt (lane 1) and PrPΔ121-123 (lane 2). The samples were treated with PNGase F to remove N-linked oligosaccharides. Alexa Fluor 680 goat anti-mouse (red) was used as secondary antibody. The positions of full length (FL) and the C1 fragment are indicated. Approximate molecular masses are indicated on the left-hand side (kDa). (E) Inhibition of the α-cleavage of PrP as detected by mAb L42. The ratio of C1 to total PrP was determined for PrPwt (black column) and PrP∆121-123 (gray column). Bars represent the mean ± standard deviation based on six experiments.
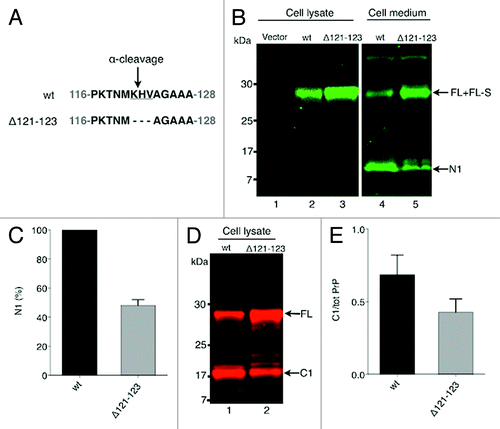
In medium conditioned for 6 h, in addition to a band migrating at the FL-PrP position, a band of around 11 kDa was seen (, lanes 4 and 5). This band corresponds to the accumulated N1 fragment released into the medium as result of the α-cleavage. The N1 fragment could only be detected in the medium and was not seen in the cell lysate (). When using the C-terminal antibodies L42 (, lanes 4–5) and 6H4 (not shown) the N1 fragment was not detected. The presence of the N1 fragment in the medium demonstrated the α-cleavage and as the N1 fragment was not detected in the cell lysate, it also suggests that the α-cleavage took place at the cell surface releasing the N1 fragment directly into the extracellular medium.
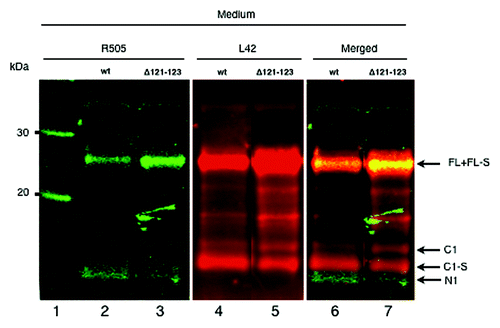
Figure 2. PrP populations with and without the GPI-anchor are shed into the cell medium from PrP expressing cells in addition to the N1 fragment. Multiplex western immunoblot detection of cell medium from PrPwt (lanes 2, 4 and 6) and PrPΔ121–123 (lanes 3, 5 and 7) expressing cells. The samples were treated with PNGase F to remove N-linked oligosaccharides. The PVDF membrane was probed with both the N-terminal polyclonal rabbit antibody R505 and the C-terminal monoclonal mouse antibody L42, followed by IRDye 800 goat anti-rabbit (green) and Alexa Fluor 680 goat anti-mouse (red). Both colors were imaged in a single scan. The N-terminal antibody (green, lanes 2 and 3) detected full-length PrP with intact GPI-anchor (FL), GPI-anchorless full-length (FL-S), and the N-terminal fragment (N1) and the C-terminal antibody (red, lanes 4 and 5) detected the FL, FL-S, the truncated C-terminal fragments with intact GPI-anchor (C1) and without GPI-anchor (C1-S). Yellow indicates merged overlapping colors (lanes 6 and 7). Approximate molecular masses are indicated on the left-hand side (kDa).
The PrPΔ121–123 had a marked effect on the α-cleavage, which was hindered in the deletion mutant as was seen when comparing the different amounts of released N1 fragment from PrPwt and PrPΔ121–123 expressing cells (, lanes 4 and 5). The amount of accumulated N1 fragment in the cell medium released from the PrPΔ121–123 expressing cells was reduced by roughly 50% compared with the PrPwt expressing cells, indicating a partial hindrance of the α-cleavage (). Furthermore, when the cell lysate from cell cultures expressing PrPwt and PrPΔ121–123 were compared using a C-terminal antibody, a decreased generation of the C1 fragment was found in PrPΔ121-123 expressing cells (). The ratio of C1 to total PrP in the cell lysate was for PrPwt and PrPΔ121-123, 0.72 and 0.37, respectively () demonstrating the effect of the deletion on the α-cleavage. In summary, deletion of three aa in the α-cleavage site hindered the α-cleavage, which points to a partial sequence specificity for the amino acids KHV in the α-cleavage process.
In the cell medium, distinct PrP populations could be found, one population with an intact GPI-anchor and one population without GPI-anchor
It has earlier been shown that PrP fragments are shed into the cell culture medium by a proteolytic cleavage in the extreme C-terminal end,Citation5 situated three amino acids from the GPI-anchor attachment site.Citation2,Citation12 The cleavage results in shedding of FL-S and C1-S (soluble fragments lacking the GPI anchor) to the medium. When a monoclonal antibody directed to the C-terminal part of PrP was used, an FL-sized band and two separate C1-sized fragments were seen in the cell medium (, lanes 4 and 5). The slower migrating C1 band migrated to the same position as the cell anchored C1 fragment and the other, more abundant band, migrated faster. This fragment did not react with the N-terminal antibody and migrated slower on the gel than N1 (, lanes 6 and 7). The slightly different running behavior on the gel and the small shift between the two C1 fragments demonstrates a truncation of PrPC, which corresponded to the C1 fragment without its GPI-anchor (C1-S). Similarly, the FL band seen in the cell medium corresponds both to FL and FL-S (without GPI-anchor; see , lanes 4 to 6). The ratio between the C1 fragments and the FL band in the cell medium were in PrPwt and PrPΔ121-123, 0.95 and 0.65, respectively. This demonstrates a 30% decrease in the C1 production from PrPΔ121–123 expressing cells compared with the PrPwt expressing cells. Taken together, these results suggested that both GPI-anchored PrP and PrP without GPI-anchor are released outside the cell.
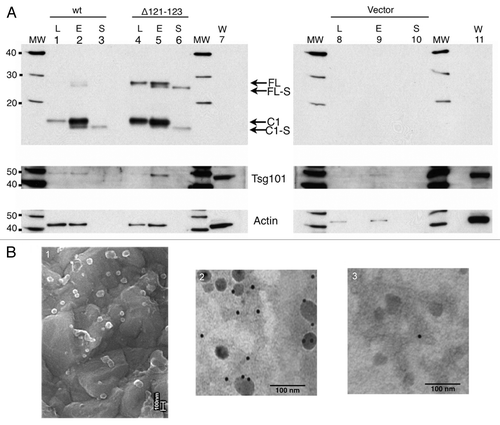
Figure 3. PrP with GPI-anchor is shed in association with exosomes. (A) Western immunoblot detection with the C-terminal antibody L42. Cell lysates (L; lanes 1, 4 and 8), 100,000 x g pelleted exosomal fractions (E; lanes 2, 5 and 9), and 100,000 x g supernatant fractions (S; lanes 3, 6 and 10) from cells transiently expressing PrPwt (lanes 1–3), PrPΔ121-123 (lanes 4–6), and vector control (lanes 8–10). Untransfected control cell lysates (W; lanes 7 and 11) are indicated. The same PVDF membrane was stripped and reprobed with antisera against Tsg101 and actin, respectively (below). The representative western blot displayed is one of three experiments. All samples were treated with PNGase F. Molecular mass markers are indicated (MW) and given in kDa. (B) The 100,000 x g pelleted exosomal fraction was analyzed by scanning electron microscopy (SEM) (panel 1) and immunogold transmission electron microscopy (TEM) (panels 2 and 3). PrP was detected by mAb L42 (panel 2). Unspecific antibody (antiBLV-gp51 (panel 3). Secondary antibody was gold-conjugated anti-mouse mAb (10 nm gold particles). Bars indicate 100 nm.
The shed PrP population with intact GPI-anchor was released in association with exosomes
When medium was conditioned for more than 4 h and analyzed by western immunoblotting the C1 fragment was detected in the medium, in addition to the faster migrating C1-S (, lanes 4 and 5). GPI-anchored proteins are membrane bound, thus, the shed GPI-anchor containing C1 was likely to be bound to the lipid bilayer of a membrane. Association of PrP with exosomes has been describedCitation14,Citation15,Citation24 and the methodology based on sequential centrifugation was used to analyze the presence of exosome bound PrPwt and PrPΔ121-123 recovered from the cell medium. Cell culture medium conditioned for 6.5 h was subjected to sequential centrifugation to isolate exosomes. The 100,000 x g supernatant and pellet were analyzed by western blot after deglycosylation to clarify the nature of the shed PrP populations. In the pelleted, exosomal fraction (E), the GPI-anchored FL and C1-populations were detected (, lanes 2 and 5), and in the supernatant (S) the soluble FL-S and C1-S were identified (, lanes 3 and 6). In fact, this separation demonstrated that the cell medium contained both PrP molecules with GPI-anchor (FL and C1) and without GPI anchor (FL-S and C1-S). Calculations based on the amount loaded on gels and the intensity of the PrP bands shed in association with pelleted exosomes compared with the proteolytically shed PrP demonstrate that the steady-state of exosome-mediated shedding is around 200 times lower than the protease-mediated shedding and seemed not to be affected by the deletion in the α-cleavage site. The exosome-associated C1 is probably anchored in the exosomes via its GPI moiety as it co-migrated with the GPI-anchored PrP present in cell lysate (L) (, compare lanes 2 and 5 with lanes 1 and 4) and since treatment of the pelleted exosomes by PIPLC released this fragment from the pelleted fraction (data not shown). Additionally, an increased population of FL PrP was again found in the separated fractions from the PrPΔ121–123 expressing cells compared with PrPwt expressing cells. The cell lysate, PrP-containing exosomal pellet, and PrP-containing supernatants were analyzed for the existence of Tsg101, a protein known to be enriched in exosomes and involved in the biogenesis of multivesicular bodies (MVBs).Citation14,Citation25 As compared with cell lysates, the PrP-containing pelleted fraction was enriched in Tsg101 (, Tsg101 panel). Actin, which is confined to cellular structures, was not found in the 100,000 x g supernatant (, actin panel). Interestingly, although the amount of PrPwt lysate loaded was at least twice that of PrPΔ121–123 lysate as judged by the actin signal, the amount of PrP detected was two to four times larger in the latter. The trace amount of C1-S that is seen in the exosomal pellet ( lane E) is most probably residual C1-S that unspecifically remained in the exosomal pellet due to the large excess of C1-S in the starting medium. However, it cannot be ruled out the possibility that there was a low amount of C1-S inside the exosomes. When the 100,000 x g pellet was analyzed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), membrane vesicles with size of around 50–100 nm with the morphology of exosomes were recovered (, panel 1). Anti-PrP immunogold-labeling resulted in labeled exosomes (, panel 2) and a non-specific antibody resulted in minimal labeling (panel 3). These findings demonstrate that PrPC in its GPI-anchored form is released into the medium in association with exosomes.
Kinetic analysis of the cleavage and shedding of PrPC in PrPwt- and PrPΔ121-123-expressing cells
In the cell medium from PrPΔ121-123 expressing cells the amount of shed FL-S fragment was larger than that from PrPwt expressing cells as measured by western immunoblotting (, compare lanes 4 and 5). This was probably the result of an increase in uncleaved PrPC in PrPΔ121-123 expressing cells due to the reduced α-cleavage at the cell membrane. A pulse chase experiment of transiently expressed PrPwt and PrPΔ121-123 cells was done in order to in real time analyze the shedding of PrP into the medium and to elucidate if deletion of the three aa encompassing the α-cleavage site also would interfere with the cleavage in the extreme C-terminal end and/or the exosomal release of PrPC. Transfected cells were pulse-labeled with 35S-methionine and the radioactivity was chased for various times. The kinetics of the α-cleavage was measured in the cell lysate as the generation of C1-fragment. Cell lysates and cell media were immunoprecipitated with mAb 6H4 and the immunoprecipitates were analyzed by SDS-PAGE followed by autoradiography (). The radioactivity of the full length and C1 sized fragments were quantified by a phosphoimager (). In the PrPwt expressing cells, C1 could be seen already at 20 min of chase and at 60 min almost half of the label was seen in C1. A delay in the generation of C1 was observed in the PrP∆121-123 cell lysate compared with PrPwt (). The ratios of C1 to total PrPC, were in the PrPwt and PrPΔ121-123 expressing cells, 0.15 and 0.08 at 20 min, 0.30 and 0.14 at 40 min and 0.45 and 0.24 at 60 min of chase, respectively (). These differences corresponded to around 60% decrease in the rate of α-cleavage in the PrPΔ121-123 expressing cells. In the cell medium radioactively labeled PrP was seen at around 40 min of chase and at 240 min of chase around 15% of total cell labeling had been shed into the medium. The time course experiments showed that there was no time delay between the shedding of total PrPwt and PrPΔ121-123 into the cell medium (), which further indicated that the extreme C-terminal cleavage was not affected by the α-cleavage site deletion. The ratio of C1-S to total shed PrPC was at 240 min of chase 0.87 and 0.72 (), respectively, in the medium from PrPwt and PrPΔ121–123 expressing cells. Since these fragments lack the GPI-anchor (, lanes 3 and 6) and since the pulse-chase experiments demonstrated shedding from the cell around 20 min after the α-cleavage, this shedding is probably due to the proteolytic cleavage at the extreme C-terminal end by a sheddase activity not related to the α-cleavage protease. Furthermore, the exosomal shedding of C1 with intact GPI-anchor was much slower and released C1 could only be demonstrated as a minor fraction at late time points (). Based on these results, the deletion of the three aa in the α-cleavage site did not appear to affect the shedding of PrPC by the extreme C-terminal cleavage or the release of exosomally associated PrPC.
Figure 4. Kinetic analysis of the shedding and cleavages of PrPC. Cell cultures expressing PrPwt or PrP∆121-123 were pulse-labeled with [35S]-methionine/cysteine for 15 min followed by chase for indicated time. Cell lysates or cell media were subjected to immunoprecipitation with mAb 6H4, treated with PNGase F, analyzed by SDS-PAGE, autoradiography and the radioactivity was quantitated by phosphoimaging. The positions of GPI anchored FL and C1, and GPI anchorless FL-S and C1-S are indicated. Apparent molecular masses based on migration of protein standards are indicated (kDa). (A) Cell lysate. In the cell lysate α-cleavage was seen after around 20 min of chase as an appearance of the C1-fragment. The strongest total labeling was reached after 120 min of chase. (B) The ratios of C1 to total PrP in the cell lysate at 20, 40 and 60 min of chase are presented in a bar graph for PrPwt (gray) and PrP∆121-123 (light gray). (C) Cell medium. Labeled PrP was shed and accumulated in the cell medium due to the extreme c-terminal cleavage as FL-S and C1-S after around 40 min of chase. (D) Fraction of cleaved PrP in C1-S and C1 to total PrP in the cell medium for PrPwt and PrP∆121-123, respectively.
![Figure 4. Kinetic analysis of the shedding and cleavages of PrPC. Cell cultures expressing PrPwt or PrP∆121-123 were pulse-labeled with [35S]-methionine/cysteine for 15 min followed by chase for indicated time. Cell lysates or cell media were subjected to immunoprecipitation with mAb 6H4, treated with PNGase F, analyzed by SDS-PAGE, autoradiography and the radioactivity was quantitated by phosphoimaging. The positions of GPI anchored FL and C1, and GPI anchorless FL-S and C1-S are indicated. Apparent molecular masses based on migration of protein standards are indicated (kDa). (A) Cell lysate. In the cell lysate α-cleavage was seen after around 20 min of chase as an appearance of the C1-fragment. The strongest total labeling was reached after 120 min of chase. (B) The ratios of C1 to total PrP in the cell lysate at 20, 40 and 60 min of chase are presented in a bar graph for PrPwt (gray) and PrP∆121-123 (light gray). (C) Cell medium. Labeled PrP was shed and accumulated in the cell medium due to the extreme c-terminal cleavage as FL-S and C1-S after around 40 min of chase. (D) Fraction of cleaved PrP in C1-S and C1 to total PrP in the cell medium for PrPwt and PrP∆121-123, respectively.](/cms/asset/01120331-f393-43d7-9f68-d058e06841f9/kprn_a_10922588_f0004.gif)
O-phenanthroline and TAPI-1 inhibit the extreme C-terminal protease-mediated shedding but not the α-cleavage or the exosomal release of PrPC
Vincent and coworkers previously reported that including the general cell-permeable zinc-metalloprotease inhibitor O-phenanthroline, or the ADAM inhibitor TAPI-1, in the cell medium drastically reduced the shedding of N1 in transfected cells and suggested that ADAM10 was one of the proteases responsible for the α-cleavage.Citation18 However, recent studies have instead suggested that ADAM10 is the C-terminal sheddase of PrPC.Citation12,Citation19,Citation20 In order to determine the effect of the inhibitors on the α-cleavage in this system, the processing of FL to C1 was measured in the cell lysates at different concentration of the inhibitors. Quantification by measuring the intensity of bands representing FL and C1 in western blot showed a very slight variation of 2–4% in the generation of C1 in both O-phenanthroline and TAPI-1 treated cells (, lanes 1–3). The effect of the inhibitors on the extreme C-terminal cleavage was measured as the amount of FL-S and C1-S shed to the cell medium. The amount of C1-S in the medium decreased dramatically and the decrease was consistent with increasing inhibitor concentrations and probably due to inhibition of the extreme C-terminal cleavage ( and ). In addition, in the conditioned medium, a band migrating to the position of C1 could be seen, preferentially in samples from cells treated with O-phenanthroline and TAPI-1 (, lanes 4–6). Thus, O-phenanthroline and TAPI-1 did not appear to inhibit the exosomal release of PrPC and the results here indicate that the amount of C1 associated with exosomes was increased in cell medium from cells treated with these metalloprotease inhibitors ( and ). This increase in the amount of exosome associated C1 could be due to a change in the route by PrPC or an effect of more PrP being present in the membrane and thus more accessible for inclusion in exosomes. These data indicate that the metalloprotease inhibitors O-phenanthroline and TAPI-1, have a very slight impact on the enzyme(s) responsible for α-cleavage in the transient expression system used here. Instead, the active protease(s) responsible for the extreme C-terminal cleavage causing shedding of FL-S and C1-S into the conditioned medium was inhibited. The amount of the N1 fragment in the medium decreased under O-phenanthroline treatment (, ) although the α-cleavage was fairly constant at 0, 60 and 120 μM O-phenanthroline giving a ratio of C/C1+FL of 0.43, 0.41 and 0.41, respectively. In the cell medium from cells treated with TAPI-1 the amount of accumulated N1 increased compared with untreated samples. Measuring the intensity of C1/C1+FL in the cell lysate showed that the α-cleavage was not affected. Therefore, the increase of the N1 fragment in the cell medium is likely an effect of strong inhibition of the extreme C-terminal cleavage at 50 μM TAPI-1 leaving more substrate (FL) PrP available for the α-cleavage.
Figure 5. Metalloprotease inhibitors interfere with the extreme C-terminal cleavage but did not hinder α-cleavage or exosomal release of PrPC. Multiplex western immunoblot detection of shed PrP. Cell lysates and cell culture media (as indicated above the lanes) conditioned for 6 h from wt PrPC expressing cells incubated in (A) 0 μM, 60 μM and 120 μM O-phenanthroline or in (B) 0 μM, 25 μM and 50 μM TAPI-1. All samples were treated with PNGase F. The PVDF membranes were probed with both the N-terminal polyclonal rabbit antibody R505 and the C-terminal monoclonal mouse antibody L42, followed by IRDye800 goat anti-rabbit (green) and Alexa Fluor 680 goat anti-mouse (red). Both colors were imaged in a single scan. The N-terminal antibody (green) detected the full-length (FL), FL-S (without GPI-anchor) and the N-terminal fragment (N1). The C-terminal antibody (red) detected the FL, the FL-S and the truncated C-terminal fragments C1 and C1-S (with and without GPI-anchor, respectively). Yellow indicates merged overlapping colors. Apparent molecular masses based on migration of protein standards are indicated (kDa).
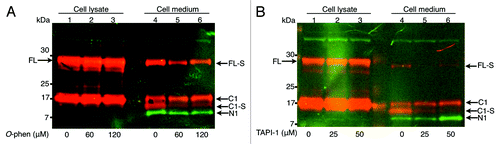
Table 1. Relative effect of O-phenanthroline on the accumulation of various PrP-fragments in the cell medium
Table 2. Relative effect of TAPI-1 on the accumulation of various PrP-fragments in the cell medium
PrPC is shed from the cell by three separate mechanisms
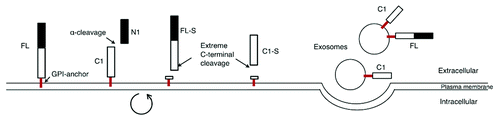
In the expression system used here, PrP was released into the medium simultaneously by three mechanisms. The first mechanism released the N1 fragment by a fast α-cleavage within the neurotoxic region of PrP. The second mechanism released the GPI-anchorless fragments FL-S and C1-S by a proteolytic cleavage in the extreme C-terminal end, which was sensitive to inhibition by metalloprotease inhibitors. The third mechanism released FL and C1 in association with exosomes. This process was much slower and most likely took place after internalization of PrPC into the cell and subsequent recycling of vesicles back to the surface for release ().Citation5
Table 3. Three separate mechanisms act concurrently to shed and release PrPC from the PrP-expressing cell culture system used in this study
Discussion
In many cell types and tissues, PrPC is processed by a specific proteolytic cleavage that generate given N- and C-terminal fragments designated N1 and C1. This cleavage, termed the α-cleavage, takes place prior to or very soon after the arrival of PrPC to the cell membrane. This leaves the N-terminally truncated PrPC attached to the cell surface via its GPI anchor and the N1 fragment is shed into the cell media or extracellular space. The ratio between the different proteolytically generated fragments varies in different tissues and cell culture systems, pointing to differences in the activity of the proteases involvedCitation5,Citation26
The membrane bound PrPC can be internalized into the cell and recycled back to the cell surface.Citation27 During this cycle, in the endocytic compartment and/or at the cell membrane, a second proteolytic cleavage in the extreme C-terminal end release soluble PrP fragments from the cell. The physiological relevance of these two endoproteolytic cleavages and proteases involved in the processes has nevertheless still not been determined. Recently, it was suggested that the cleaved fragments possess neuroprotective functions and/or interfere with Aβ-associated toxicity.Citation10,Citation28 It has also been shown that the cleaved fragments can act as an inhibitor of PrPSc formation.Citation8 These newly suggested roles for the proteolytic fragments of PrP prompt further characterization.
Of special interest is the PrPC fragments released from the cell and the focus of this study was laid on this aspect of PrP biosynthesis. Analyzing the PrPC fragments in the cell medium from PrP expressing cells will reflect different events at the cell membrane. The cell system chosen here has earlier been used to analyze different steps in the intracellular transport of PrPC and mirror well the in vivo situation and is thus a valuable model for studying the cellular processing of PrPC.Citation5 In several reports it has been demonstrated that the N1 and C1 fragments are present in human and animal brain,Citation5,Citation6,Citation29-Citation31 and found in exosomes in cerebrospinal fluid.Citation15 Thus, the diverse intracellular and extracellular PrP processing events observed in vivo appear to be faithfully recapitulated in the transfected BHK cells.
The results presented here suggest that PrPC is shed from the cell by three separate mechanisms (). The first mechanism releases an N-terminal fragment (N1) via the α-cleavage; a second by proteolytic cleavage in the extreme C-terminal end generating GPI-anchorless FL-S (or recently named N3)Citation20 and C1-S fragments; and a slower third process releasing a GPI-anchored PrPC in an exosomal fraction (). Deletion of three amino acids in the α-cleavage site reduced the α-cleavage as seen by the reduction of the N1 fragment in the cell medium. The hindrance of the cleavage was not absolute but a decrease of about 50% was seen as measured by the steady-state condition of the N1 fragment accumulation in the cell medium. The N1 fragment was not seen in the cell lysate, probably because the α-cleavage took place very soon after PrPC arrived at the cell surface () or due to degradation.
Figure 6. Schematic representation of the three different mechanisms acting concurrently to shed and release the prion protein. The first mechanism releases the N1 fragment via the α-cleavage and the second mechanism releases the FL-S and the C1-S (soluble fragments lacking the GPI-anchor) by proteolytic cleavage in the extreme C-terminal. A circular arrow represents internalization of membrane bound PrPC and recycling back to the cell surface, during which the extreme C-terminal cleavage is thought to occur. The third mechanism is a slow process that releases a GPI-anchored fraction of PrPC in association with exosomes.
In time course experiments the deletion mutation hindered the rate of α-cleavage around 63% compared with wt, indicating that the protease(s) involved in the α-cleavage possess some degree of sequence specificity. The α-cleavage has been reported to be independent of the precise sequence, change of charge and hydrophobicity near the α-cleavage site.Citation22 However, the degree of deletion in the site affects the cleavage. The cellular site at which the α-cleavage occurs has been discussed and both endosomal/lysosomal compartments, late compartments of the secretory pathway and at the cell surface have been suggested.Citation5,Citation32-Citation34 Previously, results showed that α-cleavage occurs prior to or very soon after the arrival of PrP to the cell membrane since addition of PIPLC to the medium directly released cleaved C-terminal fragment, and not FL, to the medium.Citation5 Taken together, previous results and those presented here, suggest that the protease(s) responsible for the PrP α-cleavage possess some sequence specificity and is located in a compartment close to or at the cell surface.
Time course experiments demonstrated that the α-cleavage and the extreme C-terminal cleavage were separated in time, where the extreme C-terminal cleavage took place 20–30 min after the α-cleavage. Earlier time course experimentsCitation5 with PrPwt but with other pulse times and chase periods further support the data presented here. The delay between the α-cleavage and the extreme C-terminal cleavage could be explained by the recycling of newly synthesized PrP from the cell membrane to an endocytic compartment and back before it was constitutively shed to the cell medium.Citation5,Citation27,Citation35,Citation36 These results indicate that the sheddase(s), that are responsible for the extreme C-terminal cleavage, may only be active in an endocytic compartment where PrP resides during recycling; or the extreme C-terminal cleavage site is not accessible in the “nascent” PrP traveling from the trans-golgi network to the cell surface during biosynthesis. No difference was seen between the PrPwt and PrPΔ121–123 expressing cells in the shedding rate of the soluble fragments C1-S and FL-S. Thus, the extreme C-terminal cleavage and the resulting release of soluble PrP fragments appeared to be independent of the mutation in the α-cleavage site.
In the cell culture medium conditioned for more than 6 h, a fraction migrating as GPI-anchored PrPC was found. Differential centrifugation demonstrated that this fraction was released from the cell in association with exosomes.Citation14,Citation15 Both full length PrPC and C1 fragment were present in exosomes and accounted for around 1% of the total PrP recovered in the extracellular medium. Treatment of the pelleted fraction with PIPLC released the exosome-associated FL and C1 from the pellet, indicating presence of a GPI anchor. The delay in processing of exosomally associated PrP compared with proteolytically shed PrP suggests different pathways (). As the total amount of PrP released in association with exosomes was similar in PrPwt and PrP∆121-123 expressing cell cultures, it would seem that the deletion of the α-cleavage site did not affect the exosomal release. These results again support the notion of distinct pathways of release.
It could be of great value to define what enzymatic mechanisms process the different modes of release of PrP from the cell. Several enzymes in the family of A-disintegrin-and-metalloproteinase (ADAM) have been implicated in the shedding of PrPC and/or the α-cleavage in both cell culture systems and in vivo. Suggestions have been made that these proteases could be seen as therapeutic targets. Increasing their activity could deplete the cell of full length PrP needed for conversion to PrPSc with concomitant cleavage of the proposed toxic domain 106-126 (human numbering).Citation17,Citation18,Citation37,Citation38 In the cell culture system used here the metalloprotease inhibitors O-phenanthroline and TAPI-1, were used to modulate the protease activity acting on PrPC. These inhibitors did not affect the α-cleavage as measured by the generation of the C1 fragment in the cell and thus did not inhibit the proteases involved in the α-cleavage. Likewise, other studies indicated that members of the ADAM family are not involved in the α-cleavage.Citation19,Citation20 Differently, by using the N1 fragment released into the medium as an assay for the α-cleavage, the disintegrin ADAM10 and TACE were reported to play an active role in the generation and release of N1-fragment to the cell medium.Citation18,Citation38 As was demonstrated in this report, the amount of N1 released to the medium is affected differently by O- phenanthroline and TAPI-1, respectively. In the higher concentration of TAPI-1 used, the accumulation of N1 in the medium increased compared with untreated. This is probably caused by increased availability of substrate (FL) at the cell membrane, as a result of the strong inhibition of the extreme C-terminal cleavage by the higher TAPI-1 concentration. Also, TAPI-1 seems to be a more specific inhibitor of the extreme C-terminal cleavage then O-phenanthroline. However, the shedding of the soluble C1-S and FL-S were decreased with increasing inhibitor concentration and thus hindering the extreme C-terminal cleavage. This was in line with a recent report where ADAM10 knock out mice were studied, where an intracellular accumulation and loss of shedding of PrP was observed.Citation19,Citation20 In a gain-of-function analysis it was also shown that ADAM 9 and ADAM10 are involved in the shedding of PrP,Citation12 findings which are supported by the in vitro results in the present study. The conflicting results on the proteolytic cleavage and the proteases involvedCitation12,Citation17-Citation20 might be due to not fully considering in which pathway the shed PrP fragment(s) is generated, due to cell specific differences, or that several different proteases possess α-cleavage activity. It can be of importance to observe these three different pathways in further studies with inhibitors. In addition, in this report the increase in N1 fragment recovered in the medium when cells were treated with high concentrations of TAPI-1, appeared to be explained by an increase in FL PrP in the cell, rather than by a direct effect on the α-cleavage. However, the relative influences of the different cleavages and release processes are probably very difficult to unravel. This observation is important to consider when studying the biological consequences of cleavages in PrP.
The exosome-mediated release of PrP was not hindered by O-phenanthroline or TAPI-1 but PrP was in fact increased in the exosomal fraction as a result of the treatment. This increase was probably due to the effect of these inhibitors on the extreme C-terminal cleavage, resulting in more membrane bound PrP being accessible for the exosomal pathway. Fevrier et al. showed that purified exosomes from scrapie-infected cells could infect both cell cultures and bioassay mice.Citation14 Because exosomes are enriched in glycolipids, raft lipids, and raft-associated proteins, such as GPI-anchored proteins,Citation14,Citation39 PrP is almost certainly enriched in exosomes. Since only very few molecules of PrPSc are needed to transmit prion infection,Citation40 even a minute pathway of release such as the exosome-mediated could still be important for cell-to-cell spread of prion infection. In relation to this, the proteolytic activity at the extreme C-terminal cleavage might be a target for intervention as already has been suggested for the α-cleavage. However, in one study inhibition or overexpression of ADAM 10 did not affect the production of PrPSc in scrapie-infected N2a cells, which might indicate that the extreme C-terminal cleavage is not important for Scrapie formation.Citation12
Materials and Methods
Reagents and inhibitors
Peptide: N-glycosidase F (PNGase F) and prestained protein marker (broad range 7–175 kDa) were from New England BioLabs. Proteinase K (PK) and protein molecular mass markers (MagicMark and SeeBlue Plus2) were obtained from Invitrogen. Phosphatidylinositol-specific phospholipase C (PIPLC), sodium deoxycholate, Triton X-100, sodium salicylate and phenylmethylsulfonyl fluoride (PMSF) were from Sigma-Aldrich. Dithiothreitol (DTT) came from USB Corporation. Nonidet P40 (NP40) was from Fluka AG. Complete protease inhibitor cocktail was from Roche. The metalloproteinase inhibitors O-phenanthroline (1,10-phenanthroline) and TAPI-1 were from Sigma.
Antibodies
The bovine PrP (six octarepeats) numbering scheme is applied throughout except where specifically stated otherwise. mAb 6H4 (Prionics AG, Schlieren, Switzerland) recognizes amino acid (aa) residues 155-163 of PrP.Citation41 mAb L42 (R-Biopharm AG) recognizes aa residues 156–161.Citation42,Citation43 mAb R505 (kind gift from Dr J. Langeveld, Central Veterinary Institute of Wageningen UR, Lelystad, The Netherlands) is raised against sheep PrP 100-111 (SQWNKPSKPKTN) corresponding to boPrP 108-119 (GQWNKPSKPKTN). IRDye800CW Goat anti-Rabbit (Li-Cor) and Alexa Fluor 680 donkey anti-mouse (Invitrogen) were used as fluorescent secondary antibodies. Goat polyclonal anti-Actin antiserum (I-19; Santa Cruz Biotechnology) is raised against a peptide mapping at the C-terminus of actin of human origin. Goat polyclonal anti-Tsg101 antiserum (M-19; Santa Cruz Biotechnology) is raised against a peptide mapping at the C-terminus of Tsg101 of human origin.
Cell lines, transfections and drug treatments
Baby hamster kidney-21 (BHK) cells from ATCC (ATCC number CCL-10) were grown in Glasgow medium supplemented with 5% fetal calf serum, 10% tryptose phosphate broth, 2 mM glutamine and 20 mM Hepes. Transfections were performed with a Semliki forest virus (SFV) replicon as described previously.Citation5 Transfected BHK-cells were washed with PBS and serum-free medium was added. For protease inhibitor treatments, O-phenanthroline or TAPI-1 dissolved in DMSO were added to the serum-free medium. After 7 h the serum-free cell culture media were collected, supplemented with EDTA-free protease inhibitor Cocktail, acetone-precipitated and western immunoblotted with mAb L42. Cell viability and morphology were not affected by the inhibitor concentrations chosen as judged by light microscopy.
Deletion of the α-cleavage site
Double primer pairs were designed and two different PCR reactions resulted in two fragments that were ligated to form a fragment lacking the nine nucleotides corresponding to aa 121-123 in the bovine prion protein. The primers for the first and second fragment, respectively, were as follows: 5′-CGA TCC CGG GTC ATG GTG AAA AGC CAC ATA-3′, 5′-CAT GTT GGT TTT TGG CTT ACT GGG TTT GTT CCA TTG-3′ and 5′-GCA GGA GCT GCT GCA GCT GG-3′, 5′-AAC ACC CGG GTA ATG AAA ACA GGA AGG TTG-3′ (DNA Technology A/S). The ligated fragment were cleaved with XmaI and inserted into the pGEM(XmaI) vector to generate the plasmid pGEM(XmaI)-PrP∆121-123. The deletion was confirmed by sequencing (310 Genetic Analyzer, ABI Prism) and chromatograms were analyzed with DNAStar SeqMan II version 5.52 (DNAStar Inc.). The construct was then transferred into the unique XmaI site of pSFV1 to produce the plasmid pSFV1-PrP∆121-123. The inserted fragment was confirmed by sequencing.
Metabolic labeling, immunoprecipitation and electrophoresis
Metabolic labeling and immunoprecipitation were performed essentially as described previously.Citation5 Briefly, transfected BHK cells were starved with methionine- and cysteine-free EMEM for 30 min and pulse-labeled with [35S]-methionine/cysteine (Redivue PRO-MIX; Amersham Biosciences) in methionine- and cysteine-free EMEM for 15 min. Cells were washed and chased as described earlier. Media were collected, supplemented with EDTA-free protease inhibitor Cocktail and cells were lysed with lysis buffer (0.5% Triton X-100, 0.5% Sodium deoxycholate, 5 mM TRIS-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA and 10 µg/ml PMSF). Media and lysates were then subjected to immunoprecipitation with mAb 6H4 and/or loaded onto SDS-PAGE gels. Samples were dissolved in NuPAGE LDS Sample Buffer (Invitrogen) supplemented with 0.1 M DTT and heated for 10 min at 100°C. After boiling and reduction, samples were centrifuged at 9,000 x g for 5 min at 25°C in a microcentrifuge and loaded onto 12% NuPAGE Bis-Tris Gels (Invitrogen) and electrophoresed in 2-(N-morpholino) propane sulfonic acid (MOPS) buffer (Invitrogen). Following electrophoresis the gel was soaked in a scintillator (1 M sodium salicylate) for 30 min and dried. Autoradiography was visualized on X-ray film (Hyperfilm MP; Amersham Biosciences).
Deglycosylation by PNGase F treatment
Proteins were denatured and incubated in the absence or presence of PNGase F according to the manufacturer’s protocol (New England BioLabs). After PNGase F-treatment as indicated in the specific experiments, proteins were precipitated with acetone or with trichloroacetic acid (TCA) using the PlusOne SDS-PAGE Clean-Up Kit (Amersham Biosciences, Uppsala, Sweden) according to the manufacturer’s protocol. The resulting protein pellet was prepared for electrophoresis as described above.
Western immunoblot analysis—enhanced chemiluminescence (ECL) detection
After electrophoresis, proteins were electrotransferred onto PVDF membranes (Hybond-P; Amersham Biosciences) which were then blocked for non-specific binding with 5% (wt/vol) non-fat milk in PBS with 0.05% Tween 20 (PBST). The filters were incubated with primary antibody for 1 h at room temperature. After washing with PBST, the membranes were incubated for 1 h at room temperature with peroxidase-conjugated rabbit anti-mouse, peroxidase-conjugated rabbit anti-goat immunoglobulin or peroxidase-conjugated swine anti-rabbit immunoglobulin (Dako A/S) depending on the origin species of the primary antibody. The membranes were washed again in PBST and bands were visualized on X-ray film (Hyperfilm MP; Amersham Biosciences) by enhanced chemiluminescence (ECL; Amersham Biosciences). All antibodies were diluted in 1% (wt/vol) non-fat milk in PBST.
Western immunoblot analysis—two-color western blot detection with near-infrared fluorescence
After electrophoresis, proteins were electrotransferred onto PVDF membranes (Millipore Immobilon-FL PVDF Membranes, Millipore), which were then blocked for non-specific binding with Odyssey Blocking Buffer (Li-Cor Biosciences). The filters were incubated with primary antibody diluted 1:10,000 for 1 h at room temperature. After washing with PBST, the membranes were incubated with IRDye 800CW Goat anti-Rabbit (Li-Cor Biosciences) or Alexa Fluor 680 donkey anti-mouse (Invitrogen) diluted 1:10,000 for 1 h at room temperature. The membranes were washed in PBST and PBST with 0.02% SDS and then rinsed in PBS. Bands were visualized with near-infrared fluorescent detection using the Odyssey CLx System (Li-Cor Biosciences). For IR detection of two-colors, blots were incubated with both primary antibodies (from different types of host animal), followed by IR-labeled secondary antibodies. Blots were imaged with the IR image in both 700 and 800 nm channels in a single scan.
Differential centrifugation and exosome isolation
Differential centrifugation and exosome isolation were performed essentially as described.Citation14 Cell culture media from transfected BHK cells conditioned for different times were recovered. Media were then sequentially centrifuged for 5 min at 3,000 x g, 30 min at 10,000 x g and finally for 1 h at 100,000 x g onto a 80% (w/v) sucrose cushion. The interface was collected, diluted with PBS and exosomes were recovered at 100,000 x g for 1 h. Pelleted exosomes were resuspended in PBS and prepared for further analyses as described elsewhere.
PIPLC-treatment of exosomal pellets
Conditioned cell culture media were centrifuged at 10,000 x g for 30 min followed by centrifugation at 100,000 x g for 2 h at 20°C through a 20% (w/v) sucrose cushion. Pellets were gently resuspended in 20 mM Tris pH 7.4, 150 mM NaCl supplemented with complete protease inhibitor cocktail. Samples were incubated in the presence or absence of PIPLC at 3.4 units/ml for 20 h at 4°C. Incubation mixtures were then centrifuged at 100,000 x g for 2 h at 20°C. Supernatant and pellet fractions were recovered and subjected to deglycosylation before being prepared for SDS-PAGE and immunoblotting as described above.
Electron microscopic analysis
For EM immunolabeling, grids [3.05 MM Copper grid, Formvar/carbon (TAAB)] were floated at room temperature on drops (20 μl, placed on parafilm) of the following solutions successively: (a) freshly prepared exosome preparation diluted 1:10 in PBS (3 min); (b) anti-PrP mAb L42 or unspecific mAb diluted 1:5,000 in PBS (20 min); (c) 3 washes in PBS, (5 min each); (d) anti-mouse IgG-Gold (Sigma G7652–4ML), diluted 1: 100 in PBS (20 min); (e) 3 washes in PBS, (5 min each);(f) 1% PTA pH 7,4 (3 min). Residual liquid was removed by gentle suction with filter paper. Samples were analyzed by scanning electron microscopy (SEM) at 20 kV in a Zeiss Supra 35 (Carl Zeiss SMT) and transmission electron microscopy (TEM) at 75kV in a Hitachi-710 (Hitachi).
Quantification of PrP fragments
Quantification of 35S-labeled PrP was done with a Molecular Dynamics PhosphoImager in conjunction with the ImageQuant software (v. 4.02).
Western immunoblots were scanned and the relative amounts of the bands of interest were obtained by computerized integration of peaks representing the bands using ImageJ64 software version 1.45s (by Rasband W.S., October 2011 1.45; US. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/). Scanning was done on exposures within the linear range of the photographic film.
| Abbreviations: | ||
| ADAM | = | A disintegrin and metalloproteinase |
| C1 | = | C-terminal fragment of PrP with GPI-anchor |
| C1-S | = | C-terminal fragment of PrP without GPI-anchor |
| FL | = | full-length PrP with GPI-anchor |
| FL-S | = | full-length PrP without GPI-anchor |
| GPI | = | glycosylphosphatidylinositol |
| kDa | = | kilodalton |
| mAb | = | monoclonal antibody |
| MVB | = | multivesicular bodies |
| N1 | = | N-terminal fragment |
| PNGase F, peptide | = | N-glycosidase F |
| SEM | = | Scanning electron microscopy |
| TEM | = | transmission electron microscopy |
| PrP | = | prion protein |
| PrPC | = | cellular prion protein |
| PrPSc | = | prion protein scrapie form |
Acknowledgments
We thank the EM facilities at EBC and at BMC, Uppsala for help with the electron microscopy, Dr Jan Langeveld, CVI, Lelystad, The Netherlands and Dr Malek Merza, SVANOVA, Uppsala for providing the R505 antibody and antiBLVgp51 mAb, respectively. This work was supported by grants from the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (Formas), The Royal Swedish Academy of Agriculture and Forestry (KSLA), Swedish Fund for Research Without Animal Experiments and Helge Ax:son Johnsons Foundation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987; 51:229 - 40; http://dx.doi.org/10.1016/0092-8674(87)90150-4; PMID: 2444340
- Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry 1990; 29:8879 - 84; http://dx.doi.org/10.1021/bi00490a001; PMID: 1980209
- Harris DA. Cell biological studies of the prion protein. Curr Issues Mol Biol 1999; 1:65 - 75; PMID: 11475702
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339 - 47; http://dx.doi.org/10.1016/0092-8674(93)90360-3; PMID: 8100741
- Zhao H, Klingeborn M, Simonsson M, Linné T. Proteolytic cleavage and shedding of the bovine prion protein in two cell culture systems. Virus Res 2006; 115:43 - 55; http://dx.doi.org/10.1016/j.virusres.2005.07.004; PMID: 16140411
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem 1995; 270:19173 - 80; http://dx.doi.org/10.1074/jbc.270.32.19173; PMID: 7642585
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell 1998; 93:337 - 48; http://dx.doi.org/10.1016/S0092-8674(00)81163-0; PMID: 9590169
- Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J Biol Chem 2011; 286:44234 - 42; http://dx.doi.org/10.1074/jbc.M111.286195; PMID: 22025612
- Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 2010; 13:310 - 8; http://dx.doi.org/10.1038/nn.2483; PMID: 20098419
- Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 2009; 284:35973 - 86; http://dx.doi.org/10.1074/jbc.M109.051086; PMID: 19850936
- Guillot-Sestier MV, Sunyach C, Ferreira ST, Marzolo MP, Bauer C, Thevenet A, et al. The alpha-secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric Abeta-associated cell death. J Biol Chem 2011; PMID: 22184125
- Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, et al. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem 2009; 284:22590 - 600; http://dx.doi.org/10.1074/jbc.M109.032599; PMID: 19564338
- Alais S, Simoes S, Baas D, Lehmann S, Raposo G, Darlix JL, et al. Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol Cell 2008; 100:603 - 15; http://dx.doi.org/10.1042/BC20080025; PMID: 18422484
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A 2004; 101:9683 - 8; http://dx.doi.org/10.1073/pnas.0308413101; PMID: 15210972
- Vella LJ, Sharples RA, Lawson VA, Masters CL, Cappai R, Hill AF. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J Pathol 2007; 211:582 - 90; http://dx.doi.org/10.1002/path.2145; PMID: 17334982
- Liang J, Wang W, Sorensen D, Medina S, Ilchenko S, Kiselar J, et al. Cellular prion protein regulates its own α-cleavage through ADAM8 in skeletal muscle. J Biol Chem 2012; 287:16510 - 20; http://dx.doi.org/10.1074/jbc.M112.360891; PMID: 22447932
- Cissé MA, Sunyach C, Lefranc-Jullien S, Postina R, Vincent B, Checler F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J Biol Chem 2005; 280:40624 - 31; http://dx.doi.org/10.1074/jbc.M506069200; PMID: 16236709
- Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, et al. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem 2001; 276:37743 - 6; PMID: 11477090
- Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, et al. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener 2011; 6:36; http://dx.doi.org/10.1186/1750-1326-6-36; PMID: 21619641
- Endres K, Mitteregger G, Kojro E, Kretzschmar H, Fahrenholz F. Influence of ADAM10 on prion protein processing and scrapie infectiosity in vivo. Neurobiol Dis 2009; 36:233 - 41; http://dx.doi.org/10.1016/j.nbd.2009.07.015; PMID: 19632330
- Harris DA, Huber MT, van Dijken P, Shyng SL, Chait BT, Wang R. Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry 1993; 32:1009 - 16; http://dx.doi.org/10.1021/bi00055a003; PMID: 8093841
- Oliveira-Martins JB, Yusa S, Calella AM, Bridel C, Baumann F, Dametto P, et al. Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS One 2010; 5:e9107; http://dx.doi.org/10.1371/journal.pone.0009107; PMID: 20161712
- Wegner C, Römer A, Schmalzbauer R, Lorenz H, Windl O, Kretzschmar HA. Mutant prion protein acquires resistance to protease in mouse neuroblastoma cells. J Gen Virol 2002; 83:1237 - 45; PMID: 11961279
- Leblanc P, Alais S, Porto-Carreiro I, Lehmann S, Grassi J, Raposo G, et al. Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO J 2006; 25:2674 - 85; http://dx.doi.org/10.1038/sj.emboj.7601162; PMID: 16724107
- Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 2006; Chapter 3:Unit 3 22.
- Laffont-Proust I, Faucheux BA, Hässig R, Sazdovitch V, Simon S, Grassi J, et al. The N-terminal cleavage of cellular prion protein in the human brain. FEBS Lett 2005; 579:6333 - 7; http://dx.doi.org/10.1016/j.febslet.2005.10.013; PMID: 16263114
- Shyng SL, Huber MT, Harris DA. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J Biol Chem 1993; 268:15922 - 8; PMID: 8101844
- Turnbaugh JA, Westergard L, Unterberger U, Biasini E, Harris DA. The N-terminal, polybasic region is critical for prion protein neuroprotective activity. PLoS One 2011; 6:e25675; http://dx.doi.org/10.1371/journal.pone.0025675; PMID: 21980526
- Jiménez-Huete A, Lievens PM, Vidal R, Piccardo P, Ghetti B, Tagliavini F, et al. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues. Am J Pathol 1998; 153:1561 - 72; http://dx.doi.org/10.1016/S0002-9440(10)65744-6; PMID: 9811348
- Yadavalli R, Guttmann RP, Seward T, Centers AP, Williamson RA, Telling GC. Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J Biol Chem 2004; 279:21948 - 56; http://dx.doi.org/10.1074/jbc.M400793200; PMID: 15026410
- Liu T, Zwingman T, Li R, Pan T, Wong BS, Petersen RB, et al. Differential expression of cellular prion protein in mouse brain as detected with multiple anti-PrP monoclonal antibodies. Brain Res 2001; 896:118 - 29; http://dx.doi.org/10.1016/S0006-8993(01)02050-9; PMID: 11277980
- Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 1995; 129:121 - 32; http://dx.doi.org/10.1083/jcb.129.1.121; PMID: 7698979
- Tveit H, Lund C, Olsen CM, Ersdal C, Prydz K, Harbitz I, et al. Proteolytic processing of the ovine prion protein in cell cultures. Biochem Biophys Res Commun 2005; 337:232 - 40; http://dx.doi.org/10.1016/j.bbrc.2005.09.031; PMID: 16182247
- Walmsley AR, Watt NT, Taylor DR, Perera WS, Hooper NM. alpha-cleavage of the prion protein occurs in a late compartment of the secretory pathway and is independent of lipid rafts. Mol Cell Neurosci 2009; 40:242 - 8; http://dx.doi.org/10.1016/j.mcn.2008.10.012; PMID: 19056496
- Borchelt DR, Rogers M, Stahl N, Telling G, Prusiner SB. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology 1993; 3:319 - 29; http://dx.doi.org/10.1093/glycob/3.4.319; PMID: 7691278
- Parkin ET, Watt NT, Turner AJ, Hooper NM. Dual mechanisms for shedding of the cellular prion protein. J Biol Chem 2004; 279:11170 - 8; http://dx.doi.org/10.1074/jbc.M312105200; PMID: 14711812
- Checler F, Vincent B. Alzheimer’s and prion diseases: distinct pathologies, common proteolytic denominators. Trends Neurosci 2002; 25:616 - 20; http://dx.doi.org/10.1016/S0166-2236(02)02263-4; PMID: 12446128
- Vincent B, Paitel E, Frobert Y, Lehmann S, Grassi J, Checler F. Phorbol ester-regulated cleavage of normal prion protein in HEK293 human cells and murine neurons. J Biol Chem 2000; 275:35612 - 6; http://dx.doi.org/10.1074/jbc.M004628200; PMID: 10952979
- Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol 2002; 2:569 - 79; PMID: 12154376
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, et al. The most infectious prion protein particles. Nature 2005; 437:257 - 61; http://dx.doi.org/10.1038/nature03989; PMID: 16148934
- Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R, et al. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 1997; 390:74 - 7; http://dx.doi.org/10.1038/36337; PMID: 9363892
- Langeveld JP, Jacobs JG, Erkens JH, Bossers A, van Zijderveld FG, van Keulen LJ. Rapid and discriminatory diagnosis of scrapie and BSE in retro-pharyngeal lymph nodes of sheep. BMC Vet Res 2006; 2:19; http://dx.doi.org/10.1186/1746-6148-2-19; PMID: 16764717
- Vorberg I, Buschmann A, Harmeyer S, Saalmüller A, Pfaff E, Groschup MH. A novel epitope for the specific detection of exogenous prion proteins in transgenic mice and transfected murine cell lines. Virology 1999; 255:26 - 31; http://dx.doi.org/10.1006/viro.1998.9561; PMID: 10049818