Abstract
Like patients with prion disease, Alzheimer patients suffer from a fatal, progressive form of dementia. There is growing evidence that amyloid-β (Aβ) aggregates may be transmissible similar to prions, at least under extreme experimental conditions. However, unlike mice infected with prion protein (PrP) prions, those inoculated with Aβ do not die. The transmission of Aβ and PrP thus differs conspicuously in the neurological effects they induce in their hosts, the difference being no less than a matter of life and death. Far from being a mere academic nuance, this distinction between Aβ and PrP begs the crucial questions of what, exactly, controls prion toxicity and how prion toxicity relates to prion infectivity.
Prions in neurological disease
Stanley Prusiner introduced prions in 1982 as the self-replicating forms of the prion protein that accumulate in certain transmissible diseases of the central nervous system, such as scrapie and Creutzfeldt-Jakob disease.Citation1 Although prions represent novel infectious agents lacking pathogen-encoded nucleic acids, their discovery relied upon a century-old paradigm, formalized into postulates by Robert Koch, for identifying pathological microbial agents. A key concept in Koch’s postulates is that the microbe responsible for a given disease must cause that same disease when inoculated into a susceptible host. In prion disease, the afflicted individual suffers from a progressive deterioration in neurological function that culminates, inevitably, in death. By systematically sifting through brain extracts from scrapie-infected hamsters, Prusiner found that the deadliest inoculates contained fibrillar aggregates of a proteolytic fragment of the prion protein, PrP27-30. We now know that this fragment is derived from the scrapie isoform of the prion protein, PrPSc, an aggregated, alternatively folded conformer of the cellular prion protein, PrPC.Citation2
In 2000 Lary Walker first demonstrated that intra-cerebral inoculations of brain extracts from amyloid plaque-containing brain tissue from Alzheimer patients accelerate amyloid plaque deposition and β-amyloidosis in transgenic mice expressing human Aβ proteins.Citation3 The acceleration of β-amyloidosis by inoculates containing Aβ fibrils, which form amyloid plaques, has been replicated in at least four other laboratories using inoculates from humans, several lines of plaque-forming transgenic mice and, most recently, fibrillar synthetic Aβ aggregates and synthetic Aβ dimers.Citation4-Citation7
While these results indicate that fibrillar conformers of Aβ proteins can self-replicate in susceptible hosts, it is still unclear whether such replication can be maintained over multiple serial passages from one animal to another. The latter is an integral part of the definition of a “prion.” For the sake of the following discussion, we will refer to PrPSc as the aggregated form of PrPC found in transmissible spongiform encephalopathies (TSEs), and to “prions” as the infectious agent of TSEs as measured with microbiological methods. In this frame of reference, prions are composed of PrPSc, but not all PrPSc is necessarily infectious.Citation8
Infectious agents, prions and prionoids
In 1966 Carlton Gajdusek astonished the scientific world with the claim that the fatal degenerative disease kuru was transmitted through ritualistic cannibalism among the Fore peoples of New Guinea;Citation9 the proposal that the elusive infectious agent in kuru was a prion was no less surprising. Now, however, the radical properties ascribed to prions threaten to undermine the original meaning of “infectious agent.” In the following discussion, an infectious agent transmits a disease causing deficits in the host that are the same as those in the donor and share the same pathophysiology. Simply put, infectious agents are the biological basis of ill health that can be passed between living beings.
Prions fulfill the above definition since they were initially discovered as true infectious agents using microbiological methods. However, many other proteins can aggregate into geometrically arranged structures that can seed—in vitro and in vivo—compartments containing the parent protein in a monomeric soluble state.
Simply equating the capability of seeding with the term “prion” is an oversimplification. Any inorganic crystal can seed a supersaturated solution of its cognate salt, whereas bona fide prions have caused epidemics in sheep, cows, mink, felines and humans (kuru, as well as iatrogenic and “variant” Creutzfeldt-Jakob disease). Because of their flagrant infectious traits—communicability and contagiousness—the agents of these diseases were not recognized as prions for many decades, and many preeminent scientists deemed them to be “slow viruses.”
Since none of the newly discovered seeded aggregates have yet been shown to be infectious (i.e., communicable or contagious) under natural conditions, we deem it prudent to refer to them as “prionoids.”Citation10,Citation11 Maintaining a distinction between prions and prionoids implies the existence of underlying biological processes that govern the natural transmission of diseases between organisms, including the sophisticated mechanisms by which extra-neural inoculations of prions subvert the immune system to reach and damage the brain (reviewed by Aguzzi and CalellaCitation12).
Fortunately, there is no indication that such processes exist for Aβ prionoids. However, the demonstration of inter-individual transmissibility would warrant upgrading the status of such agents to bona fide prions, as seems very likely to occur in the case of AA amyloid.Citation13
Prionoids and pathogenic proteins in neurodegenerative diseases
Prior to the era of molecular genetics, neurodegenerative diseases of the elderly were defined by the types of misfolded proteins that accumulate as insoluble deposits in the brain. The deposits consisted of highly ordered stacks of β-sheets, which is the physical definition of amyloid. Each type of amyloid resides in a characteristic nuclear, cytoplasmic or extracellular compartment. With the advent of molecular genetics came the discovery of causal genes linked to many of these neurodegenerative diseases and, in a remarkably large number of instances, a given gene encoded the very protein comprising the amyloid fibrils that characterized the neuropathology in the disease.
These diseases became known as protein misfolding disorders, because the causal genes encoded the misfolded proteins comprising the amyloid lesions. In every instance, the misfolded proteins acquire secondary structure—β-sheets—that is absent under normal conditions. Protein misfolding in neurodegenerative diseases refers to conditions in which parent proteins take on novel β-sheet secondary structure. The acquisition of novel secondary structure distinguishes protein misfolding disorders from other diseases caused by mutations that alter protein conformation, such as sickle cell anemia.
With the exception of PrPSc, there is no experimental evidence that the prions or prionoids in neurodegenerative diseases are the pathogenic proteins [star (*) proteins] inducing the neurological deterioration that devastates patients. For a hundred years, neurofibrillary tangles—the intracellular amyloid inclusions that form when tau takes on novel β-sheet structure—were believed to induce neuron death and impair cognition. However, in 2005 this hundred year-old hypothesis was disproven when it was shown that reducing soluble tau in a neurodegenerative mouse model with neurofibrillary tangles led to the cessation of neuron loss and the improvement of memory function, in spite of the startling observation that the neurofibrillary tangles kept accumulating, reminiscent of prionoids.Citation14 The negative case for β-amyloid plaques containing *proteins stems from multiple lines of evidence, including the failure of Alzheimer patients to improve following Aβ immunotherapy that nonetheless successfully removed amyloid plaques,Citation15 and the ability of immunotherapy to reverse deficits in mice without changing plaque load.Citation16,Citation17 Compared with the proteins comprising amyloid lesions, prions, and prionoids, relatively little is known about *proteins and their mechanisms of action.
By far the best understood among these disorders is familial ataxia type 1. Ataxin-1 causes familial ataxia type 1 when extraneous trinucleotide repeat sequences expand the existing polyglutamine tract to form PolyQ/ataxin-1.Citation18 Patients and mice expressing PolyQ/ataxin-1 develop nuclear inclusions and progressive ataxia. PolyQ aggregates are prionoids, since the application of PolyQ aggregates to cultured neurons nucleates the formation of intracellular PolyQ amyloid.Citation19 In mice carrying a knocked-in expanded PolyQ tract, the severity of the neurological abnormalities and neurodegeneration corresponds inversely with the number of inclusions,Citation20 and the abolition of the inclusions through the mutation of a ubiquitin ligase, which promotes the aggregation of misfolded proteins, accelerates the disease.Citation21 These studies indicate that the elimination of the inclusions will not prevent ataxia in familial ataxia type 1.
The conclusion that the removal of the inclusions would not cure familial ataxia type 1 prompted Harry Orr and Huda Zoghbi to search for the pathogenic form of PolyQ/ataxin-1 causing the neurological abnormalities in the disorder. Discovering the mechanism by which PolyQ/ataxin-1 damages neurons emerged from understanding the normal physiological roles of ataxin-1, which is a nuclear protein. The pathogenic form of PolyQ/ataxin-1 is not a misfolded form of ataxin-1; it contains no novel secondary structures, no β-sheets that are not normally present in the brain. Its pathological effects arise from alterations in its binding affinities with its normal nuclear partners, the transcriptional regulator Capicua and the regulator of RNA splicing RMB17,Citation22 leading to changes in the transcriptome that, presumably, affect neuronal function and viability. Thus, the pathogenic form of PolyQ/ataxin-1 is not a prionoid; it is neither a misfolded protein nor a soluble aggregate of the parent protein. This may prove to be a profoundly important lesson for the entire field of neurodegenerative disease research.
A puzzle and two hypotheses
Both patients with prion disease and Alzheimer disease suffer from fatal, progressive forms of dementia. However, while mice infected with PrP prions die, those inoculated with Aβ prions do not. Only two hypotheses can explain the stark contrast between the fatality rates caused by PrP and Aβ inoculations in mice.
• Hypothesis 1: All aggregated proteins (prions and prionoids) are bad.
Aggregates are pathogenic, but different aggregates exert their effects on different cellular pathways. For example, the pathogenic pathway for Aβ aggregates in humans, distinct from that of PrP prions, may not exist in mice.
• Hypothesis 2: Not all aggregates are bad.
Aggregates are not invariably pathogenic; rather, variants of parent proteins (*proteins) cause the cellular dysfunction that leads to a neurological illness (). These pathogenic variants need not be misfolded or aggregated forms of the parent proteins.
Figure 1. Prions, prionoids and pathogenic proteins in neurodegenerative diseases. PrPSc is considered to be the transmissible agent of the prion causing scrapie, Creutzfeldt-Jakob disease and related spongiform encephalopathies. Nucleating fibrillar protein aggregates (“prionoids”) are found in many neurodegenerative diseases. With the exception of PrPSc, there is little evidence in mice or humans linking prionoids in the brain to the pathophysiological processes that cause the disorders connected with these proteins. Instead, accumulating data indicate that the brain dysfunction and neurological signs associated with these illnesses are caused by non-fibrillar variants of the parent proteins (*proteins). In the case of Aβ, brain dysfunction in mice and CSF tau abnormalities in humans are strongly associated with a soluble 56-kDa assembly, Aβ*. The existence of other Aβ* molecules has not been excluded. The *proteins need not be misfolded in the sense of adopting novel secondary structure, which invariably involves β-sheets. PolyQ/ataxin-1 is the best example. Distinguishing between prionoids and *proteins, and understanding how *proteins cause neurological illness, will advance our progress in treating these profoundly devastating and fatal disorders.
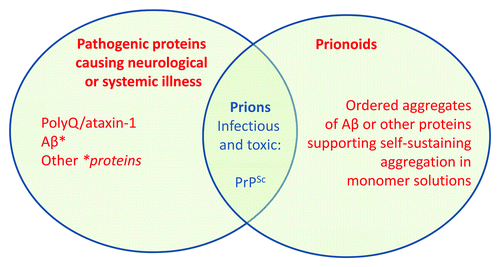
Recent advances in our understanding of the neurotoxicity of PrP and Aβ favor Hypothesis 2, as discussed below.
Neurotoxicity of PrP
In prion disease catastrophic brain dysfunction is associated with a global decrease in protein production, resulting from the dysregulation of eIF2a, a mammalian translation initiation factor.Citation23 This fascinating discovery is presumably the mechanism by which PrP prions ultimately induce neurotoxicity.
However, eIF2a is localized within the cytosol whereas infectious prions are extracellular. Therefore, we are still left wondering how prions containing pathologically aggregated PrPSc can possibly exert actions that originate from the extracellular milieu, derange protein folding in the endoplasmic reticulum, induce a surprisingly vigorous unfolded protein response, and eventually quench cytosolic translation of proteins. It is hard not to conclude that eIF2a repression likely represents a downstream effector of a pathogenic cascade that is initiated by molecularly and topologically distant events.
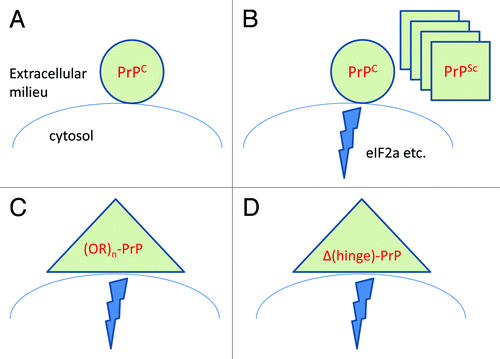
There has been recurrent discussion as to whether the self-replicating material in prion disease (the “prion”) is physically identical with the neurotoxic entity. In this context, John Collinge has recently proposed the term “PrPL” to denote a hypothetical moiety that may be neurotoxic yet differs from PrPSc.Citation24 However, the idea that PrP may produce neurological disease without the generation of infectivity dates back to 1990 when transgenic mice that spontaneously developed prion disease were created. These mice expressed PrP carrying a mutation linked to a familial prion disease, developed ataxia, lethargy and rigidity, and invariably died, but their brains contained few or no infectious prions, suggesting that “an inborn error of PrP metabolism could produce neurologic disease without the generation of infectivity.”Citation25 It is possible, and indeed very likely in our view, that PrPSc and the various non-infectious neurotoxic variants of PrP, which include PrP with supernumerary octapeptide repeatsCitation26 and PrP versions with interstitial deletions of the “hinge” region between the unstructured N-terminus and the globular domain,Citation27 activate neurotoxic pathways converging with those triggered by prion infection ().
Figure 2. The cellular prion protein is absolutely required for the toxicity of infectious prions (A),Citation39 implying that PrPSc exerts neurotoxicity by docking to PrPC (B). This toxicity may also be elicited by PrP variants occurring naturally, such as PrP carrying supernumerary octapeptide repeats (C), or experimentally constructed toxic variants such as PrP versions carrying deletions of the hinge region (D). It was recently discovered that prion infection results in a chain of events that ultimately quenches protein translation,Citation23 but it remains to be seen whether the toxicity elicited by PrP mutants (Panels C and D) utilizes the same pathway.
Neurotoxicity of Aβ
Our understanding of the neurotoxicity of Aβ lags behind that of PrP, because animal models that recapitulate all the facets of Alzheimer disease, which are needed to assay the human relevance of pathological Aβ, do not exist. In humans, the pathological transformation of Aβ initiates a process that involves the accumulation of amyloid plaques and often leads to a fatal neurodegenerative condition. In mice, the formation of pathological Aβ may induce amyloid plaque deposition, which reflects the presence of Aβ prionoids. It is astonishing that the accumulation of Aβ prionoids in the vast majority of mice does not lead to overt neurodegeneration, except in the immediate vicinity of amyloid plaques.Citation28-Citation30 In mice lacking nitric oxide synthase 2, the accumulation of Aβ prionoids is associated with neuron death in the CA3 but not the CA1 hippocampal subfield,Citation31 but in humans CA3 is spared while CA1 is not,Citation32 calling into question the relevance of this form of neuron death. Whether neuron death in this model is due to Aβ prionoids is also unknown. Fatality, when present, is strain-dependent, and can occur in the absence of amyloid formation, suggesting the existence of a neurotoxic species, which may not necessarily be congruent with the self-propagating species.Citation33 The biochemical identity and cellular effects of this form of Aβ remain unknown.
Two types of cognitive dysfunction develop in mice over producing Aβ. One type is found in mice with aggressive amyloid deposition—an excessive amount of Aβ prionoids—in which the amassed amyloid plaques and their surrounding cytopathology act like a gigantic space-occupying lesion in the brain.Citation28 No neurons remain within plaque cores, and tortuous, dystrophic neurites and dendrites partially denuded of spines reside in the 50-micron halo surrounding the cores (reviewed by Ashe and ZahsCitation34). It is therefore not surprising that, beyond a certain threshold, cognition varies inversely with plaque load. However, the density of amyloid plaques required to produce this effect is rarely reached in Alzheimer disease, which may explain why the links between plaques and neuron loss or cognition are tenuous at best.Citation35,Citation36 An excessive accumulation of Aβ prionoids in mice can result in cognitive deficits that are related to the space-occupying effects of the amyloid plaques, but this is distinct from the mechanism by which most of the neuronal dysfunction and neurodegeneration occurs in humans with β-amyloidosis or Alzheimer disease.
The other type of cognitive dysfunction occurs independently of plaques or neuron loss, and appears to be due to a non-fibrillar 56-kilodalton Aβ assembly termed Aβ*56.Citation37 Aβ*56 is a putative dodecamer most likely formed by the clustering of four Aβ trimers. Isolates of fibrillar Aβ in the brain do not contain Aβ*56 (Liu P and Ashe K, unpublished data). It is unlikely that Aβ*56 acquires novel β-sheet structure, because the compositional unit, Aβ trimers, is present even in young mice.Citation37 The absence of novel β-sheet structure in Aβ*56 argues against it being a prionoid, although formal demonstration will require transmission experiments. Aβ*56 induces no overt neurodegeneration, and yet alters cognition by disrupting long-lasting synaptic plasticity by an as yet unknown mechanism.Citation38 In humans, Aβ*56 can be measured in the CSF, where it increases with normal aging and correlates moderately strongly with the microtubule-binding protein tau (M. Handoko, M. Grant, A. Wallin, K. Blennow and K. Ashe, unpublished data). Tau is released into the CSF, through a process that remains unclear, when neurons malfunction. CSF tau correlates weakly or not at all with CSF Aβ1–42, which reflects β-amyloidosis and Aβ prionoids.
Although it was tempting to postulate that Aβ*56 triggers a sequence of events that leads to the conversion from asymptomatic aging to mild cognitive impairment or Alzheimer disease, which coincides with the onset of overt neurodegeneration and neuron loss, this prediction was not borne out in a large longitudinal study (Handoko M, Grant M, Petersen R and Ashe K, unpublished data). Thus, Aβ*56 disrupts neuronal function in mice and is linked to neuronal malfunction in humans, but is insufficient to induce overt neurodegeneration in either species.
In the absence of animal models, harboring Alzheimer-related mutations exclusively, that exhibit the full spectrum of disease, beginning with subtle neuronal dysfunction and culminating with fatal cognitive devastation, the question of whether asymptomatic β-amyloidosis requires Aβ*56 to develop into full-blown Alzheimer disease cannot be addressed experimentally. It is possible that one or more non-prionoid form of Aβ triggers neuronal dysfunction and neurodegeneration in Alzheimer disease. Discovering these pathogenic forms will depend upon the creation of high fidelity model systems of Alzheimer disease.
Conclusion
In bona fide prion diseases, a very large body of evidence links the aggregated form of PrP, PrPSc, to both prion infectivity and prion neurotoxicity. However, non-infectious, yet neurotoxic, variants of PrP occur naturally and more such variants have been constructed experimentally, indicating that the phenotypic expression typical of prion diseases can be triggered by events occurring downstream of prion infection. There is little evidence in mice or humans linking the neurological effects of Aβ to the nucleating forms of this protein, while emerging data point to a specific non-nucleating form of Aβ, Aβ*56, that produces some of the neurological signs of disease. However, Aβ*56 is not sufficient to induce the inexorable neurological deterioration that characterizes Alzheimer disease, indicating that other critical factors or forms of Aβ work in collaboration with Aβ*56 to destroy the brain. Curing prion and Alzheimer disease will depend upon developing a deeper understanding of the pathogenic forms of PrP and Aβ that cause the brain dysfunction underlying these deadly illnesses.
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Basler K, Oesch B, Scott M, Westaway D, Wälchli M, Groth DF, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986; 46:417 - 28; http://dx.doi.org/10.1016/0092-8674(86)90662-8; PMID: 2873895
- Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, et al. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci 2000; 20:3606 - 11; PMID: 10804202
- Gaspar RC, Villarreal SA, Bowles N, Hepler RW, Joyce JG, Shughrue PJ. Oligomers of beta-amyloid are sequestered into and seed new plaques in the brains of an AD mouse model. Exp Neurol 2010; 223:394 - 400; http://dx.doi.org/10.1016/j.expneurol.2009.09.001; PMID: 19744481
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313:1781 - 4; http://dx.doi.org/10.1126/science.1131864; PMID: 16990547
- Morales R, Duran-Aniotz C, Castilla J, Estrada LD, Soto C. De novo induction of amyloid-beta deposition in vivo. Mol Psychiatry 2011; 17:1347 - 53; http://dx.doi.org/10.1038/mp.2011.120; PMID: 21968933
- Stöhr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, et al. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc Natl Acad Sci U S A 2012; 109:11025 - 30; http://dx.doi.org/10.1073/pnas.1206555109; PMID: 22711819
- Aguzzi A, Weissmann C. Spongiform encephalopathies: a suspicious signature. Nature 1996; 383:666 - 7; http://dx.doi.org/10.1038/383666a0; PMID: 8878470
- Gajdusek DC, Gibbs CJ, Alpers M. Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature 1966; 209:794 - 6; http://dx.doi.org/10.1038/209794a0; PMID: 5922150
- Aguzzi A. Cell biology: Beyond the prion principle. Nature 2009; 459:924 - 5; http://dx.doi.org/10.1038/459924a; PMID: 19536253
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009; 64:783 - 90; http://dx.doi.org/10.1016/j.neuron.2009.12.016; PMID: 20064386
- Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev 2009; 89:1105 - 52; http://dx.doi.org/10.1152/physrev.00006.2009; PMID: 19789378
- Zhang B, Une Y, Fu X, Yan J, Ge F, Yao J, et al. Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proc Natl Acad Sci U S A 2008; 105:7263 - 8; http://dx.doi.org/10.1073/pnas.0800367105; PMID: 18474855
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005; 309:476 - 81; http://dx.doi.org/10.1126/science.1113694; PMID: 16020737
- Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol 2010; 9:363 - 72; http://dx.doi.org/10.1016/S1474-4422(10)70043-0; PMID: 20189881
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci 2002; 5:452 - 7; http://dx.doi.org/10.1038/nn842; PMID: 11941374
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, et al. Reversible memory loss in a mouse transgenic model of Alzheimer'sdisease. J Neurosci 2002; 22:6331 - 5; PMID: 12151510
- Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr., Servadio A, Beaudet AL, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 1993; 4:221 - 6; http://dx.doi.org/10.1038/ng0793-221; PMID: 8358429
- Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 2009; 11:219 - 25; http://dx.doi.org/10.1038/ncb1830; PMID: 19151706
- Watase K, Weeber EJ, Xu B, Antalffy B, Yuva-Paylor L, Hashimoto K, et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 2002; 34:905 - 19; http://dx.doi.org/10.1016/S0896-6273(02)00733-X; PMID: 12086639
- Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, et al. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 1999; 24:879 - 92; http://dx.doi.org/10.1016/S0896-6273(00)81035-1; PMID: 10624951
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008; 452:713 - 8; http://dx.doi.org/10.1038/nature06731; PMID: 18337722
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012; 485:507 - 11; http://dx.doi.org/10.1038/nature11058; PMID: 22622579
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540 - 2; http://dx.doi.org/10.1038/nature09768; PMID: 21350487
- Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 1990; 250:1587 - 90; http://dx.doi.org/10.1126/science.1980379; PMID: 1980379
- Chiesa R, Harris DA. Fishing for prion protein function. PLoS Biol 2009; 7:e75; http://dx.doi.org/10.1371/journal.pbio.1000075; PMID: 19338390
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, et al. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 2007; 26:538 - 47; http://dx.doi.org/10.1038/sj.emboj.7601510; PMID: 17245436
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, et al. Neuron loss in APP transgenic mice. Nature 1998; 395:755 - 6; http://dx.doi.org/10.1038/27351; PMID: 9796810
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, et al. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 1997; 17:7053 - 9; PMID: 9278541
- Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, et al. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc Natl Acad Sci U S A 2002; 99:13990 - 5; http://dx.doi.org/10.1073/pnas.222433299; PMID: 12374847
- Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, et al. Progression of amyloid pathology to Alzheimer's disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci 2008; 28:1537 - 45; http://dx.doi.org/10.1523/JNEUROSCI.5066-07.2008; PMID: 18272675
- West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 1994; 344:769 - 72; http://dx.doi.org/10.1016/S0140-6736(94)92338-8; PMID: 7916070
- Carlson GA, Borchelt DR, Dake A, Turner S, Danielson V, Coffin JD, et al. Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum Mol Genet 1997; 6:1951 - 9; http://dx.doi.org/10.1093/hmg/6.11.1951; PMID: 9302276
- Ashe KH, Zahs KR. Probing the biology of Alzheimer’s disease in mice. Neuron 2010; 66:631 - 45; http://dx.doi.org/10.1016/j.neuron.2010.04.031; PMID: 20547123
- Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 2004; 61:378 - 84; http://dx.doi.org/10.1001/archneur.61.3.378; PMID: 15023815
- Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 1997; 41:17 - 24; http://dx.doi.org/10.1002/ana.410410106; PMID: 9005861
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006; 440:352 - 7; http://dx.doi.org/10.1038/nature04533; PMID: 16541076
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 1999; 2:271 - 6; http://dx.doi.org/10.1038/6374; PMID: 10195221
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, et al. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996; 379:339 - 43; http://dx.doi.org/10.1038/379339a0; PMID: 8552188