Abstract
The concept of “prion-like” has been proposed to explain the pathogenic mechanism of the principal neurodegenerative disorders associated with protein misfolding, including Alzheimer disease (AD). Other evidence relates prion protein with AD: the cellular prion protein (PrPC) binds β amyloid oligomers, allegedly responsible for the neurodegeneration in AD, mediating their toxic effects. We and others have confirmed the high-affinity binding between β amyloid oligomers and PrPC, but we were not able to assess the functional consequences of this interaction using behavioral investigations and in vitro tests. This discrepancy rather than being resolved with the classic explanations, differencies in methodological aspects, has been reinforced by new data from different sources. Here we present data obtained with PrP antibody that not interfere with the neurotoxic activity of β amyloid oligomers. Since the potential role of the PrPC in the neuronal dysfunction induced by β amyloid oligomers is an important issue, find reasonable explanation of the inconsistent results is needed. Even more important however is the relevance of this interaction in the context of the disease, so as to develop valid therapeutic strategies.
Keywords: :
Introduction
The concept of “prion-like” has been proposed to explain the pathogenic mechanism of all the principal neurodegenerative disorders associated with protein misfolding, including Alzheimer disease (AD). The in vivo demonstration of the seeding mechanism combined with the passage from one cell to another of the pathological proteins in oligomeric form has alimented the prion-like hypothesis.Citation1-Citation4 The concept of transmission is distinguishable from that of infection,Citation5 but it has also been proposed that the pathogenic mechanism of prion diseases and the other neurodegenerative disorders overlap.Citation6,Citation7 The other information relating prion protein to AD is that the toxic effect of β amyloid (Aβ) oligomers may depend on their high affinity binding to cellular prion protein (PrPC).Citation8 The causal role of amyloid deposits in the pathogenesis of AD was formally proposed in the amyloid cascade hypothesis, 20 years ago by Hardy and Higgins (1992),Citation9 this has been recently revisited,Citation10 but the pivotal role of Aβ is substantially confirmed. This has driven therapeutic approaches focused on reducing the presence of Aβ deposits in ADCitation11 brains by various strategies. Unfortunately however clinical trials testing the anti-amyloid treatments, including the recent ones based on the anti-Aβ antibody, showed no significant effects on the progression of AD.Citation12 The timing of the intervention is the main reason for this failure, treatment being given too late to be effective. However it is also possible that the reduction of Aβ deposits, when it occurred, is not sufficient alone to affect the AD.
Aβ Oligomers
Since understanding the pathogenic mechanisms involving Aβ is essential for effective therapies, identificatifying the Aβ species responsible for the neuronal alterations in AD and their actions is a key aspect. The role of soluble small aggregates, known as oligomers, has been consolidated in the last decade as the principal cause of the neurodegeneration in AD. This concept originally arose from the studies in the late nineties, showing that the relation between amyloid fibrils and the neurotoxicity, previously postulated,Citation13 no longer hold. The presence of protofibrils and oligomers as metastable intermediates in the fibrillogenesisCitation14,Citation15 correlates better with the neurotoxicity than the stable fibrils.Citation16,Citation17 Walsh et al. (2002)Citation18 showed that neuronal dysfunction can be acutely produced by exposure of the neurons to naturally secreted Aβ oligomers that inhibit long-term potentiation (LTP), a classic experimental paradigm for synaptic plasticity. The presence at synaptic level of Aβ oligomers was specifically demonstratedCitation19 as well as the abundance of these species in the AD brain.Citation20,Citation21 Furthermore, some mutations of the amyloid precursor protein (APP) gene, associated with AD, might specifically favor the formation of Aβ oligomers.Citation22,Citation23
Since then, Aβ oligomers have been considered responsible for the neuronal toxicity in AD, and this could explain the absence of any topographic relationship between Aβ deposits and neuronal cell death, as well as the memory decline. Thus, in transgenic mice overexpressing mutated human APP gene, the cognitive behavioral impairment precedes the formation of cortical amyloid plaques.Citation24-Citation27 Although intracellular accumulation of Aβ could also explain these results, it is reasonable to assume that the formation of oligomers precedes the development of amyloid plaques and immediately produces the neurotoxic effect. The neuronal damage induced by soluble aggregates confirms that the best clinical-pathologic correlation in AD is between synaptic loss and cognitive decline.Citation28 The nature and the size of the Aβ oligomer species involved in the neuronal dysfunction and the cellular pathway mediating this effect have been widely debated . The main components of senile plaques are the peptides Aβ 1–40 and Aβ 1–42. Both have self-aggregation capacity but with a clear difference in favor of the longer sequence which in specific conditions in vitro, can spontaneously aggregate within minutes. Other Aβ peptide species present in the brain are Aβ 1–43, and Aβ 1–37 or 1–38, and although a large part of the experimental studies has been conducted using Aβ 1–42 solution, quite possibly a mixture of peptides forms the natural substrate of oligomers. The influence of uncommon peptides is described well in a recent paper where the N-terminally truncated pyroglutamylated form of Aβ, also identified in AD brain, was proposed as the seed of the nucleation of Aβ 1–42 solution.Citation29 As described in the review by Benilova et al. (2012),Citation30 to the native biological variability of Aβ species, we must add the various methodological conditions used to produce these species, or to purify them from brain tissue. Thus, we have Aβ oligomers with biological activity ranging from dimers (8KDa) to large multimeric structures (200–300 KDa). Specific neurotoxic capacity has been attributed to the dodecamer peptide Aβ 56* (56 KDa) based on its appearance in relation with the memory impairment in transgenic mice,Citation31 but in a direct comparison with trimers purified from cell culture Aβ 56* did not show a greater capacity to induce memory impairment when injected ICV in rats.Citation32 In the classical model, the senile plaques are considered to be in dynamic equilibrium with the oligomers; they gradually concentrate in the deposits which in turn act as the reservoir of oligomers that can be released. However the progression from monomeric to oligomeric and successively through protofibrils to fibrils is complex. In some cases oligomers did not generate fibrils (off pathway), in other conditions it has been proposed that oligomers forme fibrils through metastable intermediatesCitation33 in a process called “nucleated conformational conversion.” In a standard preparation of Aβ oligomers one occasionally finds species of the same size that are neither toxic nor recognized by oligomer-specific antibodies, indicating that Aβ oligomers can also possess size-independent differences in toxicity.Citation35 Similar size and different toxicity has been shown using β-sheet HypF-N oligomers and non-β-sheet Aβ oligomers. These studies suggest that the toxicity of small amyloidogenic oligomers is governed primarily by the degree of solvent exposure of hydrophobic residues and is weakly influenced by their secondary structures.Citation36,Citation37
Aβ Oligomer Neurotoxicity
Keeping in mind the differences in size, species and biological activity of synthetic or purified Aβ oligomers, there is likely to be similar variability in the biological determinants of the neurotoxic effect. To summarize the numerous data reported, the neurotoxicity of Aβ oligomers can be mediated through three different mechanisms, not mutually exclusive: unspecific perturbation of cellular and intracellular membranes; the formation of amyloid pores, as ion channels and finally, the specific interaction with membrane receptors. The challenge is to understand the pathogenic importance of these processes, not only the reproducibility of each specific event. The effects of various types of oligomer have been studied in vivo and in vitro, and impairment of cognitive function with the direct application of Aβ oligomer solutions has been shown in vivo using natural oligomers from cellsCitation38 but also with dimers isolated directly from AD brain.Citation39 Similar results have been obtained with synthetic Aβ oligomers in ratsCitation40 and miceCitation41; Aβ oligomers impair long- term potentiation (LTP)Citation18,Citation39,Citation42 and enhance long-term depression (LTD).Citation42 Both these activities involve the NMDA receptors, the receptors are partially blocked by oligomers to induce the inhibition of LTP, while the enhancement of LTD might have different mechanisms, as summarized by Selkoe and Mucke (2012).Citation43
Aβ oligomers might induce internalization or desensitization of NMDA and AMPA receptors, with the loss of dendritic spines. Direct postsynaptic activation of α7 nicotinic acetylcholine receptor (α7-nAChR) by Aβ oligomers was initially proposed,Citation44 and then the activation of extrasynaptic NMDA receptors,Citation42,Citation45 particularly NR2B-mediated NMDA currents, was shown.Citation46,Citation47 With the NMDA receptor interference induced by Aβ oligomers, it was proposed that numerous intracellular signaling pathways like calcineurin/STEP/cofilin, p38 MAPK, and GSK-3β could be activated.Citation42,Citation45,Citation48-Citation50 Aβ-induced hippocampal neuronal dysfunction occurs through NMDAR-dependent microtubule disassembly with neurite retraction and DNA fragmentation in mature hippocampal cells.Citation51 Together with α7nACh, NMDA and AMPA receptors, several others like insulin receptors,Citation52,Citation53 RAGE (the receptor for advanced glycation end products),Citation54,Citation55 the prion protein,Citation8 the Ephrin-type B2 receptor (EphB2),Citation56,Citation57 and the amylin 3-receptor subtypeCitation58 were proposed to specifically mediate the deleterious effect of Aβ oligomers. In various experimental conditions Aβ oligomers can induce tau phosphorylation.Citation29,Citation59-Citation61 This directly links the two major neuropathological hallmarks in AD: the amyloid deposits and the fibrillary tangles made up of hyperphosphorylated tau
The Role of Cellular Prion Protein
Here we focus on the interaction of Aβ oligomers with PrPC, since we have confirmed only the high-affinity binding between the twoCitation41,Citation62,Citation63 but not the functional consequences shown by Strittmatter's group.Citation8 These authors identified PrPc as ligand/receptor of Aβ oligomers, while LTP analysis in vitro and behavioral studies in vivo indicated that in the absence of PrPC Aβ oligomers induced no damage.Citation8,Citation64,Citation65 Using surface plasmon resonance (SPR), the high-affinity binding of Aβ 1–42 oligomers for PrPC was confirmed, whereas a functional role of this association has been excluded by various studies.Citation41,Citation63,Citation66 We approached this issue by directly applying Aβ oligomers in the brain, followed by behavioral examination of memory deficits. The ICV application of Aβ oligomers, 1 µM, induced cognitive impairment assessed by novel object recognition test: the treated mice did not distinguish any more between familiar and novel object. The monomeric solution as well as the Aβ fibers, in the same conditions, did not affect their memory.Citation41 In PrP knockout mice, ICV Aβ oligomers induced the memory deficit similar to that in normally expressing PrPC mice. The ICV application of antibody against Aβ (4G8), ten minutes before Aβ oligomers, completely antagonized the memory impairment.Citation41 In the same condition anti-PrP antibody (6D11) did not affect the behavioral deficit induced by Aβ oligomers, although at a higher dose the antibody had per se a deleterious effect (). This suggests that Aβ 1–42 oligomers are responsible for cognitive impairment in AD but PrPC is not required for the effect.
Figure 1. Action of anti-PrP antibody (6D11) on the effects of Aβ oligomers. (A) Object recognition test. two concentrations of 6D11 (0.08 and 0.25 µg) were administered 10 min before the ICV injection of vehicle (VEH) or Aβ oligomers (1µM). The effect of Aβ oligomers was similar in the presence or absence of anti-PrP antibody. (B) Viability of murine hippocampal cell cultures exposed for 48 h to Aβ oligomers (1 µM) with vehicle or 6D11 at 0.25, 0.50 and 2 µg/mL. The presence of PrP antibody did not interfere with the Aβ oligomers toxicity. *p < 0.01 vs respective vehicle (Tukey’s test) for methodological details see Balducci et al.Citation41
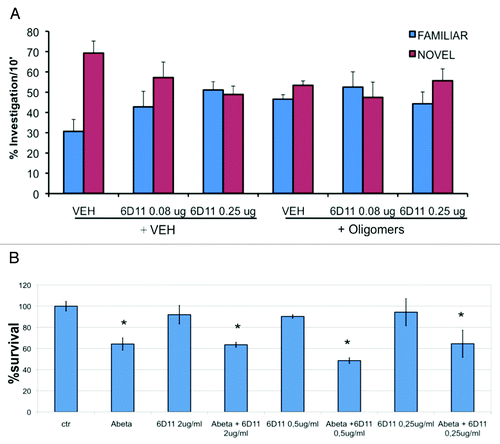
Similar results were found when the toxicity was tested in vitro. The exposure of cortical neurons to Aβ 1–42 solution at 1 µM reduced cell viability by 30–40% compared with the control condition, in cells expressing PrPC or in cells knockout for PrPC. In this case too, we tested for interference by the anti-PrP antibody, using 6D11, which should recognize the PrP domain involved in the interaction with Aβ 1–42,Citation8 but this antibody did not influence the toxicity of Aβ 1–42 (). In contrast there was a significant interaction between anti-PrP antibodies (6D11 and 6D13) and the deleterious effect of Aβ oligomers on LTP in murine brain slices or in vivo rats.Citation67 A high concentration of the PrP antibody 6D11 infused for two weeks in double-transgenic mice (APP/PS1) affected cognitive behavior in the radial arm maze test.Citation65 However, the learning curves of the different animal groups were very flat and strongly depended on the performance at the first time point, making it difficult to interpret the results.
The Strittmatter group,Citation68 has shown that the functional consequences of the Aβ 1–42 oligomers-PrPC interaction are mediated downstream by the activation of Fyn signaling. The timing (a large part of the effects are studied with exposure of only minutes) and the quantitative effect associated with the Aβo/PrPC/Fyn mechanism, in terms of synaptic dysfunction, interaction with NMDA receptors, and toxicity leave doubts about the relevance of this phenomenon. Furthermore, as the authors mention, a physical interaction of PrPC with Fyn is unlikely since PrPC is attached extracellularly to the membrane with a GPI anchor, while Fyn is inside the cell. In vivo experiments showed that the epileptic status of the APPSwe/PSenDE9 mice was strongly attenuated when PrPC was nullified. This may indicate an interaction between the expressed transgene and PrPC, but the epileptic discharge is closely related to these specific transgenic mice. Other APP single, double and triple transgenic mice, like most AD patients, do not show this condition.
A different mechanism of interaction between PrPC and Aβ oligomers was proposed by You et al. (2012).Citation69 They showed that PrPC can be a key regulator of NMDAR activity. They hypothesized that PrPC, in its copper-loaded state, binds to the NMDAR complex to allosterically reduce its glycine affinity, thereby increasing desensitization. When copper is chelated or when PrPC is absent or functionally compromised (by GPI anchor cleavage or binding to Aβ oligomers, for example), glycine affinity is enhanced, reducing receptor desensitization and producing pathologically large, steady-state currents that contribute to neuronal damage. Other studies have shown a role of PrPC in the Aβ 1–42 oligomers neurotoxicity,Citation70,Citation71 while the influence of PrPC was excluded in an in vivo investigation of cognitive behavior in J20 mice,Citation72 confirming the differences. As mentioned above, this scientific dispute should proceed to the physopathological meanings that have still to be established.
We have described here the complexity of Aβ oligomer formation and the multiple possibilities of their interaction with the cellular membranes and proteins. This issue has been addressed in specific studiesCitation73,Citation74 and discussed in a previous review.Citation75 The results indicate that PrPC is only one of the numerous entities potentially interacting with Aβ oligomers and may have an essential role in their toxicity only in specific conditions.
Conclusions
As recently pointed out by Westaway and Jhamandas (2012)Citation76 taking in account of the potential negative synergistic effect of Aβ oligomers-PrPC, there is also ample data in favor of a neuroprotective effect of PrPC.Citation77-Citation80 Rial et al. (2012)Citation81 showed that the overepression of PrPC in transgenic mice prevented cognitive dysfunction and the apoptotic neuronal cell death induced by Aβ1–40. In terms of the relevance of the findings, the neuroprotective activity of PrPC in various experimental conditions must be evaluated together with the enormous differences in the epidemiology of the diseases that seem to exclude the direct links between AD and prion-related encephalopathies as the essential role of PrPC proposed in both conditions might indicate. However, independently from the real functional consequence of the Aβ oligomers-PrPC interaction, the high affinity of this binding has been widely confirmed. Nieznaski et al. (2012)Citation82 tested the capacity of recombinant PrP 23–231 to interfere with the Aβ1–42 fibrillogenesis. At low concentrations (0.1–1µM) and low molar ratio (1/20 to 1/100) the prion protein co-incubated with Aβ1–42 inhibited the formation of oligomers and fibrils and prevented the toxicity of Aβ1–42 in cell cultures. As the authors point out, two domains (95–110 and N-terminal basic residues) of PrP are required for the anti-amyloidogenic effect and both are included in the N-terminal N1 fragment (residues 23–110/111) released from the membrane-anchored PrPC.Citation83 The N1 fragment had neuroprotective activity in vivo in rat retina ischemia model,Citation84 and also in primary cultured neurons exposed to Aβ oligomers.Citation85 Thus, although the physiopathological consequences of the Aβ1–42-PrPC interaction remain to be clarified we can reach a common point of view on the possible therapeutic prospects.
References
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313:1781 - 4; http://dx.doi.org/10.1126/science.1131864; PMID: 16990547
- Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010; 330:980 - 2; http://dx.doi.org/10.1126/science.1194516; PMID: 20966215
- Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 2010; 11:301 - 7; http://dx.doi.org/10.1038/nrm2873; PMID: 20308987
- Kanouchi T, Ohkubo T, Yokota T. Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion-like propagation?. J Neurol Neurosurg Psychiatry 2012; 83:739 - 45; http://dx.doi.org/10.1136/jnnp-2011-301826; PMID: 22544947
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009; 64:783 - 90; http://dx.doi.org/10.1016/j.neuron.2009.12.016; PMID: 20064386
- Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012; 336:1511 - 3; http://dx.doi.org/10.1126/science.1222951; PMID: 22723400
- Soto C. Transmissible proteins: expanding the prion heresy. Cell 2012; 149:968 - 77; http://dx.doi.org/10.1016/j.cell.2012.05.007; PMID: 22632966
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009; 457:1128 - 32; http://dx.doi.org/10.1038/nature07761; PMID: 19242475
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992; 256:184 - 5; http://dx.doi.org/10.1126/science.1566067; PMID: 1566067
- Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem 2009; 110:1129 - 34; http://dx.doi.org/10.1111/j.1471-4159.2009.06181.x; PMID: 19457065
- Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 2003; 43:545 - 84; http://dx.doi.org/10.1146/annurev.pharmtox.43.100901.140248; PMID: 12415125
- Callaway E. Alzheimer’s drugs take a new tack. Nature 2012; 489:13 - 4; http://dx.doi.org/10.1038/489013a; PMID: 22962697
- Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. Aggregation-related toxicity of synthetic beta-amyloid protein in hippocampal cultures. Eur J Pharmacol 1991; 207:367 - 8; http://dx.doi.org/10.1016/0922-4106(91)90014-9; PMID: 1783006
- Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol 1997; 4:119 - 25; http://dx.doi.org/10.1016/S1074-5521(97)90255-6; PMID: 9190286
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem 1997; 272:22364 - 72; http://dx.doi.org/10.1074/jbc.272.35.22364; PMID: 9268388
- Forloni G, Lucca E, Angeretti N, Della Torre P, Salmona M. Amidation of beta-amyloid peptide strongly reduced the amyloidogenic activity without alteration of the neurotoxicity. J Neurochem 1997; 69:2048 - 54; http://dx.doi.org/10.1046/j.1471-4159.1997.69052048.x; PMID: 9349550
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 1998; 95:6448 - 53; http://dx.doi.org/10.1073/pnas.95.11.6448; PMID: 9600986
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416:535 - 9; http://dx.doi.org/10.1038/416535a; PMID: 11932745
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci 2004; 24:10191 - 200; http://dx.doi.org/10.1523/JNEUROSCI.3432-04.2004; PMID: 15537891
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, et al. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A 2003; 100:10417 - 22; http://dx.doi.org/10.1073/pnas.1834302100; PMID: 12925731
- Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, et al. Globular amyloid beta-peptide oligomer - a homogenous and stable neuropathological protein in Alzheimer’s disease. J Neurochem 2005; 95:834 - 47; http://dx.doi.org/10.1111/j.1471-4159.2005.03407.x; PMID: 16135089
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci 2001; 4:887 - 93; http://dx.doi.org/10.1038/nn0901-887; PMID: 11528419
- Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol 2008; 63:377 - 87; http://dx.doi.org/10.1002/ana.21321; PMID: 18300294
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, et al. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A 1999; 96:3228 - 33; http://dx.doi.org/10.1073/pnas.96.6.3228; PMID: 10077666
- Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet 1999; 29:177 - 85; http://dx.doi.org/10.1023/A:1021691918517; PMID: 10547924
- Van Dam D, D’Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci 2003; 17:388 - 96; http://dx.doi.org/10.1046/j.1460-9568.2003.02444.x; PMID: 12542676
- Balducci C, Tonini R, Zianni E, Nazzaro C, Fiordaliso F, Salio M, et al. Cognitive deficits associated with alteration of synaptic metaplasticity precede plaque deposition in AßPP23 transgenic mice. J. Alzh Dis. 2010; 21:1367 - 81
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 1990; 27:457 - 64; http://dx.doi.org/10.1002/ana.410270502; PMID: 2360787
- Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 2012; 485:651 - 5; http://dx.doi.org/10.1038/nature11060; PMID: 22660329
- Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 2012; 15:349 - 57; http://dx.doi.org/10.1038/nn.3028; PMID: 22286176
- Lee J, Culyba EK, Powers ET, Kelly JW. Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol 2011; 7:602 - 9; http://dx.doi.org/10.1038/nchembio.624; PMID: 21804535
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006; 440:352 - 7; http://dx.doi.org/10.1038/nature04533; PMID: 16541076
- Reed MN, Hofmeister JJ, Jungbauer L, Welzel AT, Yu C, Sherman MA, et al. Cognitive effects of cell-derived and synthetically derived Aβ oligomers. Neurobiol Aging 2011; 32:1784 - 94; http://dx.doi.org/10.1016/j.neurobiolaging.2009.11.007; PMID: 20031278
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, et al. Self-assembly of Abeta(1-42) into globular neurotoxins. Biochemistry 2003; 42:12749 - 60; http://dx.doi.org/10.1021/bi030029q; PMID: 14596589
- Ladiwala AR, Litt J, Kane RS, Aucoin DS, Smith SO, Ranjan S, et al. Conformational differences between two amyloid β oligomers of similar size and dissimilar toxicity. J Biol Chem 2012; 287:24765 - 73; http://dx.doi.org/10.1074/jbc.M111.329763; PMID: 22547072
- Zampagni M, Cascella R, Casamenti F, Grossi C, Evangelisti E, Wright D, et al. A comparison of the biochemical modifications caused by toxic and non-toxic protein oligomers in cells. J Cell Mol Med 2011; 15:2106 - 16; http://dx.doi.org/10.1111/j.1582-4934.2010.01239.x; PMID: 21155974
- Stravalaci M, Bastone A, Beeg M, Cagnotto A, Colombo L, Di Fede G, et al. Specific recognition of biologically active amyloid-β oligomers by a new surface plasmon resonance-based immunoassay and an in vivo assay in Caenorhabditis elegans. J Biol Chem 2012; 287:27796 - 805; http://dx.doi.org/10.1074/jbc.M111.334979; PMID: 22736768
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 2005; 8:79 - 84; http://dx.doi.org/10.1038/nn1372; PMID: 15608634
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 2008; 14:837 - 42; http://dx.doi.org/10.1038/nm1782; PMID: 18568035
- Scopes DI, O’Hare E, Jeggo R, Whyment AD, Spanswick D, Kim EM, et al. Aβ oligomer toxicity inhibitor protects memory in models of synaptic toxicity. Br J Pharmacol 2012; 167:383 - 92; http://dx.doi.org/10.1111/j.1476-5381.2012.01973.x; PMID: 22913627
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A 2010; 107:2295 - 300; http://dx.doi.org/10.1073/pnas.0911829107; PMID: 20133875
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009; 62:788 - 801; http://dx.doi.org/10.1016/j.neuron.2009.05.012; PMID: 19555648
- Mucke L, Selkoe DJ. Neurotoxicity of Amyloid β-Protein: Synaptic and Network Dysfunction. Cold Spring Harb Perspect Med 2012; 2:a006338; PMID: 22762015
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 2005; 8:1051 - 8; http://dx.doi.org/10.1038/nn1503; PMID: 16025111
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 2007; 27:2866 - 75; http://dx.doi.org/10.1523/JNEUROSCI.4970-06.2007; PMID: 17360908
- Ferreira IL, Bajouco LM, Mota SI, Auberson YP, Oliveira CR, Rego AC. Amyloid beta peptide 1-42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012; 51:95 - 106; http://dx.doi.org/10.1016/j.ceca.2011.11.008; PMID: 22177709
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 2011; 31:6627 - 38; http://dx.doi.org/10.1523/JNEUROSCI.0203-11.2011; PMID: 21543591
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci 2004; 24:3370 - 8; http://dx.doi.org/10.1523/JNEUROSCI.1633-03.2004; PMID: 15056716
- Tackenberg C, Brandt R. Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci 2009; 29:14439 - 50; http://dx.doi.org/10.1523/JNEUROSCI.3590-09.2009; PMID: 19923278
- Sclip A, Antoniou X, Colombo A, Camici GG, Pozzi L, Cardinetti D, et al. c-Jun N-terminal kinase regulates soluble Aβ oligomers and cognitive impairment in AD mouse model. J Biol Chem 2011; 286:43871 - 80; http://dx.doi.org/10.1074/jbc.M111.297515; PMID: 22033930
- Mota SI, Ferreira IL, Pereira C, Oliveira CR, Rego AC. Amyloid-beta peptide 1-42 causes microtubule deregulation through N-methyl-D-aspartate receptors in mature hippocampal cultures. [Epub ahead of print] Curr Alzheimer Res 2012; 9:844 - 56; http://dx.doi.org/10.2174/156720512802455322; PMID: 22631440
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, et al. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A 2009; 106:1971 - 6; http://dx.doi.org/10.1073/pnas.0809158106; PMID: 19188609
- Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J Clin Invest 2012; 122:1339 - 53; http://dx.doi.org/10.1172/JCI57256; PMID: 22476196
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996; 382:685 - 91; http://dx.doi.org/10.1038/382685a0; PMID: 8751438
- Kook SY, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung IA. Aβ₁₋₄₂-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca²⁺-calcineurin signaling. J Neurosci 2012; 32:8845 - 54; http://dx.doi.org/10.1523/JNEUROSCI.6102-11.2012; PMID: 22745485
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci 2007; 27:796 - 807; http://dx.doi.org/10.1523/JNEUROSCI.3501-06.2007; PMID: 17251419
- Cissé M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, et al. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 2011; 469:47 - 52; http://dx.doi.org/10.1038/nature09635; PMID: 21113149
- Fu W, Ruangkittisakul A, MacTavish D, Shi JY, Ballanyi K, Jhamandas JH. Amyloid β (Aβ) peptide directly activates amylin-3 receptor subtype by triggering multiple intracellular signaling pathways. J Biol Chem 2012; 287:18820 - 30; http://dx.doi.org/10.1074/jbc.M111.331181; PMID: 22500019
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging 2008; 29:1334 - 47; http://dx.doi.org/10.1016/j.neurobiolaging.2007.02.029; PMID: 17403556
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A 2011; 108:5819 - 24; http://dx.doi.org/10.1073/pnas.1017033108; PMID: 21421841
- Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci 2009; 29:9078 - 89; http://dx.doi.org/10.1523/JNEUROSCI.1071-09.2009; PMID: 19605645
- Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem 2010; 285:26377 - 83; http://dx.doi.org/10.1074/jbc.M110.145516; PMID: 20576610
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, et al. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2010; 2:306 - 14; http://dx.doi.org/10.1002/emmm.201000082; PMID: 20665634
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, et al. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci 2010; 30:6367 - 74; http://dx.doi.org/10.1523/JNEUROSCI.0395-10.2010; PMID: 20445063
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, et al. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci 2010; 11:130; http://dx.doi.org/10.1186/1471-2202-11-130; PMID: 20946660
- Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature 2010; 466:E3 - 4, discussion E4-5; http://dx.doi.org/10.1038/nature09217; PMID: 20703260
- Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, et al. Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci 2011; 31:7259 - 63; http://dx.doi.org/10.1523/JNEUROSCI.6500-10.2011; PMID: 21593310
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, et al. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 2012; 15:1227 - 35; http://dx.doi.org/10.1038/nn.3178; PMID: 22820466
- You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, et al. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A 2012; 109:1737 - 42; http://dx.doi.org/10.1073/pnas.1110789109; PMID: 22307640
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, et al. Interaction between prion protein and toxic Aß assemblies can be therapeutically targeted at multiple sites. Nature Commun 2011; 2:336; http://dx.doi.org/10.1038/ncomms1341
- Bate C, Williams A. Amyloid-β-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J Biol Chem 2011; 286:37955 - 63; http://dx.doi.org/10.1074/jbc.M111.248724; PMID: 21900234
- Cissé M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci 2011; 31:10427 - 31; http://dx.doi.org/10.1523/JNEUROSCI.1459-11.2011; PMID: 21775587
- Virok DP, Simon D, Bozsó Z, Rajkó R, Datki Z, Bálint É, et al. Protein array based interactome analysis of amyloid-β indicates an inhibition of protein translation. J Proteome Res 2011; 10:1538 - 47; http://dx.doi.org/10.1021/pr1009096; PMID: 21244100
- Manzoni C, Colombo L, Bigini P, Diana V, Cagnotto A, Messa M, et al. The molecular assembly of amyloid aβ controls its neurotoxicity and binding to cellular proteins. PLoS One 2011; 6:e24909; http://dx.doi.org/10.1371/journal.pone.0024909; PMID: 21966382
- Forloni G, Balducci C. β-amyloid oligomers and prion protein: Fatal attraction?. Prion 2011; 5:10 - 5; http://dx.doi.org/10.4161/pri.5.1.14367; PMID: 21150333
- Westaway D, Jhamandas JH. The P’s and Q’s of cellular PrP-Aβ interactions. Prion 2012; 6:359 - 63; http://dx.doi.org/10.4161/pri.20675; PMID: 22874673
- Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J 2002; 21:3307 - 16; http://dx.doi.org/10.1093/emboj/cdf325; PMID: 12093732
- Martins VR, Beraldo FH, Hajj GN, Lopes MH, Lee KS, Prado MM, et al. Prion protein: orchestrating neurotrophic activities. Curr Issues Mol Biol 2010; 12:63 - 86; PMID: 19767651
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev 2008; 88:673 - 728; http://dx.doi.org/10.1152/physrev.00007.2007; PMID: 18391177
- Turnbaugh JA, Westergard L, Unterberger U, Biasini E, Harris DA. The N-terminal, polybasic region is critical for prion protein neuroprotective activity. PLoS One 2011; 6:e25675; http://dx.doi.org/10.1371/journal.pone.0025675; PMID: 21980526
- Rial D, Piermartiri TC, Duarte FS, Tasca CI, Walz R, Prediger RD. Overexpression of cellular prion protein (PrP(C)) prevents cognitive dysfunction and apoptotic neuronal cell death induced by amyloid-β (Aβ₁₋₄₀) administration in mice. Neuroscience 2012; 215:79 - 89; http://dx.doi.org/10.1016/j.neuroscience.2012.04.034; PMID: 22537845
- Nieznanski K, Choi JK, Chen S, Surewicz K, Surewicz WK. Soluble prion protein inhibits amyloid-β (Aβ) fibrillization and toxicity. J Biol Chem 2012; 287:33104 - 8; http://dx.doi.org/10.1074/jbc.C112.400614; PMID: 22915585
- Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, et al. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem 2001; 276:37743 - 6; PMID: 11477090
- Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The α-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 2009; 284:35973 - 86; http://dx.doi.org/10.1074/jbc.M109.051086; PMID: 19850936
- Guillot-Sestier MV, Sunyach C, Ferreira ST, Marzolo MP, Bauer C, Thevenet A, et al. α-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid β (Aβ)-associated cell death. J Biol Chem 2012; 287:5021 - 32; http://dx.doi.org/10.1074/jbc.M111.323626; PMID: 22184125