Abstract
The structural conversion of the prion protein PrP into a transmissible, misfolded form is the central element of prion disease, yet there is little consensus as to how it occurs. Key aspects of conversion into the diseased state remain unsettled, from details about the earliest stages of misfolding such as the involvement of partially- or fully-unfolded intermediates to the structure of the infectious state. Part of the difficulty in understanding the structural conversion arises from the complexity of the underlying energy landscapes. Single molecule methods provide a powerful tool for probing complex folding pathways as in prion misfolding, because they allow rare and transient events to be observed directly. We discuss recent work applying single-molecule probes to study misfolding in prion proteins, and what it has revealed about the folding dynamics of PrP that may underlie its unique behavior. We also discuss single-molecule studies probing the interactions that stabilize non-native structures within aggregates, pointing the way to future work that may help identify the microscopic events triggering pathogenic conversion. Although single-molecule approaches to misfolding are relatively young, they have a promising future in prion science.
Accessing Early Events in PrP Aggregation
It is widely accepted that the scrapie isoform of the prion protein, PrPSc, is able to reproduce itself by templated conversion of the native isoform, PrPC.Citation1-Citation3 Beyond this general conceptual framework, however, the microscopic details of prion propagation remain unresolved and controversial. The problem of understanding prion replication at a biophysical level can be reduced crudely to two principal questions: (1) what is the structure of PrPSc, and (2) how is PrPC converted to PrPSc? A definitive structure for PrPSc has proven elusive, although various models have been proposed based on a range of biophysical and biochemical evidence.Citation4,Citation5 Efforts to elucidate the structural properties of PrPSc have been reviewed elsewhere,Citation6 and will not be discussed here. Uncertainty about the structure of PrPSc has also made the second question difficult to address. Compounding this difficulty, PrP can aggregate into a wide variety of different forms, including various oligomers as well as amorphous aggregates and amyloids, most of which are not infectious and hence do not correspond to PrPSc.Citation3
To simplify the task of addressing the early events in PrP aggregation, we can re-frame the question in terms of what folding pathways are available to PrP, which of these lead to misfolding and aggregation, and how these pathways change in response to stimuli (such as protein-protein or ligand-protein interactions) that are known to affect conversion. Several studies have characterized oligomeric species involved in the early stages of prion protein aggregation at the ensemble level.Citation7-Citation9 However, since the initial events in conversion are likely rare, involving few molecules, it is challenging to resolve key features of this process using traditional methods, owing to ensemble averaging effects. Furthermore, the conversion from PrPC into aggregates probably involves many different pathways and numerous rare or transient conformations.Citation10 Such intermediates may not be detectable under the relevant conditions; conversely, the conditions needed for them to accumulate sufficiently to be detectable (e.g., the use of chemical denaturants to stabilize non-native states) may make the results difficult to interpret. These difficulties can be addressed by investigating the misfolding of PrP at the level of single molecules, applying techniques that have already proven very useful in providing insight into the formation of native structure in protein folding.Citation11
Studying PrP at the Single-Molecule Level
In the single-molecule regime, individual PrP monomers, oligomers or fibrils can be distinguished from one another as structural sub-populations and characterized independently. A variety of probes is available to monitor the structure, stability, and dynamics of the protein, based most commonly on fluorescence or force (; ). For example, Förster resonance energy transfer (FRET) between co-localized fluorophores is a very sensitive probe of protein conformation and dynamics,Citation12 as shown by an elegant FRET study of the prion-determining NM domain of the yeast prion protein Sup35, which found that Sup35 takes on an ensemble of rapidly-interconverting structures.Citation13 To our knowledge, however, single-molecule FRET has not yet been applied to PrP. Fluorescence probes can also monitor the formation and growth of oligomeric aggregates, for example by measuring the size-dependent diffusion of oligomers via fluorescence correlation spectroscopy (FCS) or the intermolecular FRET signals from differently-labeled proteins as they aggregate. FCS has been used to follow oligomerization in conjunction with ensemble methods, revealing a multi-step process starting with β-structured dimers.Citation14 FCS has also been used to study the interaction of PrP with antibodies, with applications for ultra-sensitive prion detection.Citation15-Citation17
Figure 1. Illustrations of single-molecule techniques. (A) In FRET, the protein is labeled with two dyes, a donor (circle) and acceptor (star), and the donor is excited. Energy transfer to the acceptor is high when the dyes are close (left), low when they are far apart (right). (B) In FCS, fluctuations in the fluorescence of a labeled molecule are measured as it diffuses through a confocal excitation volume and used to determine the diffusion time. (C) In AFM, the tip is used to pull on a molecule tethered to the surface (here, a monomer is pulled out of a fibril). (D) In optical tweezers, beads trapped by laser beams are used to pull on the ends of a protein molecule tethered between them.
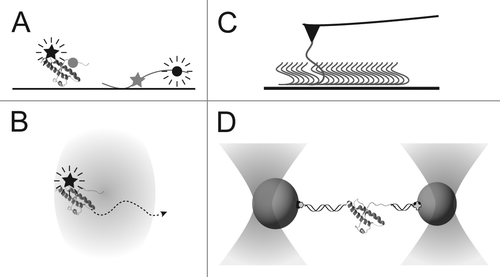
Table 1. Single-molecule techniques used in prion research. The basic principles, measurement methods and sample requirements are outlined for FRET, FCS, AFM and optical tweezers
Single-molecule studies of PrP have also made use of force spectroscopy, in which a compliant force probe like an atomic force microscope (AFM) or optical trap is used to grab onto a protein molecule, apply tension and induce structural changes. Force spectroscopy offers some particular advantages for studying misfolding and aggregation. For example, extremely stable structures such as amyloids can be disrupted with relative ease, and the use of force as a denaturant allows the solution conditions to be maintained constant while denaturing and renaturing the protein. This last point is especially important for PrP, the misfolding and aggregation of which are sensitive to conditions such as pH, temperature or chemical denaturant:Citation10,Citation18-Citation20 Since these conditions are typically used to trigger unfolding/refolding, it is challenging to isolate the effects that relate specifically to misfolding and aggregation. Misfolding can therefore be studied under solution conditions which would not normally be considered conducive to misfolding. Conversely, since the single molecules are measured in isolation, the folding dynamics of individual monomers may be studied under solution conditions in which the protein would normally aggregate rapidly.
The first force spectroscopy study of PrP used AFM to pull apart amyloid fibrils made from recombinant human PrP(90–231).Citation21 Fibrils were adsorbed to a mica surface, and the gold-coated AFM tip was bonded covalently to the thiol group of a cysteine engineered near the N terminus of the protein, thereby pulling on one of the PrP monomers comprising the fibril. Increasing the force on this monomer, part of the protein was found to stretch like an unstructured, worm-like chain (WLC) polymer before the monomer was ultimately plucked out of the fibril core at ~100–200 pN of applied force. By measuring how the force for removing the monomer changed with the pulling speed of the AFM,Citation22 the unfolding of the PrP monomer from the amyloid was shown to share characteristics typical of β-structured proteins, as opposed to the helical structure of PrPC. More interestingly, from the length of the unstructured WLC the authors were able to postulate the location of the edge of the β-sheet core of the amyloid. Their results contradicted the predictions of two structural models of PrPSc 4,5 in which substantial native-like helical structure near the C terminus is retained, but were consistent with models of PrPSc 23 and recombinant amyloidCitation24 in which the native C terminus is completely restructured.
Monomeric PrP Explores Several Folding Pathways
Single-molecule studies of fibrils provide useful clues to the structure of PrP amyloid but do not speak to the question of how the native helical structure is able to convert into a β–rich form. To address the latter question, we recently studied the structural dynamics of isolated PrP monomers with force spectroscopy, in this case using optical tweezersCitation12 as the force probe. Force was applied to the termini of hamster PrP(90–231) (), and the end-to-end extension of the protein was measured with Å-scale spatial resolution and sub-ms scale temporal resolution,Citation25,Citation26 either as a function of the applied force while ramping the force () or as a function of time at constant force ().
Figure 2. Single-molecule force spectroscopy of a single PrP molecule. (A) Experimental scheme using optical traps. Cysteine labeled PrP is attached to DNA handles linked in turn to beads held by optical traps. (B) Force-extension curves show that as the force increases, the handles stretch until the PrP structure unfolds suddenly as a two-state system. WLC fitsCitation26 (dashed lines) to the folded and unfolded parts of the curves reveal a contour length change matching the result expected for PrPC.
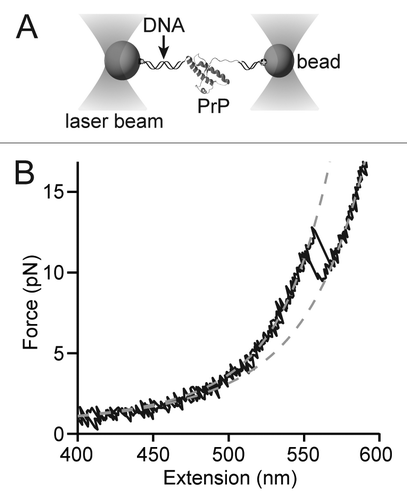
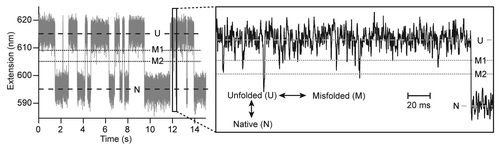
Figure 3. Constant-force trajectories reveal the presence of misfolded states. When PrP is held under constant tension (gray), the extension fluctuates on the second timescale between levels (dashed lines) corresponding to native and unfolded states. On the ms-timescale (black), frequent downward spikes in the extension correspond to formation of misfolded states (dotted lines).
One focus of our work was characterizing the native folding pathway of PrP. A partially-folded on-pathway intermediate has long been proposed to mediate misfolding,Citation27 and partially-native structures are a feature of several models of PrPSc.Citation4,Citation5 However, the experimental evidence for on-pathway intermediates is conflicting: some studies found only two-state folding, e.g., using stopped-flow chemical denaturantCitation28 or temperature jumpCitation29 at pH 7–8, whereas others were consistent with the presence of intermediates at both neutral and acidic pH.Citation30 Using the unique ability of single-molecule methods to observe intermediates directly and determine what pathway they are on,Citation31 we found that at neutral pH and without chemical denaturant there was no intermediate detectable on the native folding pathway having a lifetime longer than 50–100 μs, the limit of our resolution.Citation26 This observation does not completely rule out the possibility of a partially-native intermediate, since an intermediate involving restructuring that leaves the end-to-end extension unchanged, such as rearrangement of helix 1,Citation32 would not be detected in our measurement (although it could be detected by changing the experimental geometry to pull on helix 1 instead of the terminus). It does suggest, however, that the intermediates observed previously may be a function of the particular measurement conditions, such as the presence of chemical denaturants, rather than a general feature of PrP folding.
Looking at the native pathway in more detail, we reconstructed the profile of the energy landscape governing the folding using pulling curves.Citation33 We found a single barrier roughly halfway between the folded and unfolded state, indicating a fairly extended transition stateCitation25 (the unstable, high-energy species separating the folded and unfolded states), in contrast to the transition state located close to the folded state that was found when pulling PrP from an amyloid.Citation21 From the measured extension of the transition state, we estimated that roughly half of the structured part of native PrP was unfolded in the transition state. Based on this result, in combination with the picture of an expanded transition state consisting of native-like contacts between helices 2 and 3 that emerged from phi-value analysis,Citation29 we speculated that the transition state could consist of a structured hydrophobic core such as helix 2, strand 2, and most of helix 3 (with an uncertain degree of native contacts). The same folding barrier is likely being probed with single-molecule force spectroscopy as with the more traditional ensemble approaches, since the folding rates calculated from first principles applying the kinetic theory of KramersCitation34 to the reconstructed landscape agreed well with the rates found by other methods.Citation25
The most distinctive finding from force spectroscopy of monomeric PrP was the discovery of at least three different types of partially-folded intermediates off the native folding pathway. All were unstable (6–11 kcal/mol less stable than PrPC 26), but two formed with higher frequency than the native state during refolding (). Interestingly, this misfolding did not require low pH. PrPSc is often considered to develop in a low pH environment such as endosomes,Citation1 since PrPSc localizes thereCitation35 and low pH encourages misfolding.Citation19,Citation36 However, PrPSc also localizes to the cell surface,Citation35 and it is not evident a priori that the environments in which most PrPSc is found in the cell—likely those leading to the fastest growth of PrPSc—are necessarily the same as those in which the first PrPSc seeds form. Given that PrP explores non-native states at neutral pH, it is possible that seed formation could take place in non-acidic environments such as the endoplasmic reticulum (e.g., after being unfolded during membrane translocation), the cytosol (e.g., after retro-translocationCitation37), or the cell surface.Citation38
The misfolding of monomeric PrP originated solely from the unfolded state,Citation26,Citation31 suggesting that the role of the unfolded state may be under-appreciated.Citation39 Although the misfolded states were quite unstable in wt-PrP, if they are involved in aggregation then they might be stabilized by aggregation-enhancing mutations. This speculation was supported by measurements of the C179A/C214A mutant, which lacks an internal disulfide bond and is highly aggregation-prone in bulk. The most common misfolded states were significantly enhanced in the mutant, suggesting a role as intermediates on the aggregation pathway. Interestingly, these results also showed that, contrary to ensemble folding studies,Citation40 the disulfide bond is not required to attain the native fold: isolated molecules of mutant and wild-type PrP formed the same stable structure, whose length was consistent with PrPC to within ångtröms, with similar rates and stability.Citation26 Indeed, the disulfide bond was almost certainly not formed even for the wild-type PrP, owing to the non-oxidizing conditions of the measurement.Citation26 The mutant’s aggregation propensity is thus likely driven by the differences in the refolding from the unfolded state.
Equally interesting is what was not observed. First, the amyloidogenic part of the unstructured regionCitation41 did not form even transient structure on its own, indicating that any structure in this region must be induced by conditions not present in the measurements, such as interactions with other molecules. Second, we found no evidence for a stable misfolded form of monomeric PrP, as proposed recently.Citation42 Since measurements like those in are in equilibrium, all possible structures are explored during the trajectories, but PrPC was the only stable, long-lived structure observed.Citation26 Hence any alternate structure must either require the truncated residues 23–90 or else specific experimental conditions not found in the single-molecule measurements.
Studying Aggregation in Oligomers at the Single-Molecule Level
While studies of the dynamics of individual PrP molecules give insight into what properties of the protein may drive its unusual behavior, they don’t directly address structural conversion, which requires focusing on the interactions between monomers that lead to aggregates. One approach is to study minimal oligomers such as dimers and trimers. Several lines of evidence support the notion that PrP dimers or trimers play a role in the conversion of PrPC to PrPSc, even though larger oligomers may be more infectious.Citation43 Recombinant PrP forms dimers at low pHCitation18,Citation20 and upon dilution from 0.2 to 0.05% SDS;Citation44,Citation45 brain-derived PrPC can also dimerize in vitro, at least partially.Citation46 Synthetic PrP dimers were shown to be toxic to neurons both in vitroCitation47 and in vivo.Citation48 The crystal structure of human PrP revealed a domain-swapped dimerCitation49 which inspired a subsequent model for oligomerization based on domain swapping.Citation50 Trimers, on the other hand, are the building-blocks of both the spiralCitation4 and β-helixCitation5 model of prion fibrils.
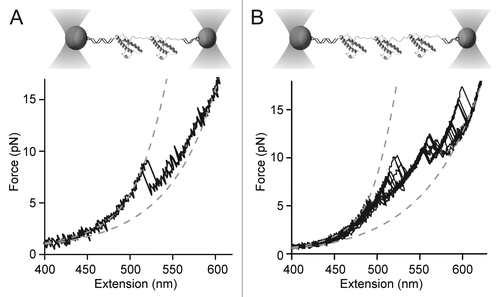
Preliminary measurements of single PrP dimers and trimers that we made using optical tweezers show intriguing results. Oligomers formed by joining monomers together end-to-end via a disulfide bridge were unfolded and refolded similarly to the monomers. Such tandem-repeat oligomers have been measured frequently with force spectroscopy,Citation11 typically showing independent native folding of each monomeric domain, with only infrequent misfolding.Citation51 In the case of PrP, however, dimer unfolding never showed independently-folded native domains. Instead, a single non-native structure unfolded via multiple intermediates, as revealed by the sequence of length changes during the many “rips” in the pulling curves (). PrP trimers also unfolded from non-native structures via multiple intermediates (). The fact that the initial structures were non-native could be determined from the total contour length change (ΔLc) during the unfolding, which relates to the number of structured amino acids. For dimers, ΔLc ~81 nm, corresponding to ~240 structured amino acids, whereas for trimers, ΔLc ~150 nm, corresponding to ~430 structured amino acids. Since hamster PrPC contains only 104 structured amino acids,Citation52 this result implies that the domains interact to form new structures incorporating the formerly unstructured part of the monomer. Whether these structures are the same as those formed by the toxic tandem-repeat dimers studied previouslyCitation48 and how they may be connected to pathogenesis remains to be determined, but single-molecule studies of small oligomers evidently provide rich opportunities for probing structural conversion in PrP.
Figure 4. Force spectroscopy of PrP oligomers. (A) Dimers linked at their termini form non-native structures with multiple intermediates. (B) Trimers form even more complex non-native structures. In each case, WLC fits (dashed lines) indicate non-native contour-length changes.
Future Directions
Single-molecule approaches can be technically challenging and have only recently begun to be applied to prion misfolding, but they provide powerful tools for deciphering the microscopic mechanisms driving the structural conversion of PrP. Several opportunities in particular stand out for future work. First, single-molecule experiments deliver a richly-detailed set of constraints that can be used to build or improve structural models of misfolded PrP as well as to guide models or simulations of the conversion process: distance and energy constraints for the possible structures (including intermediates), the pathways available (native and non-native), transition rates, even information about the barriers. Integration of single-molecule and modeling approaches should therefore prove a very fruitful source of future insight. Second, continued study of small oligomers at the single-molecule level promises to yield detailed information about the pathways for conversion into neurotoxic structures. Single-molecule methods can also play an important role in understanding the structural properties of PrP amyloid, for example by extending the initial force spectroscopy measurements to apply the techniques recently used to map out inter- and intra-molecular interactions in yeast prion amyloid.Citation53 Yet another area of potential application is in determining the mechanism of action of anti-prion agents. Observations of the effects of known anti-prion agents (e.g., tetrapyrrolesCitation54,Citation55 or curcuminCitation56) on the folding of single PrP monomers and oligomers may provide critical insights into their still-unknown mechanisms of action, potentially leading to more effective anti-prion therapies. Single-molecule approaches have much to offer prion studies, and we foresee their increasing deployment to help solve the central scientific questions posed by prions.
| Abbreviations: | ||
| FRET | = | Förster resonance energy transfer |
| FCS | = | fluorescence correlation spectroscopy |
| AFM | = | atomic force microscope |
| WLC | = | worm-like chain |
Acknowledgments
This work was supported by PrioNet Canada, the Alberta Prion Research Institute, Canadian Institute for Health Research, and the nanoWorks program of Alberta Innovates.
References
- Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 2009; 78:177 - 204; http://dx.doi.org/10.1146/annurev.biochem.78.082907.145410; PMID: 19231987
- Cobb NJ, Surewicz WK. Prion diseases and their biochemical mechanisms. Biochemistry 2009; 48:2574 - 85; http://dx.doi.org/10.1021/bi900108v; PMID: 19239250
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol 2011; 3:a006833; http://dx.doi.org/10.1101/cshperspect.a006833; PMID: 21421910
- DeMarco ML, Daggett V. From conversion to aggregation: protofibril formation of the prion protein. Proc Natl Acad Sci U S A 2004; 101:2293 - 8; http://dx.doi.org/10.1073/pnas.0307178101; PMID: 14983003
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed β-helices into trimers. Proc Natl Acad Sci U S A 2004; 101:8342 - 7; http://dx.doi.org/10.1073/pnas.0402254101; PMID: 15155909
- Diaz-Espinoza R, Soto C. High-resolution structure of infectious prion protein: the final frontier. Nat Struct Mol Biol 2012; 19:370 - 7; http://dx.doi.org/10.1038/nsmb.2266; PMID: 22472622
- Jain S, Udgaonkar JB. Evidence for stepwise formation of amyloid fibrils by the mouse prion protein. J Mol Biol 2008; 382:1228 - 41; http://dx.doi.org/10.1016/j.jmb.2008.07.052; PMID: 18687339
- Redecke L, von Bergen M, Clos J, Konarev PV, Svergun DI, Fittschen UE, et al. Structural characterization of β-sheeted oligomers formed on the pathway of oxidative prion protein aggregation in vitro. J Struct Biol 2007; 157:308 - 20; http://dx.doi.org/10.1016/j.jsb.2006.06.013; PMID: 17023178
- Sokolowski F, Modler AJ, Masuch R, Zirwer D, Baier M, Lutsch G, et al. Formation of critical oligomers is a key event during conformational transition of recombinant syrian hamster prion protein. J Biol Chem 2003; 278:40481 - 92; http://dx.doi.org/10.1074/jbc.M304391200; PMID: 12917432
- Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem 2002; 277:21140 - 8; http://dx.doi.org/10.1074/jbc.M111402200; PMID: 11912192
- Borgia A, Williams PM, Clarke J. Single-molecule studies of protein folding. Annu Rev Biochem 2008; 77:101 - 25; http://dx.doi.org/10.1146/annurev.biochem.77.060706.093102; PMID: 18412537
- Greenleaf WJ, Woodside MT, Block SM. High-resolution, single-molecule measurements of biomolecular motion. Annu Rev Biophys Biomol Struct 2007; 36:171 - 90; http://dx.doi.org/10.1146/annurev.biophys.36.101106.101451; PMID: 17328679
- Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc Natl Acad Sci U S A 2007; 104:2649 - 54; http://dx.doi.org/10.1073/pnas.0611503104; PMID: 17299036
- Post K, Pitschke M, Schäfer O, Wille H, Appel TR, Kirsch D, et al. Rapid acquisition of beta-sheet structure in the prion protein prior to multimer formation. Biol Chem 1998; 379:1307 - 17; http://dx.doi.org/10.1515/bchm.1998.379.11.1307; PMID: 9865603
- Bieschke J, Giese A, Schulz-Schaeffer W, Zerr I, Poser S, Eigen M, et al. Ultrasensitive detection of pathological prion protein aggregates by dual-color scanning for intensely fluorescent targets. Proc Natl Acad Sci U S A 2000; 97:5468 - 73; http://dx.doi.org/10.1073/pnas.97.10.5468; PMID: 10805803
- Birkmann E, Schäfer O, Weinmann N, Dumpitak C, Beekes M, Jackman R, et al. Detection of prion particles in samples of BSE and scrapie by fluorescence correlation spectroscopy without proteinase K digestion. Biol Chem 2006; 387:95 - 102; http://dx.doi.org/10.1515/BC.2006.013; PMID: 16497169
- Fujii F, Horiuchi M, Ueno M, Sakata H, Nagao I, Tamura M, et al. Detection of prion protein immune complex for bovine spongiform encephalopathy diagnosis using fluorescence correlation spectroscopy and fluorescence cross-correlation spectroscopy. Anal Biochem 2007; 370:131 - 41; http://dx.doi.org/10.1016/j.ab.2007.07.018; PMID: 17825783
- Gerber R, Tahiri-Alaoui A, Hore PJ, James W. Conformational pH dependence of intermediate states during oligomerization of the human prion protein. Protein Sci 2008; 17:537 - 44; http://dx.doi.org/10.1110/ps.073163308; PMID: 18218718
- Hornemann S, Glockshuber R. A scrapie-like unfolding intermediate of the prion protein domain PrP(121-231) induced by acidic pH. Proc Natl Acad Sci U S A 1998; 95:6010 - 4; http://dx.doi.org/10.1073/pnas.95.11.6010; PMID: 9600908
- O’Sullivan DB, Jones CE, Abdelraheim SR, Thompsett AR, Brazier MW, Toms H, et al. NMR characterization of the pH 4 beta-intermediate of the prion protein: the N-terminal half of the protein remains unstructured and retains a high degree of flexibility. Biochem J 2007; 401:533 - 40; http://dx.doi.org/10.1042/BJ20060668; PMID: 16958619
- Ganchev DN, Cobb NJ, Surewicz K, Surewicz WK. Nanomechanical properties of human prion protein amyloid as probed by force spectroscopy. Biophys J 2008; 95:2909 - 15; http://dx.doi.org/10.1529/biophysj.108.133108; PMID: 18539633
- Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys J 1997; 72:1541 - 55; http://dx.doi.org/10.1016/S0006-3495(97)78802-7; PMID: 9083660
- Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol 2011; 18:504 - 6; http://dx.doi.org/10.1038/nsmb.2035; PMID: 21441913
- Cobb NJ, Sönnichsen FD, McHaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: a parallel, in-register β-structure. Proc Natl Acad Sci U S A 2007; 104:18946 - 51; http://dx.doi.org/10.1073/pnas.0706522104; PMID: 18025469
- Yu H, Gupta AN, Liu X, Neupane K, Brigley AM, Sosova I, et al. Energy landscape analysis of native folding of the prion protein yields the diffusion constant, transition path time, and rates. Proc Natl Acad Sci U S A 2012; 109:14452 - 7; http://dx.doi.org/10.1073/pnas.1206190109; PMID: 22908253
- Yu H, Liu X, Neupane K, Gupta AN, Brigley AM, Solanki A, et al. Direct observation of multiple misfolding pathways in a single prion protein molecule. Proc Natl Acad Sci U S A 2012; 109:5283 - 8; http://dx.doi.org/10.1073/pnas.1107736109; PMID: 22421432
- Cohen FE, Pan KM, Huang Z, Baldwin M, Fletterick RJ, Prusiner SB. Structural clues to prion replication. Science 1994; 264:530 - 1; http://dx.doi.org/10.1126/science.7909169; PMID: 7909169
- Wildegger G, Liemann S, Glockshuber R. Extremely rapid folding of the C-terminal domain of the prion protein without kinetic intermediates. Nat Struct Biol 1999; 6:550 - 3; http://dx.doi.org/10.1038/9323; PMID: 10360358
- Hart T, Hosszu LLP, Trevitt CR, Jackson GS, Waltho JP, Collinge J, et al. Folding kinetics of the human prion protein probed by temperature jump. Proc Natl Acad Sci U S A 2009; 106:5651 - 6; http://dx.doi.org/10.1073/pnas.0811457106; PMID: 19321423
- Jenkins DC, Sylvester ID, Pinheiro TJT. The elusive intermediate on the folding pathway of the prion protein. FEBS J 2008; 275:1323 - 35; http://dx.doi.org/10.1111/j.1742-4658.2008.06293.x; PMID: 18279390
- Hoffmann A, Woodside MT. Signal-pair correlation analysis of single-molecule trajectories. Angew Chem Int Ed Engl 2011; 50:12643 - 6; http://dx.doi.org/10.1002/anie.201104033; PMID: 22057589
- De Simone A, Zagari A, Derreumaux P. Structural and hydration properties of the partially unfolded states of the prion protein. Biophys J 2007; 93:1284 - 92; http://dx.doi.org/10.1529/biophysj.107.108613; PMID: 17483173
- Gupta AN, Vincent A, Neupane K, Yu H, Wang F, Woodside MT. Experimental validation of free-energy-landscape reconstruction from non-equilibrium single-molecule force spectroscopy measurements. Nat Phys 2011; 7:631 - 4; http://dx.doi.org/10.1038/nphys2022
- Hänggi P, Talkner P, Borkovec M. Reaction-Rate Theory - 50 Years after Kramers. Rev Mod Phys 1990; 62:251 - 341; http://dx.doi.org/10.1103/RevModPhys.62.251
- Veith NM, Plattner H, Stuermer CAO, Schulz-Schaeffer WJ, Bürkle A. Immunolocalisation of PrPSc in scrapie-infected N2a mouse neuroblastoma cells by light and electron microscopy. Eur J Cell Biol 2009; 88:45 - 63; http://dx.doi.org/10.1016/j.ejcb.2008.08.001; PMID: 18834644
- Bjorndahl TC, Zhou GP, Liu X, Perez-Pineiro R, Semenchenko V, Saleem F, et al. Detailed biophysical characterization of the acid-induced PrP(c) to PrP(β) conversion process. Biochemistry 2011; 50:1162 - 73; http://dx.doi.org/10.1021/bi101435c; PMID: 21189021
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 2002; 298:1781 - 5; http://dx.doi.org/10.1126/science.1073725; PMID: 12386337
- Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem 1991; 266:18217 - 23; PMID: 1680859
- Gerum C, Silvers R, Wirmer-Bartoschek J, Schwalbe H. Unfolded-State Structure and Dynamics Influence the Fibril Formation of Human Prion Protein. Angew Chem 2009; 121:9616 - 20; http://dx.doi.org/10.1002/ange.200903771
- Maiti NR, Surewicz WK. The role of disulfide bridge in the folding and stability of the recombinant human prion protein. J Biol Chem 2001; 276:2427 - 31; http://dx.doi.org/10.1074/jbc.M007862200; PMID: 11069909
- Kuwata K, Matumoto T, Cheng H, Nagayama K, James TL, Roder H. NMR-detected hydrogen exchange and molecular dynamics simulations provide structural insight into fibril formation of prion protein fragment 106-126. Proc Natl Acad Sci U S A 2003; 100:14790 - 5; . http://dx.doi.org/10.1073/pnas.2433563100; PMID: 14657385
- Zhou M, Ottenberg G, Sferrazza GF, Lasmézas CI. Highly neurotoxic monomeric α-helical prion protein. Proc Natl Acad Sci U S A 2012; 109:3113 - 8; http://dx.doi.org/10.1073/pnas.1118090109; PMID: 22323583
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, et al. The most infectious prion protein particles. Nature 2005; 437:257 - 61; http://dx.doi.org/10.1038/nature03989; PMID: 16148934
- Jansen K, Schäfer O, Birkmann E, Post K, Serban H, Prusiner SB, et al. Structural intermediates in the putative pathway from the cellular prion protein to the pathogenic form. Biol Chem 2001; 382:683 - 91; http://dx.doi.org/10.1515/BC.2001.081; PMID: 11405232
- Kaimann T, Metzger S, Kuhlmann K, Brandt B, Birkmann E, Höltje HD, et al. Molecular model of an α-helical prion protein dimer and its monomeric subunits as derived from chemical cross-linking and molecular modeling calculations. J Mol Biol 2008; 376:582 - 96; http://dx.doi.org/10.1016/j.jmb.2007.11.035; PMID: 18158160
- Meyer RK, Lustig A, Oesch B, Fatzer R, Zurbriggen A, Vandevelde M. A monomer-dimer equilibrium of a cellular prion protein (PrPC) not observed with recombinant PrP. J Biol Chem 2000; 275:38081 - 7; http://dx.doi.org/10.1074/jbc.M007114200; PMID: 10967124
- Roostaee A, Côté S, Roucou X. Aggregation and amyloid fibril formation induced by chemical dimerization of recombinant prion protein in physiological-like conditions. J Biol Chem 2009; 284:30907 - 16; http://dx.doi.org/10.1074/jbc.M109.057950; PMID: 19710507
- Simoneau S, Rezaei H, Salès N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 2007; 3:e125; http://dx.doi.org/10.1371/journal.ppat.0030125; PMID: 17784787
- Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol 2001; 8:770 - 4; http://dx.doi.org/10.1038/nsb0901-770; PMID: 11524679
- Lee S, Eisenberg D. Seeded conversion of recombinant prion protein to a disulfide-bonded oligomer by a reduction-oxidation process. Nat Struct Biol 2003; 10:725 - 30; http://dx.doi.org/10.1038/nsb961; PMID: 12897768
- Oberhauser AF, Marszalek PE, Carrion-Vazquez M, Fernandez JM. Single protein misfolding events captured by atomic force microscopy. Nat Struct Biol 1999; 6:1025 - 8; http://dx.doi.org/10.1038/14907; PMID: 10542093
- James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang H, Donne DG, et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A 1997; 94:10086 - 91; http://dx.doi.org/10.1073/pnas.94.19.10086; PMID: 9294167
- Dong J, Castro CE, Boyce MC, Lang MJ, Lindquist S. Optical trapping with high forces reveals unexpected behaviors of prion fibrils. Nat Struct Mol Biol 2010; 17:1422 - 30; http://dx.doi.org/10.1038/nsmb.1954; PMID: 21113168
- Dee DR, Gupta AN, Anikovskiy M, Sosova I, Grandi E, Rivera L, et al. Phthalocyanine tetrasulfonates bind to multiple sites on natively-folded prion protein. Biochim Biophys Acta 2012; 1824:826 - 32; http://dx.doi.org/10.1016/j.bbapap.2012.03.011; PMID: 22480824
- Caughey WS, Raymond LD, Horiuchi M, Caughey B. Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc Natl Acad Sci U S A 1998; 95:12117 - 22; http://dx.doi.org/10.1073/pnas.95.21.12117; PMID: 9770449
- Caughey B, Raymond LD, Raymond GJ, Maxson L, Silveira J, Baron GS. Inhibition of protease-resistant prion protein accumulation in vitro by curcumin. J Virol 2003; 77:5499 - 502; http://dx.doi.org/10.1128/JVI.77.9.5499-5502.2003; PMID: 12692251