Abstract
In all cell types, protein homeostasis, or “proteostasis,” is maintained by sophisticated quality control networks that regulate protein synthesis, folding, trafficking, aggregation, disaggregation, and degradation. In one notable example, Escherichia coli employ a proteostasis system that determines whether substrates of the twin-arginine translocation (Tat) pathway are correctly folded and thus suitable for transport across the tightly sealed cytoplasmic membrane. Herein, we review growing evidence that the Tat translocase itself discriminates folded proteins from those that are misfolded and/or aggregated, preferentially exporting only the former. Genetic suppressors that inactivate this mechanism have recently been isolated and provide direct evidence for the participation of the Tat translocase in structural proofreading of its protein substrates. We also discuss how this discriminatory “folding sensor” has been exploited for the discovery of structural probes (e.g., sequence mutations, pharmacologic chaperones, intracellular antibodies) that modulate the folding and solubility of virtually any protein-of-interest, including those associated with aggregation diseases (e.g., α-synuclein, amyloid-β protein). Taken together, these studies highlight the utility of engineered bacteria for rapidly and inexpensively uncovering potent anti-aggregation factors.
Maintenance of proteome integrity (proteostasis) is essential for cellular and organismal survival, and represents a major challenge across all kingdoms of life. Proteostasis involves highly integrated cellular networks that generate and protect the protein fold.Citation1 Even in simple organisms, such as Escherichia coli, a sophisticated protein quality control (QC) system works to preserve proteostasis by orchestrating protein synthesis, folding, degradation and trafficking. At the heart of this network are molecular chaperones and proteases that prevent or reverse protein misfolding and aggregation, enhance the de novo folding efficiency of newly synthesized proteins, and remove misfolded and aggregated proteins by degradation.Citation2
In addition to keeping protein aggregation in check, chaperones also have essential roles in assisting the membrane transport of newly synthesized polypeptides. A significant fraction (~34%) of the E. coli proteome is localized partially or completely outside of the cytosol,Citation3 which requires insertion into or passage across at least one hydrophobic lipid bilayer membrane. In many instances, the process of membrane translocation is dependent on proper structural integrity of the protein to be transported. For example, the translocase of the Sec protein export pathway provides an aqueous channel that is approximately the same width as a polypeptide chain (estimated as 15–20 Å on the basis of the crystal structure).Citation4 Given such a narrow pore, the translocase can tolerate polypeptides that form an α-helix but not tertiary structure; hence, Sec substrates must be transported in an unfolded state.Citation4,Citation5 The task of preventing premature folding of Sec substrates prior to translocation is performed in part by a chaperone network, which in E. coli consists of GroEL, SecB and trigger factor.Citation6,Citation7 These chaperones bind Sec substrates during or just after translation and provide an important QC layer to the Sec pathway by effectively maintaining the polypeptide chains in a conformation suitable for transport and preventing illicit interactions between these unfolded polypeptides which could lead to aggregation.
In stark contrast to the threading of unfolded substrates through the Sec translocase, the twin-arginine translocation (Tat) pathway has the unique ability to transport structurally diverse proteins that have already folded in the cytoplasm prior to membrane translocation (reviewed in ref.Citation8 and elsewhere). The difficulty of this task is underscored by the fact that only one other protein transport system in nature, namely the peroxisomal import pathway, is known to exhibit this capability with a similarly diverse set of substrate proteins. The remarkable feat of transporting prefolded Tat substrates is performed by a translocase that is completely distinct from the Sec machinery. In E. coli, this translocase consists of three integral membrane proteins, one from each of the TatA, TatB and TatC families. While the details of how this translocase works are incompletely understood, single-particle electron microscopyCitation9,Citation10 along with the solution NMR structure of TatACitation11 and the recently reported crystal structure of TatC solved at a resolution of 3.5 Å12 are helping to shed new light on the protein transport mechanism. Even without a complete mechanistic description, the Tat translocase’s versatility is firmly established on the basis of the size and types of proteins that utilize the Tat pathway. For example, in addition to soluble periplasmic proteins, the Tat translocase exports lipoproteinsCitation13 and membrane proteins that are integrated into the inner and outer membranes.Citation14,Citation15 The majority of these substrates are complex cofactor-containing redox proteins, such as the model [NiFe] hydrogenase-2 system, that function in anaerobic respiration and whose cofactors must be correctly assembled before transport can proceed. Furthermore, whereas the Sec translocase exports only unfolded substrates that have a nearly constant cross-sectional area, the Tat translocase is able to transport substrates with a range of diameters (typically between 20 and 70 Å but also much smaller in some engineered systems). In fact, some of the largest Tat substrates are not single polypeptides but rather hetero-oligomeric complexes in which only one of the protein subunits possesses an N-terminal Tat-targeting signal peptide and the other protein subunit is co-translocated by virtue of its assembly with the signal peptide-bearing partner in the cytoplasm. The prototypical example of this “hitchhiker” mode of export is the [NiFe] hydrogenase-2 complex whereby the HybC subunit is co-translocated with its partner HybO in a process mediated by the signal peptide of the latter subunit.Citation16
It should be stressed that, in most instances, use of the Tat pathway appears to have evolved because of an inherent incompatibility between the substrate and the Sec export mechanism. This can occur if, as discussed above, the substrate needs to assemble subunitsCitation16 or incorporate cofactorsCitation17 in the cytoplasm prior to export. Because the synthesis and maturation of certain cofactors (e.g., metal-sulfur clusters, cofactors containing a nucleotide moiety) occur in the cytoplasm, Tat targeting allows proteins to acquire their cofactor before transport across the cytoplasmic membrane. Why certain other proteins such as SufI, an archetypal Tat substrate of unknown function, and the periplasmic amidases AmiA and AmiC, which are neither exported as multimers nor contain cofactors, have evolved to utilize the Tat pathway is far less obvious. It has been widely speculated that these substrates transit the Tat pathway because they require cytoplasmic factors that assist with protein folding or maturation, have difficulties in achieving a biologically active conformation outside of the cytoplasm, and/or are unable to maintain an unfolded conformation prior to export.Citation8
Some cofactor-containing Tat substrates undergo chaperone-mediated “proofreading” to ensure that they are correctly assembled (and any co-exported partner proteins are bound as in the case of HybOC) before they interact with the translocase. Proofreading associated with the Tat system involves dedicated chaperones such as DmsD, HybE, NapD and TorD that coordinate cofactor insertion and substrate assembly with substrate translocationCitation18,Citation19 and general molecular chaperones (e.g., DnaK, SlyD) that affect the stability and targeting of certain substrates.Citation20,Citation21 One notable example is the HybE chaperone, which effectively blocks the export of unassembled HybO lacking its partner HybC by interacting with the precursor forms of both HybO and HybC but not with the mature form of each subunit.Citation19 Likewise, the NapD chaperone binds tightly and specifically to a region of the twin-arginine signal peptide of NapA that overlaps significantly with the twin-arginine targeting motif.Citation22 As a result of its tight binding, NapD invokes a strong “antitransport” activity such that transport of NapA via the Tat pathway is retarded. Given these and other related examples, it is clear that chaperone binding is an essential proofreading step that prevents native Tat substrates from prematurely interacting with the Tat translocase.
In addition to carrying out chaperone-mediated proofreading, the Tat pathway has an intrinsic capacity to differentiate between folded and mis/unfolded substrate proteins by a process termed folding QC. Indeed, growing evidence indicates that in vivo only correctly folded proteins are exported by the Tat translocase,Citation16,Citation23-Citation27 with only a few rare exceptions reported in plant thylakoidsCitation28,Citation29 or in bacteria where the levels of the TatABC proteins are artificially increased by 50-fold.Citation30 In one striking example, E. coli alkaline phosphatase (PhoA) modified with a functional Tat signal peptide was only exported when its native disulfide bonds had been formed to generate the correctly folded molecule.Citation23 In the absence of these bonds, Tat-targeted PhoA was not exported out of the cytoplasm. Hence, not only can the Tat pathway accommodate folded proteins, but it can also discriminate against misfolded proteins. Other proteins whose folding is dependent on the formation of disulfide bonds, such as single-chain Fv (scFv) and FAB antibody fragments, are discriminated in a similar fashion. In fact, the rate of scFv folding is a critical determinant of Tat export efficiency, with faster folding scFv antibodies undergoing more efficient translocation than their slower folding counterparts.Citation31 Likewise, thioredoxin-1, a protein that exhibits very fast folding kinetics, is exported by the Tat translocase with very high efficiency.Citation31 This is in stark contrast to the very inefficient export of thioredoxin-1 when it is fused to a signal peptide that directs post-translational Sec export.Citation32 These observations have led to speculation that Tat export favors folding properties that are diametrically opposite of those required for Sec export.
An interesting observation made by two separate groups is that Tat-targeted PhoA, which fails to be translocated, is still able to reach the Tat translocase.Citation33,Citation34 This implies that discrimination of the PhoA folding state occurs after targeting to the translocase. In support of this hypothesis, the molecular contacts between misfolded PhoA and the TatBC components of the translocase were notably different from the contacts observed between TatBC and correctly folded PhoA.Citation34 It is possible that these differential contacts reflect active discrimination of folded and mis/unfolded substrates by the Tat translocase. If this interpretation is correct, then folding QC would be an inherent property of the Tat translocase. To test this hypothesis, we recently performed a search for genetic suppressors that inactivate Tat translocase-mediated QC and permit export of the otherwise export-defective proteins.Citation25 We identified several genetic suppressors that export a misfolded protein called α3B, a designed three-helix-bundle protein that lacks a uniquely folded structure and is thus not tolerated by the wild-type (wt) translocase. Importantly, the isolation of suppressors that inactivated the Tat QC mechanism provides direct evidence for the participation of the Tat translocase in structural proofreading of substrate proteins and reveals epitopes in the translocase that are important for this process. Based on the clustering of suppressor mutations in the membrane-extrinsic domain of TatB (residues 90–140) and the first cytoplasmic loop of TatC between predicted transmembrane helices II and III (TM2 and TM3; residues 94–105), it is tempting to speculate that hydrophobic surface patches on nonnative proteins might be recognized by a “folding sensor” formed by these cytoplasmic domains of TatB and TatC. Indeed, a conceptual model based on the recent crystal structure of TatC suggests that the amphipathic helix of TatB could run across the cytoplasmic face of TatC from the TatB transmembrane helix binding site at TM5 to the signal peptide-binding site at TM1.Citation12
Even though a complete molecular-level understanding of Tat QC is far from complete, this hasn’t prevented the opportunistic use of this mechanism to study and engineer protein folding in living cells. For example, we have translated the folding sensor of the Tat translocase into an enabling technology for quantifying protein stability and identifying mutations that stabilize proteins.Citation24 This involved the creation of a genetic selection that directly links the in vivo stability of proteins to antibiotic resistance. The design principle for this genetic assay is based on a tripartite fusion whereby a test protein is inserted between an N-terminal Tat export signal (ssTorA) and a C-terminal TEM1 β-lactamase (Bla) reporter enzyme. This selection system provides a unique strategy for directly quantifying protein stability in the bacterial cytoplasm, as we showed recently for a wide array of prokaryotic and eukaryotic test proteins.Citation24,Citation35 The assay has also been used to directly select for more stable variants of a given test protein simply by demanding bacterial growth on an antibiotic.Citation24,Citation36,Citation37 Such selections offer a new route to understanding the fundamental basis of protein stability and expression in a cellular context and require no knowledge of the test protein’s structure or function. In one notable example, we used this strategy to isolate mutations that reduced the aggregation of the 42-residue amyloid-β-peptideCitation24 (Aβ42; ), which is the primary component of amyloid fibrils found in the brains of Alzheimer patients. Cells expressing a tripartite fusion with the aggregation-prone wt Aβ42 sequence as the test protein were sensitive to low levels of antibiotic; however, cells expressing library-selected Aβ42 variants, which carried amino acid substitutions that decreased Aβ42 aggregation, were highly resistant to antibiotic. The most frequently selected mutations (e.g., F19S, L34P) were consistent with previous models implicating hydrophobic regions as determinants of Aβ42 aggregation.
Figure 1. Use of the Tat folding sensor for uncovering structural probes of protein misfolding and aggregation. (A) Chimeras comprised of aggregation-prone test proteins (e.g., ssTorA-Aβ42-Bla) expressed in the cytoplasm readily aggregate via self-assembly of the test proteins. This aggregation is “sensed” by the Tat translocase, and export of these aggregated substrates is blocked. (B) In the presence of stabilizing mutations or exogenous factors such as chemical or protein chaperones that bind to the test protein and prevent aggregation, chimeras such as ssTorA-Aβ42-Bla remain soluble and are efficiently exported to the periplasm where Bla hydrolyzes Amp. The extent to which protein chimeras are exported, and thus the amount of Bla that is transferred to the periplasm, determines the resistance phenotype of the host cells. In this fashion, in vivo stability of proteins is directly linked to antibiotic resistance. The approach facilitates the identification of mutations and other factors (e.g., chemical probes, antibodies) that stabilize test proteins, all without any prior structural or functional knowledge of the test protein.
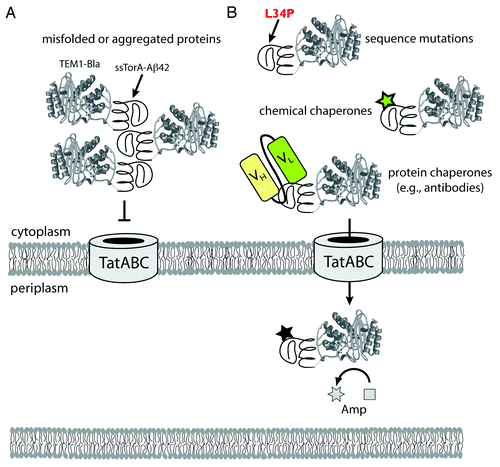
The same genetic assay can also be used to identify small-molecule compounds that similarly modulate protein aggregation (). To demonstrate this concept, we screened a library of triazine derivatives and isolated several nontoxic, cell-permeable, chaperone-like compounds that promoted efficient Tat-dependent export of the ssTorA-Aβ42-Bla chimera.Citation38 Each of these compounds was subsequently shown to be a bona fide inhibitor of Aβ42 aggregation using a standard thioflavin T fibrillization assay, indicating that similar principles govern stability in vivo and in vitro. Taken together, these results highlight the utility of the Tat folding sensor as a unique tool for quantifying protein stability in living cells, engineering stability-enhanced proteins, and isolating factors (e.g., mutations, chemical probes) that stabilize otherwise aggregation-prone proteins.
The Tat folding sensor together with the hitchhiker mechanism can be exploited for genetic selection of well-expressed, stable, antigen-specific, and intracellular functional antibodies.Citation39,Citation40 This strategy, termed FLI-TRAP (functional ligand-binding identification by Tat-based recognition of associating proteins), is based on the unique ability of the Tat translocase to efficiently co-localize non-covalent complexes of 2-folded polypeptides to the periplasmic space of E. coli.Citation16 The design principle for this genetic assay involves two engineered fusion proteins: an N-terminal Tat signal peptide fused to a receptor protein, and the corresponding ligand fused to Bla. The efficiency with which different ligand-Bla chimeras are co-localized to the periplasm is linked to both the stability of the chimeras and the relative binding affinity of the receptor-ligand system. Thus, antibiotic resistance can be used as a phenotypic readout for identifying and optimizing receptor-ligand (e.g., antibody-antigen) interactions in living E. coli cells.
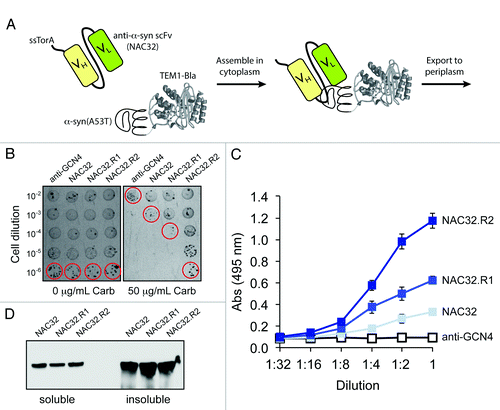
Because the Tat translocase precludes export of aggregated proteins and protein complexes, this strategy can be used to isolate binding proteins that prevent or reverse the aggregation of the target ligand (). To test this notion, we extended FLI-TRAP for selection of intracellular antibodies (intrabodies) that target the α-synuclein (α-syn) protein (). We chose this target because intrabody-mediated prevention of abnormal misfolding and aggregation of α-syn in vulnerable neurons has been proposed as a viable therapeutic strategy for reducing pathogenesis in Parkinson disease. For the antibody component of the assay, we generated a fusion between the ssTorA signal peptide and a previously isolated anti-α-syn scFv intrabody called NAC32,Citation41 which recognizes the nonamyloid component (NAC) region of α-syn that shows strong tendencies to form β-sheet structures. For the antigen component of the assay, a second chimera was generated by fusing the Bla enzyme with α-syn(A53T),Citation41 a highly toxic version of α-syn. Co-expression of these chimeras in E. coli cells conferred a low level of antibiotic resistance, which was slightly above the background resistance conferred by co-expression of a non-specific anti-GCN4 scFv intrabody (). To identify mutations that either improved NAC32 stability or increased affinity of NAC32 for α-syn(A53T), we screened an error-prone PCR library of wt NAC32 sequences for clones that conferred greater antibiotic resistance to cells. Following one round of mutagenesis and selection, clone NAC32.R1 was isolated which carried three mutations (C24S/G141D/S207I; Fig. S1). A second round of mutagenesis and selection yielded clone NAC32.R2, which acquired four additional mutations (V119S/S143T/Q186L/T200S; Fig. S1) that bestowed significantly greater resistance to cells. This increased resistance was attributed to the > 3-fold improvement in antigen binding activity (). The expression of this clone was nearly identical to that observed for the wt NAC32 intrabody (), implying that the binding affinity of NAC32.R2 for α-syn(A53T) was increased. Importantly, our NAC32 study shows that FLI-TRAP, and the underlying Tat folding sensor, is a powerful new tool for isolating specific binders with high affinity and selectivity for aggregation-prone target proteins. We anticipate that this technology platform will spawn a new generation of engineered antibody fragments for use as possible therapeutic leads or in the development of small molecules toward relevant epitopes, most importantly when there are no specific small-molecule lead candidates available.
Figure 2. Use of the Tat folding sensor combined with hitchhiker mechanism to select intracellular antibodies. (A) Schematic representation of engineered assay for co-translocation of interacting receptor-ligand pairs via the Tat translocase. The Tat signal peptide chosen was ssTorA, the reporter enzyme was Bla, and the receptor-ligand pair was the anti-α-syn scFv intrabody NAC32 and the α-syn(A53T) protein. (B) Selective plating of E. coli cells co-expressing α-syn(A53T)-Bla with ssTorA-NAC32 or library-selected NAC32 derivatives. Overnight cultures were serially diluted in liquid LB and plated on LB agar supplemented with carbenicillin (Carb). Clone NAC32.R1 was derived from wt NAC32 following an initial round of error-prone PCR mutagenesis and selection on 50 μg/mL Carb. A second round of error-prone PCR mutagenesis, using NAC32.R1 as template, and selection on 100 μg/mL Carb was used to isolate NAC32.R2. For sequences of these clones, see Figure S1. (C) Antigen binding activity and (D) western blot analysis of NAC32 and library-selected derivatives. NAC32 and its derivatives were expressed from plasmid pET-28a without the ssTorA signal peptide and with a C-terminal FLAG epitope tag. Binding activity in cell lysates was measured by ELISA with microtiter plates coated with α-syn(A53T) as antigen. Western blot analysis of soluble and insoluble fractions was according to standard protocols. Detection was with anti-FLAG antibodies.
Additional material
Download Zip (475.4 KB)Related Research Data
References
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science 2008; 319:916 - 9; http://dx.doi.org/10.1126/science.1141448; PMID: 18276881
- Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 2009; 16:574 - 81; http://dx.doi.org/10.1038/nsmb.1591; PMID: 19491934
- Horler RS, Butcher A, Papangelopoulos N, Ashton PD, Thomas GH. EchoLOCATION: an in silico analysis of the subcellular locations of Escherichia coli proteins and comparison with experimentally derived locations. Bioinformatics 2009; 25:163 - 6; http://dx.doi.org/10.1093/bioinformatics/btn596; PMID: 19015139
- Van den Berg B, Clemons WM Jr., Collinson I, Modis Y, Hartmann E, Harrison SC, et al. X-ray structure of a protein-conducting channel. Nature 2004; 427:36 - 44; http://dx.doi.org/10.1038/nature02218; PMID: 14661030
- Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007; 450:663 - 9; http://dx.doi.org/10.1038/nature06384; PMID: 18046402
- Collier DN, Bankaitis VA, Weiss JB, Bassford PJ Jr.. The antifolding activity of SecB promotes the export of the E. coli maltose-binding protein. Cell 1988; 53:273 - 83; http://dx.doi.org/10.1016/0092-8674(88)90389-3; PMID: 2834066
- Lecker S, Lill R, Ziegelhoffer T, Georgopoulos C, Bassford PJ Jr., Kumamoto CA, et al. Three pure chaperone proteins of Escherichia coli--SecB, trigger factor and GroEL--form soluble complexes with precursor proteins in vitro. EMBO J 1989; 8:2703 - 9; PMID: 2531087
- Palmer T, Berks BC. The twin-arginine translocation (Tat) protein export pathway. Nat Rev Microbiol 2012; 10:483 - 96; PMID: 22683878
- Tarry MJ, Schäfer E, Chen S, Buchanan G, Greene NP, Lea SM, et al. Structural analysis of substrate binding by the TatBC component of the twin-arginine protein transport system. Proc Natl Acad Sci U S A 2009; 106:13284 - 9; http://dx.doi.org/10.1073/pnas.0901566106; PMID: 19666509
- Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, et al. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci U S A 2005; 102:10482 - 6; http://dx.doi.org/10.1073/pnas.0503558102; PMID: 16027357
- Hu Y, Zhao E, Li H, Xia B, Jin C. Solution NMR structure of the TatA component of the twin-arginine protein transport system from gram-positive bacterium Bacillus subtilis. J Am Chem Soc 2010; 132:15942 - 4; http://dx.doi.org/10.1021/ja1053785; PMID: 20726548
- Rollauer SE, Tarry MJ, Graham JE, Jääskeläinen M, Jäger F, Johnson S, et al. Structure of the TatC core of the twin-arginine protein transport system. Nature 2012; 492:210 - 4; http://dx.doi.org/10.1038/nature11683; PMID: 23201679
- Gralnick JA, Vali H, Lies DP, Newman DK. Extracellular respiration of dimethyl sulfoxide by Shewanella oneidensis strain MR-1. Proc Natl Acad Sci U S A 2006; 103:4669 - 74; http://dx.doi.org/10.1073/pnas.0505959103; PMID: 16537430
- Rondelet A, Condemine G. SurA is involved in the targeting to the outer membrane of a Tat signal sequence-anchored protein. J Bacteriol 2012; 194:6131 - 42; http://dx.doi.org/10.1128/JB.01419-12; PMID: 22961852
- Hatzixanthis K, Palmer T, Sargent F. A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase. Mol Microbiol 2003; 49:1377 - 90; http://dx.doi.org/10.1046/j.1365-2958.2003.03642.x; PMID: 12940994
- Rodrigue A, Chanal A, Beck K, Müller M, Wu LF. Co-translocation of a periplasmic enzyme complex by a hitchhiker mechanism through the bacterial tat pathway. J Biol Chem 1999; 274:13223 - 8; http://dx.doi.org/10.1074/jbc.274.19.13223; PMID: 10224080
- Matos CF, Robinson C, Di Cola A. The Tat system proofreads FeS protein substrates and directly initiates the disposal of rejected molecules. EMBO J 2008; 27:2055 - 63; http://dx.doi.org/10.1038/emboj.2008.132; PMID: 18615097
- Oresnik IJ, Ladner CL, Turner RJ. Identification of a twin-arginine leader-binding protein. Mol Microbiol 2001; 40:323 - 31; http://dx.doi.org/10.1046/j.1365-2958.2001.02391.x; PMID: 11309116
- Dubini A, Sargent F. Assembly of Tat-dependent [NiFe] hydrogenases: identification of precursor-binding accessory proteins. FEBS Lett 2003; 549:141 - 6; http://dx.doi.org/10.1016/S0014-5793(03)00802-0; PMID: 12914940
- Pérez-Rodríguez R, Fisher AC, Perlmutter JD, Hicks MG, Chanal A, Santini CL, et al. An essential role for the DnaK molecular chaperone in stabilizing over-expressed substrate proteins of the bacterial twin-arginine translocation pathway. J Mol Biol 2007; 367:715 - 30; http://dx.doi.org/10.1016/j.jmb.2007.01.027; PMID: 17280684
- Graubner W, Schierhorn A, Brüser T. DnaK plays a pivotal role in Tat targeting of CueO and functions beside SlyD as a general Tat signal binding chaperone. J Biol Chem 2007; 282:7116 - 24; http://dx.doi.org/10.1074/jbc.M608235200; PMID: 17215254
- Grahl S, Maillard J, Spronk CA, Vuister GW, Sargent F. Overlapping transport and chaperone-binding functions within a bacterial twin-arginine signal peptide. Mol Microbiol 2012; 83:1254 - 67; http://dx.doi.org/10.1111/j.1365-2958.2012.08005.x; PMID: 22329966
- DeLisa MP, Tullman D, Georgiou G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A 2003; 100:6115 - 20; http://dx.doi.org/10.1073/pnas.0937838100; PMID: 12721369
- Fisher AC, Kim W, DeLisa MP. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci 2006; 15:449 - 58; http://dx.doi.org/10.1110/ps.051902606; PMID: 16452624
- Rocco MA, Waraho-Zhmayev D, DeLisa MP. Twin-arginine translocase mutations that suppress folding quality control and permit export of misfolded substrate proteins. Proc Natl Acad Sci U S A 2012; 109:13392 - 7; http://dx.doi.org/10.1073/pnas.1210140109; PMID: 22847444
- Brüser T, Yano T, Brune DC, Daldal F. Membrane targeting of a folded and cofactor-containing protein. Eur J Biochem 2003; 270:1211 - 21; http://dx.doi.org/10.1046/j.1432-1033.2003.03481.x; PMID: 12631279
- Sanders C, Wethkamp N, Lill H. Transport of cytochrome c derivatives by the bacterial Tat protein translocation system. Mol Microbiol 2001; 41:241 - 6; http://dx.doi.org/10.1046/j.1365-2958.2001.02514.x; PMID: 11454216
- Cline K, McCaffery M. Evidence for a dynamic and transient pathway through the TAT protein transport machinery. EMBO J 2007; 26:3039 - 49; http://dx.doi.org/10.1038/sj.emboj.7601759; PMID: 17568769
- Hynds PJ, Robinson D, Robinson C. The sec-independent twin-arginine translocation system can transport both tightly folded and malfolded proteins across the thylakoid membrane. J Biol Chem 1998; 273:34868 - 74; http://dx.doi.org/10.1074/jbc.273.52.34868; PMID: 9857014
- Richter S, Lindenstrauss U, Lücke C, Bayliss R, Brüser T. Functional Tat transport of unstructured, small, hydrophilic proteins. J Biol Chem 2007; 282:33257 - 64; http://dx.doi.org/10.1074/jbc.M703303200; PMID: 17848553
- Ribnicky B, Van Blarcom T, Georgiou G. A scFv antibody mutant isolated in a genetic screen for improved export via the twin arginine transporter pathway exhibits faster folding. J Mol Biol 2007; 369:631 - 9; http://dx.doi.org/10.1016/j.jmb.2007.03.068; PMID: 17462668
- Huber D, Cha MI, Debarbieux L, Planson AG, Cruz N, López G, et al. A selection for mutants that interfere with folding of Escherichia coli thioredoxin-1 in vivo. Proc Natl Acad Sci U S A 2005; 102:18872 - 7; http://dx.doi.org/10.1073/pnas.0509583102; PMID: 16357193
- Richter S, Brüser T. Targeting of unfolded PhoA to the TAT translocon of Escherichia coli. J Biol Chem 2005; 280:42723 - 30; http://dx.doi.org/10.1074/jbc.M509570200; PMID: 16263723
- Panahandeh S, Maurer C, Moser M, DeLisa MP, Müller M. Following the path of a twin-arginine precursor along the TatABC translocase of Escherichia coli. J Biol Chem 2008; 283:33267 - 75; http://dx.doi.org/10.1074/jbc.M804225200; PMID: 18836181
- Lim HK, Mansell TJ, Linderman SW, Fisher AC, Dyson MR, DeLisa MP. Mining mammalian genomes for folding competent proteins using Tat-dependent genetic selection in Escherichia coli. Protein Sci 2009; 18:2537 - 49; http://dx.doi.org/10.1002/pro.262; PMID: 19830686
- Fisher AC, DeLisa MP. Efficient isolation of soluble intracellular single-chain antibodies using the twin-arginine translocation machinery. J Mol Biol 2009; 385:299 - 311; http://dx.doi.org/10.1016/j.jmb.2008.10.051; PMID: 18992254
- Fisher AC, Rocco MA, DeLisa MP. Genetic selection of solubility-enhanced proteins using the twin-arginine translocation system. Methods Mol Biol 2011; 705:53 - 67; http://dx.doi.org/10.1007/978-1-61737-967-3_4; PMID: 21125380
- Lee LL, Ha H, Chang YT, DeLisa MP. Discovery of amyloid-beta aggregation inhibitors using an engineered assay for intracellular protein folding and solubility. Protein Sci 2009; 18:277 - 86; http://dx.doi.org/10.1002/pro.33; PMID: 19177561
- Waraho D, DeLisa MP. Versatile selection technology for intracellular protein-protein interactions mediated by a unique bacterial hitchhiker transport mechanism. Proc Natl Acad Sci U S A 2009; 106:3692 - 7; http://dx.doi.org/10.1073/pnas.0704048106; PMID: 19234130
- Waraho D, DeLisa MP. Identifying and optimizing intracellular protein-protein interactions using bacterial genetic selection. Methods Mol Biol 2012; 813:125 - 43; http://dx.doi.org/10.1007/978-1-61779-412-4_7; PMID: 22083739
- Lynch SM, Zhou C, Messer A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J Mol Biol 2008; 377:136 - 47; http://dx.doi.org/10.1016/j.jmb.2007.11.096; PMID: 18237741