Abstract
Prion diseases are associated with misfolding of the natively α-helical prion protein into isoforms that are rich in cross β-structure. However, both the mechanism by which pathological conformations are produced and their structural properties remain unclear. Using a combination of nuclear magnetic resonance spectroscopy, computation, hydroxyl radical probing combined with mass-spectrometry and site-directed mutagenesis, we showed that prion stop mutants that accumulate in amyloidogenic plaque-forming aggregates fold into a β-helix. The polymorphic residue 129 is located in the hydrophobic core of the β-helix in line with a critical role of the 129 region in the packing of protein chains into prion particles. Together with electron microscopy our data support a trimeric left-handed β-helix model in which the trimer interface is formed by residues L125, Y128 and L130. Different prion types or strains might be related to different aggregate structures or filament assemblies.
Keywords: :
A hallmark of various human and animal diseases known as transmissible spongiform encephalopathies is the misfolding of the prion protein into β-sheet rich aggregates.Citation1 Prion diseases can be caused by either spontaneous aggregation of the endogenous prion protein [sporadic Creutzfeldt-Jakob disease (CJD)] or by mutations within the prnp gene (familial CJD, Gerstmann-Sträussler-Scheinker (GSS)-like phenotype, Fatal familial insomnia).Citation2 The mutations include naturally occurring truncated variants, Y145Stop and Q160Stop that result in a GSS-like phenotype. Patients carrying the Q160Stop mutation are prone to earlier onset of disease.Citation3 A consistent feature of the GSS-syndrome is extensive accumulation of amyloid fibrils.Citation4 In addition, it was found that the prion stop mutants aggregate in vitro under non-denaturing conditions.Citation5,Citation6 The high molecular weight of the aggregates, however, makes elucidation of their structure difficult.
Recently we showed that the Y145Stop and Q160Stop stop mutants of the human prion protein (humPrP) share a common structure in the fibrillar state.Citation7,Citation8 The amyloid core of both mutants is formed by a small, hydrogen-bonded region comprising residues 109–142.Citation7,Citation9 Residues 144–156, which form helix1 in the native structure of humPrP,Citation10 are not part of this core, in agreement with antibody staining.Citation6 Moreover, solvent exchange measurements on the designed mutant N174Stop demonstrated that residues 160QVYY163, which form β-strand 2 in the native PrP structure, do not convert to hydrogen-bonded structure upon aggregation of this mutant into amyloid fibrils. The identified amyloid core is in line with a PrP fragment of ~7 kDa, which spans residues 81–82 to 144–153 and which was detected in aggregates purified from the Y145Stop and Q227Stop variants,Citation11,Citation12 as well as from GSS-brains.Citation13
The amyloid core of stop mutants of humPrP is folded into a β-helix conformation forming a tight hydrophobic core ().Citation7,Citation8 The β-helix comprises three β-strands. The N-terminal strand is formed by residues 111HMAGA115. The end of this strand together with the following turn consists of the palindromic sequence 113AGAAAAGA120. Subsequently, a small β-strand made up of residues 120AVVG123 follows. The second turn is formed by the poly-glycine motif 124GLGG127 that harbours the hydrophobic residue L125 at its center. The β-helix fold is then completed by a third β-strand comprising 128YMLGSAMSR136. At P137 this strand twists such that residues 138IIHF141 extend the β-helix. The location of the β-helix maps to the most hydrophobic part of the Q160Stop mutant (). Deeply buried in the hydrophobic core is the polymorphic residue M129 (). In line with the burial of the side chain of residue 129, substitution of methionine by asparagine completely inhibited both spontaneous and seeded aggregation, while valine at position 129 resulted in a strong enhancement of aggregation.Citation7 In addition, when the heterozygous situation was modeled by mixing M129 and V129 humPrP(108–143) peptides at equimolar ratios, a longer nucleation phase than in any of the two pure peptides was observed. Thus, humans that are heterozygote at codon 129 might be relatively protected from prion diseases. A left-handed β-helix structure was also suggested for infectious prion (PrPSc) on the basis of electron crystallography of two-dimensional crystals of N-terminally truncated PrPSc.Citation14,Citation15
Figure 1. Amyloid core structure of stop mutants of the human prion protein. (A) Backbone fold of the amyloid core obtained by a combination of solid-state NMR chemical shifts and CS-Rosetta modeling.Citation7,Citation9,Citation27 Residues 111–141 form a left-handed β-helix. The deeply buried side chain of M129 and the side chains of residues I138 and I139 are highlighted. In addition, the location of the palindromic sequence is marked. Backbone amide protons of residues 121–139, which were found to have high solvent protection values (> 70%) in amyloid fibrils of Q145Stop and Q160Stop mutants,Citation7 are colored black. (B) View along the axis of a filament that is based on the core structure shown in (A). (C) Side view of the filament shown in (B). (D) Hydrophobicity profile of the Q160Stop mutant of humPrP.
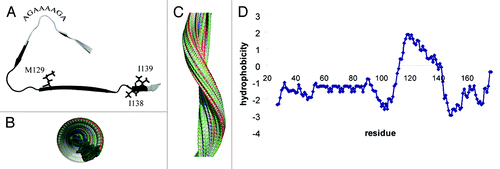
The maximum diameter within the β-helix motif shown in is approximately 5 nm. Electron microscopy, on the other hand, estimated the diameter of amyloid fibrils of the Y145X and Q160X stop mutants and of the peptide encompassing residues 108–143 (humPrP(108–143)) as 7–9 nm.Citation6,Citation7,Citation16 A diameter of 7–9 nm could only be reached for a single filament of humPrP(108–143) when the peptide would be fully extended, in disagreement to the identified β-helix structure. Thus, it is likely that multiple filaments come together in amyloid fibrils of prion stop mutants as suggested for N-terminally truncated PrPSc.Citation14,Citation15
Scanning transmission electron microscopy can potentially provide information about the number of filaments in amyloid fibrils.Citation17 However, even if the number of filaments can reliably be identified, the specific arrangement into multimers would remain unknown. Identification of the arrangement would require a sufficiently large number of accurate intermolecular distance restraints for example from solid-state NMR spectroscopy. To obtain insight into potential multimeric arrangements of the β-helix in the absence of additional experimental restraints, rigid body docking of either two or three filaments composed of stacked β-helix motifs was performed (). The docking solutions obtained from the ClusPro Web server (http://www.nrc.bu.edu/cluster) were ranked according to electrostatic, hydrophobic and van der Waals interactions.Citation18 Subsequently the solutions were grouped into 10 clusters. A potential two-filament arrangement that is consistent with a dimeric filament interface formed by residues Y128 and L130 is shown in . Note, that only a small number of the obtained docking solutions are physically possible in a full filament, as for many of the docking solutions a steric clash was present when the filament was extended beyond 20 β-helix motifs (data not shown). The maximum diameter in the dimeric β-helix is approximately 7 nm, thus at the lower end of the dimensions expected on the basis of electron microscopy.
Figure 2. Potential two- and three-filament arrangements of the β-helix of stop mutants of the human prion protein. The filament arrangements were obtained by ClusPro rigid body docking.Citation18 The view is along the fibril axis. (A) Dimeric β-helix. Shown is a representative structure that is consistent with a multimerization interface formed by residues Y128 and L130. (B) Trimeric β-helix. Shown is a representative structure that was obtained by rigid-body docking and is typical for trimeric left-handed β-helices. (C) Differences in oxidation of single side chains between amyloid fibrils and monomers of humPrP(108–143)—as determined from hydroxyl radical probing detected by mass spectrometryCitation7—are mapped onto a surface representation of the trimeric β-helix. Cleavage sites identified by limited proteolysis of PrPSc 19 are marked by stars. (D) Solvent protection levels of backbone amide protons determined by amide proton/deuterium exchange coupled to solution-state NMR spectroscopy.Citation7 Residues are color-coded according to experimental back-exchange ratios: Dark blue (I24h/I0 < 0.8), cyan (I24h/I0 = 0.8–1.0) and light blue (I24h/I0 > 1.0). Residues for which signal intensities could not be analyzed due to signal overlap are shown in gray. The hydrophobic residues L125, Y128 and L130 form the trimer interface.
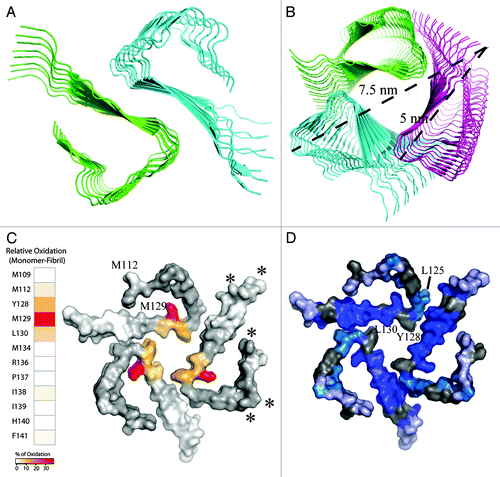
shows a representative structure of one of the clusters obtained from rigid-body docking of three filaments. Indeed, this was the only cluster obtained from rigid-body docking that showed an arrangement which is typical for a trimeric left-handed β-helix (). The interface in the β-helix trimer is formed by the hydrophobic residues L125, Y128 and L130 (). The maximum diameter of the β-helix trimer is approximately 7.5 nm. The experimental solvent accessibilities of single side chains, which we determined by hydroxyl radical probing coupled to mass spectrometry, are in good agreement with the structure of the trimeric β-helix (). The most pronounced reduction in solvent accessibility upon aggregation into amyloid fibrils was observed for the side chains of Y128, M129 and L130.Citation7 All three residues are part of the same β-strand (), indicating that both sides of this β-strand are less solvent accessible in amyloid fibrils. The reduced solvent accessibility is consistent with a hydrophobic trimer interface involving residues Y128 and L130 (). In clear contrast, oxidation of the N-terminal methionine M109 did not differ in the monomeric and fibrillar state. In addition, only small or no differences in oxidation levels were detected for I138 and I139, in line with the location of these two residues in the β-strand protruding from the β-helix (). Importantly, the strongly reduced reactivity of the M129 side chain upon incorporation into amyloid fibrils supports its burial in the amyloid core structure ().
In contrast to the side chain of M129, the side chain of M112 is located on the surface of the trimeric β-helix. Its surface localization is supported by hydroxyl radical probing, which revealed pronounced reactivity of the M112 side chain in amyloid fibrils. The solvent exposure of one face of the β-strand that harbors M112, 111HMAGA115, was further supported by the solvent exchange properties of backbone amide protons:Citation7 amide protons in the β-strand 111HMAGA115 had significantly lower solvent protection values than residues 121–139 which are involved in the trimer interface ( and ). Notably, the distinct differences in the solvent protection of residues 109–120 and 121–139 could not be explained on the basis of a dimeric filament arrangement as shown in .
As shown above, the trimeric β-helix is in agreement with a large body of data. In addition, internal cleavage sites at positions 117, 119, 135, 139, 142 and 154, which were identified by limited proteolysis of PrPSc isolated from brains of hamsters infected with 263K and drowsy prions,Citation19 are located at positions that are at least partially accessible according to the trimeric β-helix model (). The trimeric left-handed β-helix structure is also consistent with electron crystallography of two-dimensional crystals of N-terminally truncated PrPSc with the trimer core located in the peptide segment 89–140.Citation14,Citation15 At the same time, it should be kept in mind that studies on the amyloid-β peptide have revealed different multimeric filament assemblies despite a highly similar structure of the structure of a single filament. The different multimeric arrangements resulted in structural polymorphism.Citation20 Thus, other dimeric, trimeric or even higher oligomeric filament arrangements of the left-handed β-helix might be possible in prion aggregates.
While our data established a common core structure of stop mutants of humPrP, solvent exchange and site-directed spin-labeling of amyloid fibrils of recombinant humPrP(90–231) suggested that the core of amyloid fibrils maps to the C-terminal part of PrP encompassing residues 160–230.Citation21 However, humPrP(90–231) fibrils have an atypically short proteinase K resistant core of 8–12 kDa compared with brain-derived aggregated PrP, for which the PK resistant core usually starts around residue ~90.Citation22 humPrP(90–231) aggregates that were seeded with PrPSc have an N-terminally extended core with a significant degree of order within residues 117–133,Citation23 i.e., within the β-helix region of the amyloid core structure of . Moreover, residues 112–119 that form the N-terminal β-strand and the first loop of the β-helix () were reported as important for replication of the infectious agent.Citation24,Citation25 In brain-derived PrPSc the entire region from residue ~80–90 to the C-terminus changes its conformation to a hydrogen-bonded structure.Citation26 The data might be brought in agreement by a model in which the left-handed β-helix formed by residues 109–141 forms part of a larger aggregate structure in PrPSc.
In summary, we showed that fibrils of the Y145Stop and Q160Stop mutants of the human prion protein share a common amyloid structure. Thus, differences in the presentation of clinical symptoms in patients and the progress of the disease in patients carrying nonsense mutations might be caused by the kinetic characteristics of the aggregation process rather than differences in the structure of resulting aggregates. This, however, does not exclude that different prion types or prion strains might be related to different aggregate structures or differences in the multimeric arrangement of filaments that have the same basic core structure.
| Abbreviations: | ||
| CJD | = | Creutzfeldt-Jakob disease |
| GSS | = | Gerstmann-Sträussler-Scheinker |
| PrP | = | prion protein |
| humPrP | = | human prion protein |
| PrPSc | = | misfolded PrP |
Acknowledgments
I thank Romina V. Hofele and Luis Fonseca for preparation of Figures 2C and 2D, respectively, and Dr Walter J. Schulz-Schäffer for fruitful discussions. This work was supported by the Cluster of Excellence and DFG Research Center “Nanoscale Microscopy and Molecular Physiology of the Brain.”
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 2008; 3:11 - 40; http://dx.doi.org/10.1146/annurev.pathmechdis.3.121806.154326; PMID: 18233951
- Parchi P, Gambetti P. Human prion diseases. Curr Opin Neurol 1995; 8:286 - 93; http://dx.doi.org/10.1097/00019052-199508000-00007; PMID: 7582044
- Finckh U, Müller-Thomsen T, Mann U, Eggers C, Marksteiner J, Meins W, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet 2000; 66:110 - 7; http://dx.doi.org/10.1086/302702; PMID: 10631141
- Jayadev S, Nochlin D, Poorkaj P, Steinbart EJ, Mastrianni JA, Montine TJ, et al. Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol 2011; 69:712 - 20; http://dx.doi.org/10.1002/ana.22264; PMID: 21416485
- Kundu B, Maiti NR, Jones EM, Surewicz KA, Vanik DL, Surewicz WK. Nucleation-dependent conformational conversion of the Y145Stop variant of human prion protein: structural clues for prion propagation. Proc Natl Acad Sci U S A 2003; 100:12069 - 74; http://dx.doi.org/10.1073/pnas.2033281100; PMID: 14519851
- Watzlawik J, Skora L, Frense D, Griesinger C, Zweckstetter M, Schulz-Schaeffer WJ, et al. Prion protein helix1 promotes aggregation but is not converted into beta-sheet. J Biol Chem 2006; 281:30242 - 50; http://dx.doi.org/10.1074/jbc.M605141200; PMID: 17012240
- Skora L, Fonseca-Ornelas L, Hofele RV, Riedel D, Giller K, Watzlawik J, et al. Burial of the polymorphic residue 129 in amyloid fibrils of prion stop mutants. J Biol Chem 2013; 288:2994 - 3002; http://dx.doi.org/10.1074/jbc.M112.423715; PMID: 23209282
- Skora L, Zweckstetter M. Determination of amyloid core structure using chemical shifts. Protein Sci 2012; 21:1948 - 53; http://dx.doi.org/10.1002/pro.2170; PMID: 23033250
- Helmus JJ, Surewicz K, Nadaud PS, Surewicz WK, Jaroniec CP. Molecular conformation and dynamics of the Y145Stop variant of human prion protein in amyloid fibrils. Proc Natl Acad Sci U S A 2008; 105:6284 - 9; http://dx.doi.org/10.1073/pnas.0711716105; PMID: 18436646
- Wüthrich K, Riek R. Three-dimensional structures of prion proteins. Adv Protein Chem 2001; 57:55 - 82; http://dx.doi.org/10.1016/S0065-3233(01)57018-7; PMID: 11447697
- Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F, et al. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci U S A 1996; 93:744 - 8; http://dx.doi.org/10.1073/pnas.93.2.744; PMID: 8570627
- Jansen C, Parchi P, Capellari S, Vermeij AJ, Corrado P, Baas F, et al. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol 2010; 119:189 - 97; http://dx.doi.org/10.1007/s00401-009-0609-x; PMID: 19911184
- Tagliavini F, Prelli F, Ghiso J, Bugiani O, Serban D, Prusiner SB, et al. Amyloid protein of Gerstmann-Sträussler-Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N-terminal glycine at codon 58. EMBO J 1991; 10:513 - 9; PMID: 1672107
- Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Natl Acad Sci U S A 2004; 101:8342 - 7; http://dx.doi.org/10.1073/pnas.0402254101; PMID: 15155909
- Wille H, Govaerts C, Borovinskiy A, Latawiec D, Downing KH, Cohen FE, et al. Electron crystallography of the scrapie prion protein complexed with heavy metals. Arch Biochem Biophys 2007; 467:239 - 48; http://dx.doi.org/10.1016/j.abb.2007.08.010; PMID: 17935686
- Salmona M, Morbin M, Massignan T, Colombo L, Mazzoleni G, Capobianco R, et al. Structural properties of Gerstmann-Straussler-Scheinker disease amyloid protein. J Biol Chem 2003; 278:48146 - 53; http://dx.doi.org/10.1074/jbc.M307295200; PMID: 12970341
- Goldsbury C, Baxa U, Simon MN, Steven AC, Engel A, Wall JS, et al. Amyloid structure and assembly: insights from scanning transmission electron microscopy. J Struct Biol 2011; 173:1 - 13; http://dx.doi.org/10.1016/j.jsb.2010.09.018; PMID: 20868754
- Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res 2004; 32:Web Server issue W96-9; http://dx.doi.org/10.1093/nar/gkh354; PMID: 15215358
- Sajnani G, Pastrana MA, Dynin I, Onisko B, Requena JR. Scrapie prion protein structural constraints obtained by limited proteolysis and mass spectrometry. J Mol Biol 2008; 382:88 - 98; http://dx.doi.org/10.1016/j.jmb.2008.06.070; PMID: 18621059
- Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci U S A 2008; 105:18349 - 54; http://dx.doi.org/10.1073/pnas.0806270105; PMID: 19015532
- Lu X, Wintrode PL, Surewicz WK. Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc Natl Acad Sci U S A 2007; 104:1510 - 5; http://dx.doi.org/10.1073/pnas.0608447104; PMID: 17242357
- Bocharova OV, Breydo L, Salnikov VV, Gill AC, Baskakov IV. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob Disease. Protein Sci 2005; 14:1222 - 32; http://dx.doi.org/10.1110/ps.041186605; PMID: 15802644
- Smirnovas V, Kim JI, Lu X, Atarashi R, Caughey B, Surewicz WK. Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J Biol Chem 2009; 284:24233 - 41; http://dx.doi.org/10.1074/jbc.M109.036558; PMID: 19596861
- Wopfner F, Weidenhöfer G, Schneider R, von Brunn A, Gilch S, Schwarz TF, et al. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol 1999; 289:1163 - 78; http://dx.doi.org/10.1006/jmbi.1999.2831; PMID: 10373359
- Hölscher C, Delius H, Bürkle A. Overexpression of nonconvertible PrPc delta114-121 in scrapie-infected mouse neuroblastoma cells leads to trans-dominant inhibition of wild-type PrP(Sc) accumulation. J Virol 1998; 72:1153 - 9; PMID: 9445012
- Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol 2011; 18:504 - 6; http://dx.doi.org/10.1038/nsmb.2035; PMID: 21441913
- Simons KT, Bonneau R, Ruczinski I, Baker D. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins 1999; Suppl 3 171 - 6; http://dx.doi.org/10.1002/(SICI)1097-0134(1999)37:3+<171::AID-PROT21>3.0.CO;2-Z; PMID: 10526365