Abstract
While the conversion of the normal form of prion protein to a conformationally distinct pathogenic form is recognized to be the primary cause of prion disease, it is not clear how this leads to spongiform change, neuronal dysfunction and death. Mahogunin ring finger-1 (Mgrn1) and Attractin (Atrn) null mutant mice accumulate vacuoles throughout the brain that appear very similar to those associated with prion disease, but they do not accumulate the protease-resistant scrapie form of the prion protein or become sick. A study demonstrating an interaction between cytosolically-exposed prion protein and MGRN1 suggested that disruption of MGRN1 function may contribute to prion disease pathogenesis, but we recently showed that neither loss of MGRN1 nor MGRN1 overexpression influences the onset or progression of prion disease following intracerebral inoculation with Rocky Mountain Laboratory prions. Here, we show that loss of ATRN also has no effect on prion disease onset or progression and discuss possible mechanisms that could cause vacuolation of the central nervous system in Mgrn1 and Atrn null mutant mice and whether the same pathways might contribute to this intriguing phenotype in prion disease.
Introduction
One of the most striking and intriguing neuropathological hallmarks of prion disease is the widespread appearance of vacuoles in the central nervous system (CNS). Other features, such as neuronal loss and gliosis, are associated with many neurodegenerative disorders, but spongiform change is found in fewer, seemingly unrelated disorders, including prion disease, viral infection (i.e., HIV encephalopathy) and metabolic diseases.Citation1-Citation4 In transmissible spongiform encephalopathies, vacuoles are most commonly observed within the neuropil and contain membrane fragments and amorphous material. Both the mechanism by which they arise and their role in prion disease remain unclear. The lysosomal pathway is thought to be involved since spongiform degeneration is reduced in the brains of aged prion-infected mink homozygous for the Aleutian allele, which is a loss-of-function mutation in the lysosomal trafficking regulator (LYST) gene.Citation5,Citation6 Interestingly, disease incubation time, clinical signs and gliosis are not altered, suggesting that the effect of the LYST mutation on the lysosomal pathway is specific to the formation of vacuoles and that this phenotype can be separated from other aspects of prion disease.
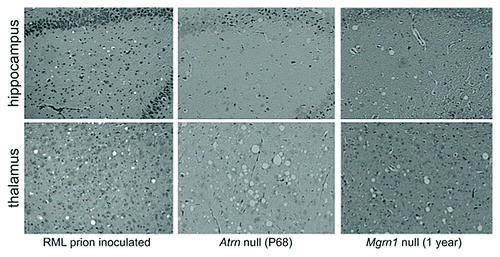
Vacuolation and gliosis of the CNS without accumulation of the protease-resistant scrapie form of the prion protein (PrPSc) occurs in several mouse mutants, including Mahogunin ring finger-1 (Mgrn1) and Attractin (Atrn) null animals.Citation7 These mice develop widespread spongiform encephalopathy with a similar anatomical distribution and histological appearance to that associated with prion diseases () but do not become clinically ill. If there is a shared mechanism underlying this phenotype in prion diseases and Mgrn1 and Atrn mutant mice, then understanding the normal function of the MGRN1 and ATRN proteins in the brain and the pathways disrupted by their absence will provide insight into the cause of vacuolation in prion disease.
Figure 1. Histologically similar spongiform degeneration of the CNS with a similar anatomical distribution is observed in RML prion-inoculated mice and mice lacking ATRN or MGRN1. Top row: CA3 region of hippocampus; bottom row: thalamus.
RML Prions Act Through MGRN1 and ATRN-Independent Pathways
ATRN is a widely expressed type I transmembrane protein, the CNS function of which remains unknown.Citation8 MGRN1 is a RING-domain family E3 ubiquitin ligase, the only target of which identified to date is the endosomal sorting complex required for transport-I (ESCRT-I) protein, tumor susceptibility gene 101 protein (TSG101).Citation9,Citation10 TSG101 is required for the sorting of ubiquitinated proteins into multivesicular bodies (MVBs) and MVB formation.Citation11 Its depletion from mammalian cells results in the accumulation of vacuolar, multicisternal endosomes,Citation12 which is likely to have an indirect effect on membrane homeostasis. Loss of MGRN1 causes a partial loss of TSG101 function that leads to abnormalities in early and late endosomes and prolonged epidermal growth factor receptor signaling. In cultured cells, cytosolically-exposed PrP was shown to interact with and functionally sequester MGRN1, resulting in endosomal defects similar to those observed in cells in which Mgrn1 expression was knocked down by siRNA.Citation13 These defects could be rescued by Mgrn1 overexpression. Cytosolically-exposed PrP has been detected in both transmissible and inherited forms of prion disease,Citation14,Citation15 suggesting that CNS vacuolation associated with these diseases might be caused by disrupted MGRN1 function. If this were the case, a complete absence of MGRN1 might be expected to accelerate the onset of pathogenesis, while overexpression of Mgrn1 could be protective.
We tested whether MGRN1 plays a role in the pathogenesis of transmissible prion disease by inoculating Mgrn1 null mutant (Mgrn1md-nc/md-nc) mice and transgenic mice that overexpress Mgrn1 with Rocky Mountain Laboratory (RML) prions. MGRN1 levels had no effect on PrPSc accumulation or the onset, progression, symptoms or histopathology of disease.Citation16 This suggests that cytosolically-exposed PrP either is not produced or does not play a significant role in the pathogenesis of prion disease caused by RML-prions. The fact that disease progression was not altered in mice lacking MGRN1 also suggests that partial disruption of TSG101-dependent endosomal trafficking does not significantly impact the conversion of PrPC to PrPSc. This is consistent with a report that siRNA knockdown of ALIX (an ESCRT-associated protein) does not affect the production of PrPSc or the intracellular localization of PrPC or PrPSc in RML prion-infected GT1 or CAD cells.Citation17
Mice homozygous for null alleles of Atrn also develop CNS vacuolation but at a much earlier age than Mgrn1 null mutants.Citation18 If ATRN and MGRN1 act in the same pathway, this would suggest that loss of ATRN causes a more severe disruption of that pathway and that prion disease might therefore be more likely to progress more rapidly in Atrn null mutant mice than in wild-type or Mgrn1 null mutant mice. To test this, female Atrn null (Atrnmg-3J/mg-3J) and wild-type control mice were inoculated with RML prions at 37–52 d of age. No significant genotype-dependent differences were observed in incubation or survival time (), indicating that RML-prions do not act through an ATRN-dependent pathway to cause disease. Although the role of ATRN in the CNS is not known, it appears to be required for normal membrane homeostasis since Atrn null mutant mice show age-dependent loss of detergent-resistant glycolipid-enriched membrane domains (sometimes referred to as membrane rafts).Citation19 When brain protein lysates from adult Atrn null mutant mice are centrifuged through a discontinuous sucrose gradient, proteins normally found in detergent-resistant membrane fractions (i.e., flotillin-1) relocalize to high-density membrane fractions (). The distribution of membrane raft proteins was normal in brain lysates from both young (post-natal day 5) Atrn null mutant mice and adult animals homozygous for a hypomorphic allele (Atrnmg-L), neither of which have histologically-detectable vacuoles in their brain (; refs. Citation18 and Citation19). Localization of PrPC to lipid rafts has been implicated in its conversion to PrPSc,Citation20 but the existence of these membrane domains remains controversial.Citation21 The fact that neither the onset nor progression of disease is delayed in Atrn null mutant mice inoculated with RML-prions at an age by which they already show spongiform change and have begun to lose membrane raft domains suggests that the conversion of PrPC to PrPSc can occur even as the ability of cells in the CNS to maintain membrane rafts is diminishing.
Figure 2. Kaplan-Meier plot showing health status following RML prion inoculation of Atrn null (C3HeB/FeJ-Atrnmg-3J/mg-3J) and control (C3HeB/FeJ) mice. A total of 8 (5 female, 3 male) mutant and 10 (6 female, 4 male) control mice were inoculated intracerebrally with RML-prions at 37−52 d of age and monitored daily for general health status and thrice weekly for neurological symptoms. There was no significant difference in the time to onset of clinical symptoms of Atrn-deficient mice relative to controls.
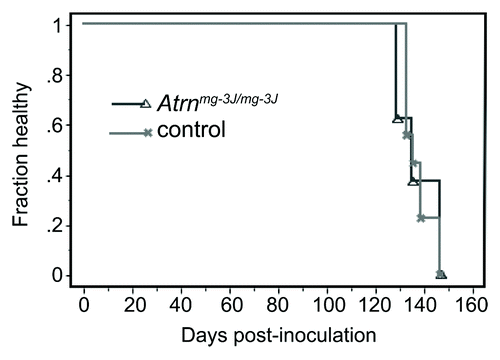
Figure 3.Atrn but not Mgrn1 null mutant mice show age-dependent loss of membrane raft domains. Brain proteins were extracted in MES buffered saline containing 0.1% Triton-X100 and protease inhibitors from animals of the indicated genotypes and ages. Lysates were centrifuged through a discontinuous sucrose gradient as described in ref.Citation40 and fractions analyzed for the presence of the raft marker flotillin-1 by western blotting. Detergent-resistant membrane rafts float to the interface between 5% and 35% sucrose (detergent-resistant fraction, DRF). In samples from the brains of wild-type C3H/HeJ mice, young (postnatal day 5, P5) Atrn null mutants, adult mice homozygous for the hypomorphic Atrnmg-L allele and aged (one year old) Mgrn1 null mutant mice, the vast majority of flotillin-1 localized to the DRF. In samples from the brains of adult Atrn null mutant mice (postnatal day 68 (P68) shown), there is progressive redistribution of flotillin-1 from the DRF to the detergent-soluble fraction (DSF), consistent with loss of membrane rafts.

Are All Vacuoles Created Equal?
MGRN1 and ATRN are unlikely to act downstream of PrPSc to mediate prion disease pathogenesis since neither loss of MGRN1 or ATRN nor overexpression of MGRN1 altered the course of disease in RML-prion inoculated animals. The fact that Mgrn1 and Atrn null mice develop spongiform change without overt neurological symptoms or illness suggests vacuoles may be a secondary phenotype, but understanding how they form could reveal primary mechanisms and perturbed pathways in prion disease, and it is possible that loss of MGRN1 or ATRN and the presence of PrPSc (and/or loss of PrPC) act on the same downstream pathway(s) to cause spongiform change. Loss of MGRN1 has been associated with disrupted TSG101-dependent endo-lysosomal trafficking but it is not known whether ATRN plays a role in this pathway, while age-dependent loss of lipid raft domains associated with loss of ATRN is not observed in the brains of Mgrn1 null mutant mice (). A phenotype that is shared by Atrn and Mgrn1 null mutant mice is elevated oxidative stress and mitochondrial dysfunction, apparent in the CNS by 1-mo of age.Citation22 Interestingly, mitochondrial diseases and mutations that disrupt mitochondrial function can cause spongiform encephalopathy,Citation23-Citation29 and mitochondrial dysfunction has been observed in the brains of scrapie-infected miceCitation30,Citation31 and hamsters,Citation32 as well as in mice lacking PrPC.Citation33 These observations suggest that reduced energy production due to mitochondrial dysfunction may contribute to CNS vacuolation in a variety of disorders, although in the case of prion disease this would most likely be a secondary effect of either loss of PrPC or accumulation of PrPSc in neuronal membranes. An alternative mechanism could involve disrupted membrane functions due to the accumulation of PrPSc, in prion diseases, or an effect of loss of ATRN or MGRN1 on membrane homeostasis. Consistent with this hypothesis, widespread CNS vacuolation is observed in mice homozygous for loss-of-function mutations in FIG4 homolog gene (Fig4) or Vac14,Citation34,Citation35 the products of which form a complex that regulates phosphatidylinositol 3,5-bisphosphate (PI[3,5]P2) levels and select membrane trafficking pathways.
There is evidence for both neuronal- and oligodendroglial-dependent forms of spongiform degeneration, suggesting that either interactions between these two cell types is important or different mechanisms are responsible for vacuolation involving distinct CNS cell types in different diseases. Neuronal depletion of endogenous PrPC in mice with established CNS prion disease (following inoculation with RML prions) prevented illness and reversed early spongiform change.Citation36 Similarly, neuron-specific expression of Figure 4 or Ndufs4 (which encodes NADH dehydrogenase [ubiquinone] Fe-S protein 4) was sufficient to rescue the vacuolation observed in the respective null mutant mice.Citation28,Citation37 On the other hand, transgenic expression of HIV type I proteins in mice under control of the oligodendrocyte-specific myelin basic protein promoter caused vacuolar lesions in the spinal cord, similar to those found in AIDS patients,Citation38 and mice homozygous for mutations in oligodendroglial-specific genes Kcnj10, which encodes a potassium channel, and Gjb1 and Gjc2 (connexin32; connexin47 double mutants) develop vacuoles predominantly in white matter tracts.Citation39 In the latter study, there was a positive correlation between the size and number of vacuoles and axonal activity, suggesting disrupted oligodendroglial-neuron interactions may play a role in vacuole formation. This could also explain why most forms of spongiform degeneration are associated with myelin defects. Further studies are needed to determine whether vacuoles themselves have a direct detrimental effect on CNS function and whether they form through perturbation of the same pathway in different diseases.
| Abbreviations: | ||
| CNS | = | central nervous system |
| HIV | = | human immunodeficiency virus |
| LYST | = | lysosomal trafficking regulator |
| PrP | = | prion protein |
| PrPC | = | cellular (normal) prion protein |
| PrPSc | = | scrapie form of prion protein |
| Mgrn1 | = | Mahogunin ring finger-1 gene |
| MGRN1 | = | mahogunin ring finger-1 protein |
| Atrn | = | Attractin gene |
| ATRN | = | attractin protein |
| ESCRT-I | = | endosomal sorting complex required for transport-I |
| TSG101 | = | tumor susceptibility gene 101 protein |
| MVBs | = | multivesicular bodies |
| siRNA | = | small interfering ribonucleic acid |
| RML | = | Rocky Mountain Laboratory |
| ALIX | = | ALG-2-interacting protein X (officially known as Programmed cell death 6-interacting protein, PDCD6IP) |
| Fig4 | = | FIG4 homolog gene |
| Vac14 | = | Vac14 homolog gene |
| PI(3,5)P2 | = | phosphatidylinositol 3,5-bisphosphate |
| Ndufs4 | = | NADH dehydrogenase (ubiquinone) Fe-S protein 4 gene |
| AIDS | = | acquired immunodeficiency syndrome |
| Kcnj10 | = | potassium inwardly-rectifying channel, subfamily J, member 10 gene |
| Gjb1 | = | gap junction protein, beta 1 gene |
| Gjc2 | = | gap junction protein, gamma 2 |
Acknowledgments
RML-prion inoculated mouse brain histology images shown in were provided by Dr Stephen DeArmond. RML-prion inoculation of Atrn null mutant mice was funded by National Institutes of Health program project grant NS041997 and performed in collaboration with Dr Gregory S. Barsh; that data are shown with his permission.
Submitted
04/10/2013
Accepted
05/15/2013
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Kumar S, Mattan NS, de Vellis J. Canavan disease: a white matter disorder. Ment Retard Dev Disabil Res Rev 2006; 12:157 - 65; http://dx.doi.org/10.1002/mrdd.20108; PMID: 16807907
- Martínez AJ, Sell M, Mitrovics T, Stoltenburg-Didinger G, Iglesias-Rozas JR, Giraldo-Velásquez MA, et al. The neuropathology and epidemiology of AIDS. A Berlin experience. A review of 200 cases. Pathol Res Pract 1995; 191:427 - 43; http://dx.doi.org/10.1016/S0344-0338(11)80730-2; PMID: 7479362
- Budka H. Neuropathology of prion diseases. Br Med Bull 2003; 66:121 - 30; http://dx.doi.org/10.1093/bmb/66.1.121; PMID: 14522854
- Brown GK, Squier MV. Neuropathology and pathogenesis of mitochondrial diseases. J Inherit Metab Dis 1996; 19:553 - 72; http://dx.doi.org/10.1007/BF01799116; PMID: 8884579
- Marsh RF, Sipe JC, Morse SS, Hanson RP. Transmissible mink encephalopathy. Reduced spongiform degeneration in aged mink of the Chediak-Higashi genotype. Lab Invest 1976; 34:381 - 6; PMID: 131218
- Anistoroaei R, Krogh AK, Christensen K. A frameshift mutation in the LYST gene is responsible for the Aleutian color and the associated Chédiak-Higashi syndrome in American mink. Anim Genet 2013; 44:178 - 83; http://dx.doi.org/10.1111/j.1365-2052.2012.02391.x; PMID: 22762706
- He L, Lu XY, Jolly AF, Eldridge AG, Watson SJ, Jackson PK, et al. Spongiform degeneration in mahoganoid mutant mice. Science 2003; 299:710 - 2; http://dx.doi.org/10.1126/science.1079694; PMID: 12560552
- Gunn TM, Miller KA, He L, Hyman RW, Davis RW, Azarani A, et al. The mouse mahogany locus encodes a transmembrane form of human attractin. Nature 1999; 398:152 - 6; http://dx.doi.org/10.1038/18217; PMID: 10086356
- Jiao J, Sun K, Walker WP, Bagher P, Cota CD, Gunn TM. Abnormal regulation of TSG101 in mice with spongiform neurodegeneration. Biochim Biophys Acta 2009; 1792:1027 - 35; http://dx.doi.org/10.1016/j.bbadis.2009.08.009; PMID: 19703557
- Kim BY, Olzmann JA, Barsh GS, Chin LS, Li L. Spongiform neurodegeneration-associated E3 ligase Mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol Biol Cell 2007; 18:1129 - 42; http://dx.doi.org/10.1091/mbc.E06-09-0787; PMID: 17229889
- Babst M, Odorizzi G, Estepa EJ, Emr SD. Mammalian tumor susceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic 2000; 1:248 - 58; http://dx.doi.org/10.1034/j.1600-0854.2000.010307.x; PMID: 11208108
- Doyotte A, Russell MR, Hopkins CR, Woodman PG. Depletion of TSG101 forms a mammalian “Class E” compartment: a multicisternal early endosome with multiple sorting defects. J Cell Sci 2005; 118:3003 - 17; http://dx.doi.org/10.1242/jcs.02421; PMID: 16014378
- Chakrabarti O, Hegde RS. Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell 2009; 137:1136 - 47; http://dx.doi.org/10.1016/j.cell.2009.03.042; PMID: 19524515
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 2002; 298:1781 - 5; http://dx.doi.org/10.1126/science.1073725; PMID: 12386337
- Hegde RS, Tremblay P, Groth D, DeArmond SJ, Prusiner SB, Lingappa VR. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature 1999; 402:822 - 6; http://dx.doi.org/10.1038/45574; PMID: 10617204
- Gunn TM, Silvius D, Bagher P, Sun K, Walker KK. MGRN1-dependent pigment-type switching requires its ubiquitination activity but not its interaction with TSG101 or NEDD4. Pigment Cell Melanoma Res 2013; 26:263 - 8; http://dx.doi.org/10.1111/pcmr.12059; PMID: 23253940
- Marijanovic Z, Caputo A, Campana V, Zurzolo C. Identification of an intracellular site of prion conversion. PLoS Pathog 2009; 5:e1000426; http://dx.doi.org/10.1371/journal.ppat.1000426; PMID: 19424437
- Gunn TM, Inui T, Kitada K, Ito S, Wakamatsu K, He L, et al. Molecular and phenotypic analysis of Attractin mutant mice. Genetics 2001; 158:1683 - 95; PMID: 11514456
- Azouz A, Gunn TM, Duke-Cohan JS. Juvenile-onset loss of lipid-raft domains in attractin-deficient mice. Exp Cell Res 2007; 313:761 - 71; http://dx.doi.org/10.1016/j.yexcr.2006.11.018; PMID: 17196964
- Taylor DR, Hooper NM. Role of lipid rafts in the processing of the pathogenic prion and Alzheimer’s amyloid-beta proteins. Semin Cell Dev Biol 2007; 18:638 - 48; http://dx.doi.org/10.1016/j.semcdb.2007.07.008; PMID: 17822928
- Leslie M. Mysteries of the cell. Do lipid rafts exist?. Science 2011; 334:1046 - 7; http://dx.doi.org/10.1126/science.334.6059.1046-b; PMID: 22116853
- Sun K, Johnson BS, Gunn TM. Mitochondrial dysfunction precedes neurodegeneration in mahogunin (Mgrn1) mutant mice. Neurobiol Aging 2007; 28:1840 - 52; http://dx.doi.org/10.1016/j.neurobiolaging.2007.07.012; PMID: 17720281
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, et al. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet 1998; 18:159 - 63; http://dx.doi.org/10.1038/ng0298-159; PMID: 9462746
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004; 119:121 - 35; http://dx.doi.org/10.1016/j.cell.2004.09.013; PMID: 15454086
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 2005; 3:e101; http://dx.doi.org/10.1371/journal.pbio.0030101; PMID: 15760270
- Kaul R, Gao GP, Balamurugan K, Matalon R. Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nat Genet 1993; 5:118 - 23; http://dx.doi.org/10.1038/ng1093-118; PMID: 8252036
- Egger J, Wynne-Williams CJ, Erdohazi M. Mitochondrial cytopathy or Leigh’s syndrome? Mitochondrial abnormalities in Spongiform encephalopathies. Neuropediatrics 1982; 13:219 - 24; http://dx.doi.org/10.1055/s-2008-1059627; PMID: 7155306
- Quintana A, Kruse SE, Kapur RP, Sanz E, Palmiter RD. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc Natl Acad Sci U S A 2010; 107:10996 - 1001; http://dx.doi.org/10.1073/pnas.1006214107; PMID: 20534480
- Puccio H, Simon D, Cossée M, Criqui-Filipe P, Tiziano F, Melki J, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet 2001; 27:181 - 6; http://dx.doi.org/10.1038/84818; PMID: 11175786
- Park JH, Kim BH, Park SJ, Jin JK, Jeon YC, Wen GY, et al. Association of endothelial nitric oxide synthase and mitochondrial dysfunction in the hippocampus of scrapie-infected mice. Hippocampus 2011; 21:319 - 33; http://dx.doi.org/10.1002/hipo.20753; PMID: 20082297
- Sisková Z, Mahad DJ, Pudney C, Campbell G, Cadogan M, Asuni A, et al. Morphological and functional abnormalities in mitochondria associated with synaptic degeneration in prion disease. Am J Pathol 2010; 177:1411 - 21; http://dx.doi.org/10.2353/ajpath.2010.091037; PMID: 20651247
- Choi SI, Ju WK, Choi EK, Kim J, Lea HZ, Carp RI, et al. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathol 1998; 96:279 - 86; http://dx.doi.org/10.1007/s004010050895; PMID: 9754961
- Miele G, Jeffrey M, Turnbull D, Manson J, Clinton M. Ablation of cellular prion protein expression affects mitochondrial numbers and morphology. Biochem Biophys Res Commun 2002; 291:372 - 7; http://dx.doi.org/10.1006/bbrc.2002.6460; PMID: 11846415
- Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007; 448:68 - 72; http://dx.doi.org/10.1038/nature05876; PMID: 17572665
- Zhang Y, Zolov SN, Chow CY, Slutsky SG, Richardson SC, Piper RC, et al. Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci U S A 2007; 104:17518 - 23; http://dx.doi.org/10.1073/pnas.0702275104; PMID: 17956977
- Mallucci G, Dickinson A, Linehan J, Klöhn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003; 302:871 - 4; http://dx.doi.org/10.1126/science.1090187; PMID: 14593181
- Ferguson CJ, Lenk GM, Jones JM, Grant AE, Winters JJ, Dowling JJ, et al. Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum Mol Genet 2012; 21:3525 - 34; http://dx.doi.org/10.1093/hmg/dds179; PMID: 22581779
- Goudreau G, Carpenter S, Beaulieu N, Jolicoeur P. Vacuolar myelopathy in transgenic mice expressing human immunodeficiency virus type 1 proteins under the regulation of the myelin basic protein gene promoter. Nat Med 1996; 2:655 - 61; http://dx.doi.org/10.1038/nm0696-655; PMID: 8640556
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, et al. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci 2006; 26:10984 - 91; http://dx.doi.org/10.1523/JNEUROSCI.0304-06.2006; PMID: 17065440
- Cota CD, Liu RR, Sumberac TM, Jung S, Vencato D, Millet YH, et al. Genetic and phenotypic studies of the dark-like mutant mouse. Genesis 2008; 46:562 - 73; http://dx.doi.org/10.1002/dvg.20432; PMID: 18821597