Abstract
A common feature of neurodegenerative diseases is the accumulation of disease-specific, aggregated protein species in the nervous system. Transmissible spongiform encephalopathies are universally fatal neurodegenerative diseases involving the transconformation and aggregation of prion proteins. At the cellular level macroautophagy has been identified as an efficient pathway for the clearance of these toxic protein aggregates. Hence, recent research has focused on the pharmacological manipulation of autophagy as a potential treatment for neurodegenerative diseases. Independent of their effects on the estrogen receptor, tamoxifen and its metabolite 4-hydroxytamoxifen are well known inducers of autophagy. However, we recently reported that the ability of 4-hydroxytamoxifen to clear prion infection is independent of autophagy. In contrast, we provide a model whereby perturbation of cholesterol metabolism, and not autophagy, is the main mechanism whereby 4-hydroxytamoxifen is able to exert its anti-prion effects. Thus, while tamoxifen, a widely available pharmaceutical, may have applications in prion therapy, prions may also represent a special case and may require different pharmacological interventions than other proteinopathies.
The Role of Protein Aggregates and Autophagy in Neurodegenerative Disease
A near universal feature of neurodegenerative diseases is the accumulation of aggregated proteins in the nervous system.Citation1,Citation2 Parkinson Disease, Huntington’s Disease, Alzheimer Disease, frontal-temporal lobe dementia, and prion diseases are universally fatal and involve the cruel psycho-motor deterioration of sufferers. Post-mortem pathological examination reveals that their brains are burdened with aggregated protein species unique to each disease. At the cellular level, aggregated proteins pose a particular problem to cells insofar as their disposal is difficult. Indeed, the ubiquitin-proteasome system (UPS) for degrading cytosolic proteins appears to be ineffective for detangling large protein aggregates and unfolding individual proteins for degradation.Citation3 This problem is particularly dire for the cells of non-renewable tissues, such as neurons, which cannot dilute the aggregated protein burden by cell division, nor undergo apoptosis and be replaced.Citation1,Citation2 However, there is one pathway that seems to be able to efficiently dispose of large protein aggregates - macroautophagy. Macroautophagy (hereafter referred to as autophagy) is the process whereby portions of the cytosol and damaged organelles are engulfed in autophagosomes; subsequently, these structures fuse with lysosomes (forming autolysosomes) to degrade their contents and recycle essential cell building blocks. The reliance of neurons on this pathway is borne out by experiments demonstrating that genetic knockout of essential autophagy genes in mice, atg5 or atg7, results in the accumulation of protein aggregates in the nervous system accompanied by neurodegeneration and death within months of birth.Citation4,Citation5 The autophagy pathway is thought to have evolved in eukaryotic organisms in order to recycle building blocks and salvage energy during times of nutrient deprivation. In higher organisms it may serve the additional function of enabling non-renewable tissues to rid themselves of the toxic protein aggregates that accumulate as the result of metabolic stress.Citation1 It may be the case that evolution has taken advantage of the chronic nutrient deprivation inevitably suffered by our ancestors to perform housekeeping functions, such as the clearance of toxic protein aggregates, as a by-product of salvaging nutrients and building blocks. The existence of large segments of society in the developed world that have never suffered chronic nutrient deprivation might be a contributing factor to the rise of Alzheimer Disease and other dementias.Citation6 With a looming wave of neurodegenerative diseases on the horizon, researchers have begun to wonder if autophagy could be exploited pharmacologically to stem the tide. Hence, a flurry of research activity has recently been undertaken to explore the potential of chemical agents to clear disease-specific aggregated protein species by stimulating autophagy.
Prion Protein Aggregates—A Special Case?
Transmissible spongiform encephalopathies are infectious, neurodegenerative diseases involving the abnormal folding of the cellular prion protein PrPC into a pathological conformer, PrPSc.Citation7 This malconformed state also leads to its accumulation as aggregates in both the cytoplasm of affected neurons and the interstitial spaces within the brains of afflicted individuals. It has been proposed that the amount of protease-resistant, aggregated prion protein is maintained by a balance between PrPSc formation by conversion of native PrPC and its destruction in lysosomes, which could be mediated by the autophagic pathway, as shown in the case of other neurodegenerative diseases of protein aggregation.Citation8 However, the role of autophagy in prion diseases in highly debated.Citation9 The appearance of multi-vesicular bodies and autophagic vacuoles in both prion-infected, neuronal cells in cultureCitation10 and in brain biopsies from prion-infected patientsCitation11,Citation12 suggested a protective role in the disease. Alternatively, it was proposed that autophagy may contribute to the spongiform changes that are a pathological hallmark of prion-affected brains, and may be activated by apoptosis.Citation11-Citation13
We have recently explored the question of the biological role of autophagy in prion infection and disease and looked more closely into the molecular interplay between autophagy, prion propagation, trafficking and clearance using a tandem fluorescent reporter, LC3 (tf-LC3), containing both GFP and RFP.Citation14 With tf-LC3 it is possible to distinguish autophagosomes from autolysosomes on the basis of the differential sensitivity of the green and red fluorophores to the acidic conditions in the latter compartment.Citation15 Using this tool we found that chronic scrapie infection enhances autophagic fluxCitation14 but not autophagy per se, which is already active in neuronal cell culture as previously shown in primary neurons.Citation16,Citation17 Despite the increase in autophagic flux, we observed that PrPSc was mostly absent from autophagic vesicles, even when lysosomal degradation was impaired. This raised the possibility that PrPSc was not processed by the autophagic machinery.
To further investigate the effects of autophagy on prion infection we tested tamoxifen along with a panel of other autophagy inducers, including rapamycin and lithium chloride. Surprisingly, tamoxifen was the only compound to consistently cause a substantial decrease in proteinase-K-resistant PrPSc. Although it has pleiotropic effects, tamoxifen is a well-known inducer of autophagy.Citation18,Citation19 Puzzled by these results we decided to test the active metabolite of tamoxifen, 4-hydroxytamoxifen (OHT), which had been reported to be a somewhat weaker inducer of autophagy based on its reduced induction of Beclin-1.Citation20 Contrary to expectations, we consistently found OHT to be a more potent anti-prion agent than tamoxifen insofar as its ability to clear proteinase-K-resistant prion infection from cultured cells. This result suggested that autophagy was not responsible for the anti-prion effects of tamoxifen and OHT.Citation14 We tested this hypothesis directly using a gene-silencing approach. Inhibition of autophagy using anti-atg7 siRNA did not affect the ability of OHT to reduce PrPSc content in infected cells, suggesting that autophagy was not involved in this process. Furthermore, prolonged knockdown of Atg7 did not lead to an accumulation of PrPSc above the levels observed in untreated cells, as would be expected if it were being degraded by autophagy.
Having ruled out autophagy we began to investigate other possible mechanisms to explain the anti-prion effects of tamoxifen and OHT. Interestingly, we observed that prion degradation seemed to depend upon degradation in lysosomes. However, it is important to note that delivery to lysosomes did not appear to occur by the autophagic route given that no increase in co-localization between autophagosomes and prion protein occurred upon induction by OHT. The localization of prions in lysosomes is not surprising in and of itself, for this is a well known site for prion distribution.Citation21 However, enrichment of PrP in lysosomes under OHT treatment coincided with the accumulation of cholesterol in these organelles (). This is interesting for a number of reasons.
Figure 1. Schematic presentation of PrP trafficking in infected cells and upon OHT treatment. In infected cells PrPC and PrPSc interact at the plasma membrane in cholesterol-rich lipid domains called lipid rafts. Upon internalization, both PrPC and PrPSc can recycle via the endosomal recycling compartment (ERC) or can be routed for degradation in lysosomes. Subcellular cholesterol distribution influences PrPSc trafficking in the endocytic pathway. In untreated, infected cells, the majority of PrP recycles through the cholesterol-rich ERC, supporting conversion of PrPC to PrPSc. Treatment with 4- hydroxytamoxifen (OHT) induces cholesterol accumulation in enlarged late endosomes (LE). PrPSc production and degradation defines the cellular load of infectious prions. We propose that 4-hydroxytamoxifen-induced changes in PrPSc trafficking favor PrPSc degradation. EE, early endosomes; LYS, lysosomes. (Adapted with permission from Marzo et al., 2013).Citation14
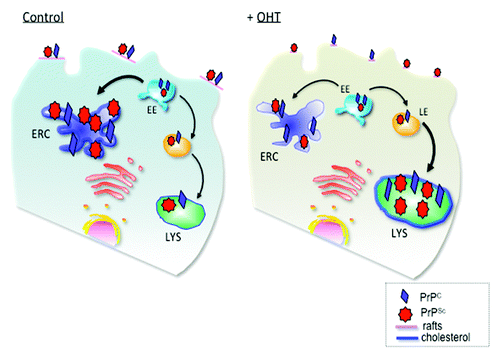
First, in contrast to other neurodegenerative diseases the prion protein is not exposed to the cytosolCitation22; it is a GPI-anchored, extrinsic membrane protein found on the exofacial leaflet of membranes. For this reason it may be able to evade detection by the autophagic machinery in the cytosol. Under normal circumstances it can be distributed to the Golgi, the endosomal recycling compartment (ERC), lysosomes and the plasma membrane.Citation23 When we treated cells with rapamycin, the classic autophagy-inducing agent, we did observe a slight reduction of proteinase-K-resistant PrPSc. It could be that a population of PrP molecules was degraded due to autophagy by virtue of being in the wrong subcellular locale at the wrong time. However the majority of PrP molecules may evade the autophagic machinery and escape degradation explaining the weak effect compared with tamoxifen and OHT.
Second, attenuation of prion protein expression/generation by pharmacological manipulation of cholesterol and glycosphingolipid metabolism has been well established.Citation24-Citation26 For example, inhibition of cholesterol synthesis with statins has been shown to delay the onset of symptoms and prolong survival times in scrapie-infected mice.Citation27 Furthermore, treatment with U18666A perturbs cholesterol trafficking causing a Niemann-Pick-like phenotype resulting in lysosomal accumulation of cholesterol and depletion of prion protein.Citation25 Tamoxifen/OHT treatment shares many features with U18666A treatment. Like U18666A, both tamoxifen and OHT inhibit cholesterol synthesis and perturb cholesterol traffickingCitation20 (). Therefore, it seems the anti-prion effects of tamoxifen/OHT are due to a classic mechanism of interference with cholesterol metabolism and not to autophagy as was first hypothesized.
Figure 2. Possible mechanisms of prion clearance by tamoxifen and OHT. Above dashed line: Tamoxifen and OHT may impair trafficking of cholesterol out of lysosomes. The high cholesterol environment favors PrPSc degradation and/or impairs PrPSc transconformation from PrPC substrate molecules (left lysosome). In contrast, without drug treatment normal cholesterol trafficking supports an environment that prevents PrPSc degradation and/or favors PrPSc formation (right lysosome). Below dashed line: Tamoxifen and OHT inhibit cholesterol biosynthesis at different steps. While tamoxifen treatment leads to zymosterol accumulation, OHT leads to desmosterol accumulation.Citation20 Desmosterol may be a more potent inhibitor of PrPSc formation or promoter of its degradation than zymosterol as indicated by the differently weighted lines impacting on PrPSc formation.
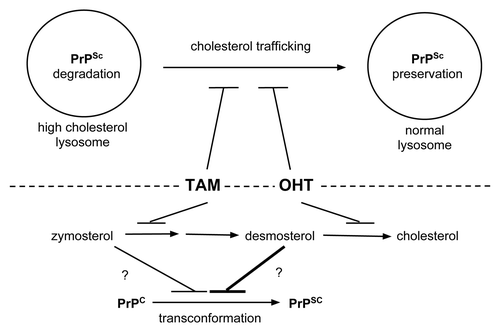
As described above, one of the properties shared by tamoxifen and OHT is the ability to inhibit cholesterol biosynthesis.Citation20 Interestingly, these closely related compounds induce the accumulation of distinct biosynthetic intermediates of cholesterol in treated cells (). Specifically, while tamoxifen leads to the preferential accumulation of zymosterol, OHT treatment causes the preferential accumulation of desmosterol. Interestingly, U18666A, a potent anti-prion compoundCitation23 also results in desmosterol accumulation by way of desmosterol reductase inhibition.Citation28 We used the polyene, macrolide filipin to detect the subcellular location of cholesterol by fluorescent microscopy.Citation14 Since filipin has been shown to bind sterols promiscuously,Citation29 it is unclear whether the sterol content we detected in lysosomes was cholesterol or one of these other biosynthetic intermediates. Therefore, it could be that an abundance of these intermediates in lysosomal membranes, in place of cholesterol, could perturb prion synthesis or promote its degradation. This possibility is supported by our results demonstrating that although inhibition of lysosomal proteases by NH4Cl treatment rescued prions from degradation induced by OHT treatment, the effect was not complete. This result might indicate that in addition to promoting prion degradation, tamoxifen and OHT may also foster a sterol-rich lysosomal membrane environment that inhibits the conversion of PrPC to PrPSc.
To further complicate the picture, cholesterol depletion itself has also been shown to induce autophagy.Citation20 Thus, the major anti-prion effects of tamoxifen and OHT are likely due to their inhibition of cholesterol biosynthesis and perturbation of lipid trafficking; in contrast, minor anti-prion activity may be due to autophagy, induced as a by-product of interference with cholesterol biosynthesis. Unlike other neurodegenerative diseases of protein aggregation, prions diseases may represent a special case, responding to perturbation of cholesterol metabolism rather than to autophagy. That being said, tamoxifen has also been shown to play a protective role in Alzheimer Disease and this effect was shown to be partly due to its effects on cholesterol biosynthesis.Citation30 In light of these data, it would be interesting to investigate the effect of tamoxifen and OHT on other proteinopathies.
Conclusion
We consistently observed tamoxifen and especially its metabolite OHT to be superior to known autophagy inducing agents (including rapamycin and lithium) in their ability to clear cellular prion infection. While autophagy induction may be a reliable means of degrading aggregated proteins of cytosolic origin such as huntingtin, tau, Aβ, or α-synuclein it may be less effective for prions, which are sequestered from the cytosol. We found the ability of tamoxifen and OHT to reduce prion burden to be independent of autophagy. Furthermore, we demonstrated that the anti-prion effect was due in part to degradation in lysosomes and that these lysosomes were laden with sterols similar to what was observed for another anti-prion compound: U18666A. Thus, while autophagy may be a worthy avenue for discovering therapies for other neurodegenerative diseases of protein aggregation, prions may represent a unique case. As the search continues for a prion therapeutic, it may be more fruitful to investigate the cholesterol biosynthesis and trafficking networks as classic pathways known to be involved in the generation and degradation of prion protein aggregates rather than those of autophagy.
Acknowledgments
We would like to thank all the members of the Zurzolo Lab for helpful discussion. DB was funded by a post-doctoral fellowship from the Canadian Louis Pasteur Foundation. The work in CZ lab was supported by European Union FP7 (Priority, Grant 222887), by ANR (Blanc, Priontraf 2009) and (DISCover, 2009 NEUR 00203) and by Pasteur-Weizmann Foundation (2010–2012).
Submitted
06/25/13
Revised
07/15/13
Accepted
07/18/13
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Cherra SJ 3rd, Dagda RK, Chu CT. Review: autophagy and neurodegeneration: survival at a cost?. Neuropathol Appl Neurobiol 2010; 36:125 - 32; PMID: 20202120
- Yao H, Zhao D, Khan SH, Yang L. Role of autophagy in prion protein-induced neurodegenerative diseases. Acta Biochim Biophys Sin (Shanghai) 2013; 45:494 - 502; http://dx.doi.org/10.1093/abbs/gmt022; PMID: 23459558
- Knaevelsrud H, Simonsen A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett 2010; 584:2635 - 45; http://dx.doi.org/10.1016/j.febslet.2010.04.041; PMID: 20412801
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Wang J, Ho L, Qin W, Rocher AB, Seror I, Humala N, et al. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J 2005; 19:659 - 61; PMID: 15650008
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/10.1073/pnas.95.23.13363; PMID: 9811807
- Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, et al. Chaperone-mediated autophagy at a glance. J Cell Sci 2011; 124:495 - 9; http://dx.doi.org/10.1242/jcs.073874; PMID: 21282471
- Heiseke A, Aguib Y, Schatzl HM. Autophagy, prion infection and their mutual interactions. Curr Issues Mol Biol 2010; 12:87 - 97; PMID: 19767652
- Schätzl HM, Laszlo L, Holtzman DM, Tatzelt J, DeArmond SJ, Weiner RI, et al. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J Virol 1997; 71:8821 - 31; PMID: 9343242
- Liberski PP, Brown DR, Sikorska B, Caughey B, Brown P. Cell death and autophagy in prion diseases (transmissible spongiform encephalopathies). Folia Neuropathol 2008; 46:1 - 25; PMID: 18368623
- Sikorska B, Liberski PP, Giraud P, Kopp N, Brown P. Autophagy is a part of ultrastructural synaptic pathology in Creutzfeldt-Jakob disease: a brain biopsy study. Int J Biochem Cell Biol 2004; 36:2563 - 73; http://dx.doi.org/10.1016/j.biocel.2004.04.014; PMID: 15325593
- Liberski PP, Jeffrey M. Tubulovesicular structures--the ultrastructural hallmark for transmissible spongiform encephalopathies or prion diseases. Folia Neuropathol 2004; 42:Suppl B 96 - 108; PMID: 16903145
- Marzo L, Marijanovic Z, Browman D, Chamoun Z, Caputo A, Zurzolo C. 4-hydroxytamoxifen leads to PrPSc clearance by conveying both PrPC and PrPSc to lysosomes independently of autophagy. J Cell Sci 2013; 126:1345 - 54; http://dx.doi.org/10.1242/jcs.114801; PMID: 23418355
- Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol 2009; 452:1 - 12; http://dx.doi.org/10.1016/S0076-6879(08)03601-X; PMID: 19200872
- Mitra S, Tsvetkov AS, Finkbeiner S. Protein turnover and inclusion body formation. Autophagy 2009; 5:1037 - 8; http://dx.doi.org/10.4161/auto.5.7.9291; PMID: 19838079
- Son JH, Shim JH, Kim KH, Ha JY, Han JY. Neuronal autophagy and neurodegenerative diseases. Exp Mol Med 2012; 44:89 - 98; http://dx.doi.org/10.3858/emm.2012.44.2.031; PMID: 22257884
- Bursch W, Ellinger A, Kienzl H, Török L, Pandey S, Sikorska M, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis 1996; 17:1595 - 607; http://dx.doi.org/10.1093/carcin/17.8.1595; PMID: 8761415
- Cho KS, Yoon YH, Choi JA, Lee S-J, Koh J-Y. Induction of autophagy and cell death by tamoxifen in cultured retinal pigment epithelial and photoreceptor cells. Invest Ophthalmol Vis Sci 2012; 53:5344 - 53; http://dx.doi.org/10.1167/iovs.12-9827; PMID: 22786900
- de Medina P, Payré B, Boubekeur N, Bertrand-Michel J, Tercé F, Silvente-Poirot S, et al. Ligands of the antiestrogen-binding site induce active cell death and autophagy in human breast cancer cells through the modulation of cholesterol metabolism. Cell Death Differ 2009; 16:1372 - 84; http://dx.doi.org/10.1038/cdd.2009.62; PMID: 19521424
- Peters PJ, Mironov A Jr., Peretz D, van Donselaar E, Leclerc E, Erpel S, et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J Cell Biol 2003; 162:703 - 17; http://dx.doi.org/10.1083/jcb.200304140; PMID: 12925711
- Lewis V, Hooper NM. The role of lipid rafts in prion protein biology. Front Biosci 2011; 16:151 - 68; http://dx.doi.org/10.2741/3681; PMID: 21196164
- Marijanovic Z, Caputo A, Campana V, Zurzolo C. Identification of an intracellular site of prion conversion. PLoS Pathog 2009; 5:e1000426; http://dx.doi.org/10.1371/journal.ppat.1000426; PMID: 19424437
- Hagiwara K, Nakamura Y, Nishijima M, Yamakawa Y. Prevention of prion propagation by dehydrocholesterol reductase inhibitors in cultured cells and a therapeutic trial in mice. Biol Pharm Bull 2007; 30:835 - 8; http://dx.doi.org/10.1248/bpb.30.835; PMID: 17409533
- Klingenstein R, Löber S, Kujala P, Godsave S, Leliveld SR, Gmeiner P, et al. Tricyclic antidepressants, quinacrine and a novel, synthetic chimera thereof clear prions by destabilizing detergent-resistant membrane compartments. J Neurochem 2006; 98:748 - 59; http://dx.doi.org/10.1111/j.1471-4159.2006.03889.x; PMID: 16749906
- Naslavsky N, Shmeeda H, Friedlander G, Yanai A, Futerman AH, Barenholz Y, et al. Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. J Biol Chem 1999; 274:20763 - 71; http://dx.doi.org/10.1074/jbc.274.30.20763; PMID: 10409615
- Mok SWF, Thelen KM, Riemer C, Bamme T, Gültner S, Lütjohann D, et al. Simvastatin prolongs survival times in prion infections of the central nervous system. Biochem Biophys Res Commun 2006; 348:697 - 702; http://dx.doi.org/10.1016/j.bbrc.2006.07.123; PMID: 16890918
- Cenedella RJ. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids 2009; 44:477 - 87; http://dx.doi.org/10.1007/s11745-009-3305-7; PMID: 19440746
- Arthur JR, Heinecke KA, Seyfried TN. Filipin recognizes both GM1 and cholesterol in GM1 gangliosidosis mouse brain. J Lipid Res 2011; 52:1345 - 51; http://dx.doi.org/10.1194/jlr.M012633; PMID: 21508255
- Peri A, Benvenuti S, Luciani P, Deledda C, Cellai I. Membrane cholesterol as a mediator of the neuroprotective effects of estrogens. Neuroscience 2011; 191:107 - 17; http://dx.doi.org/10.1016/j.neuroscience.2011.03.011; PMID: 21396986